
Exploration of the Activation Mechanism of the Epigenetic Regulator MLL3: A QM/MM Study

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
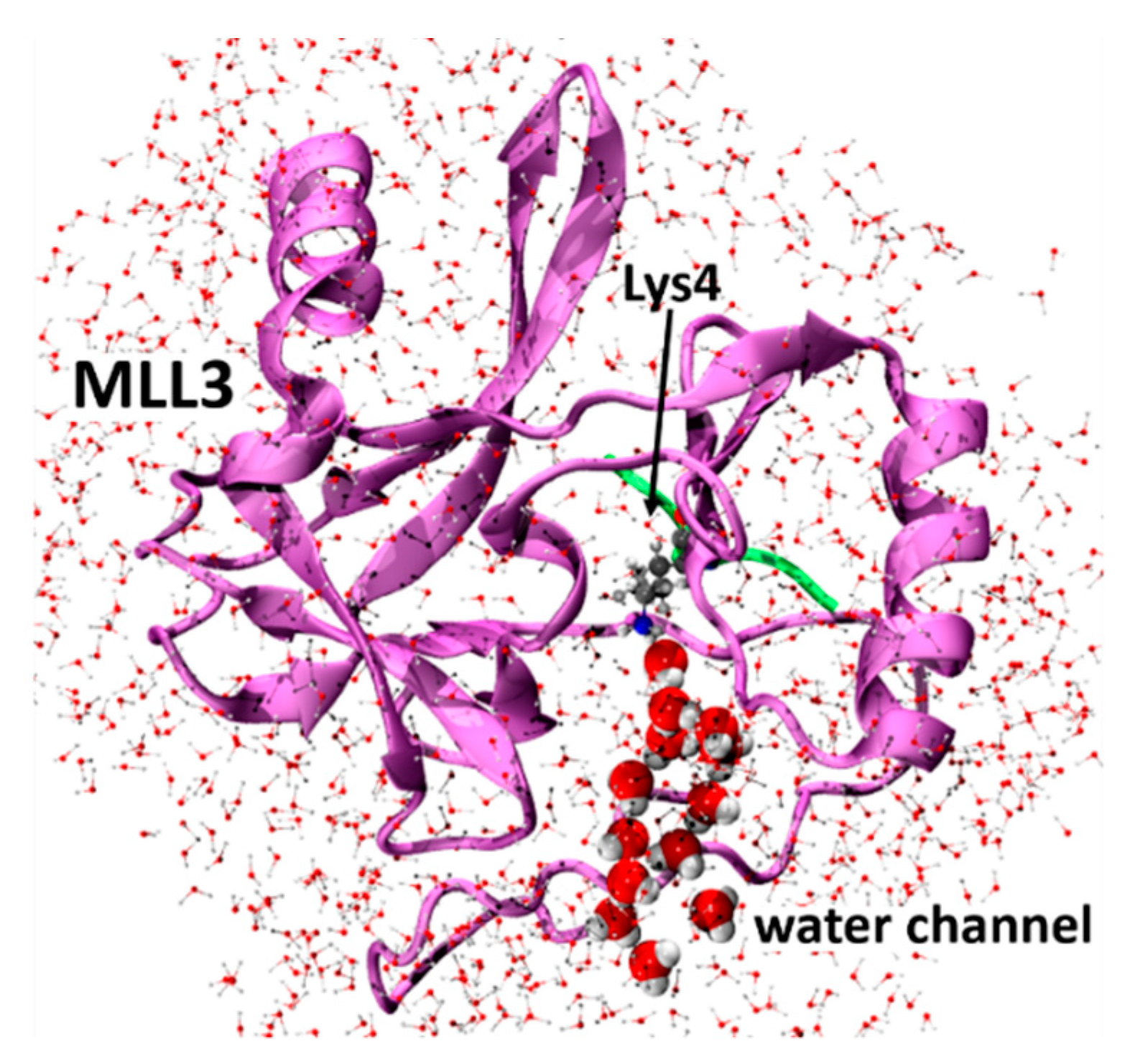
2.1. Model Preparation and MD Simulations
2.2. QM/MM Calculations
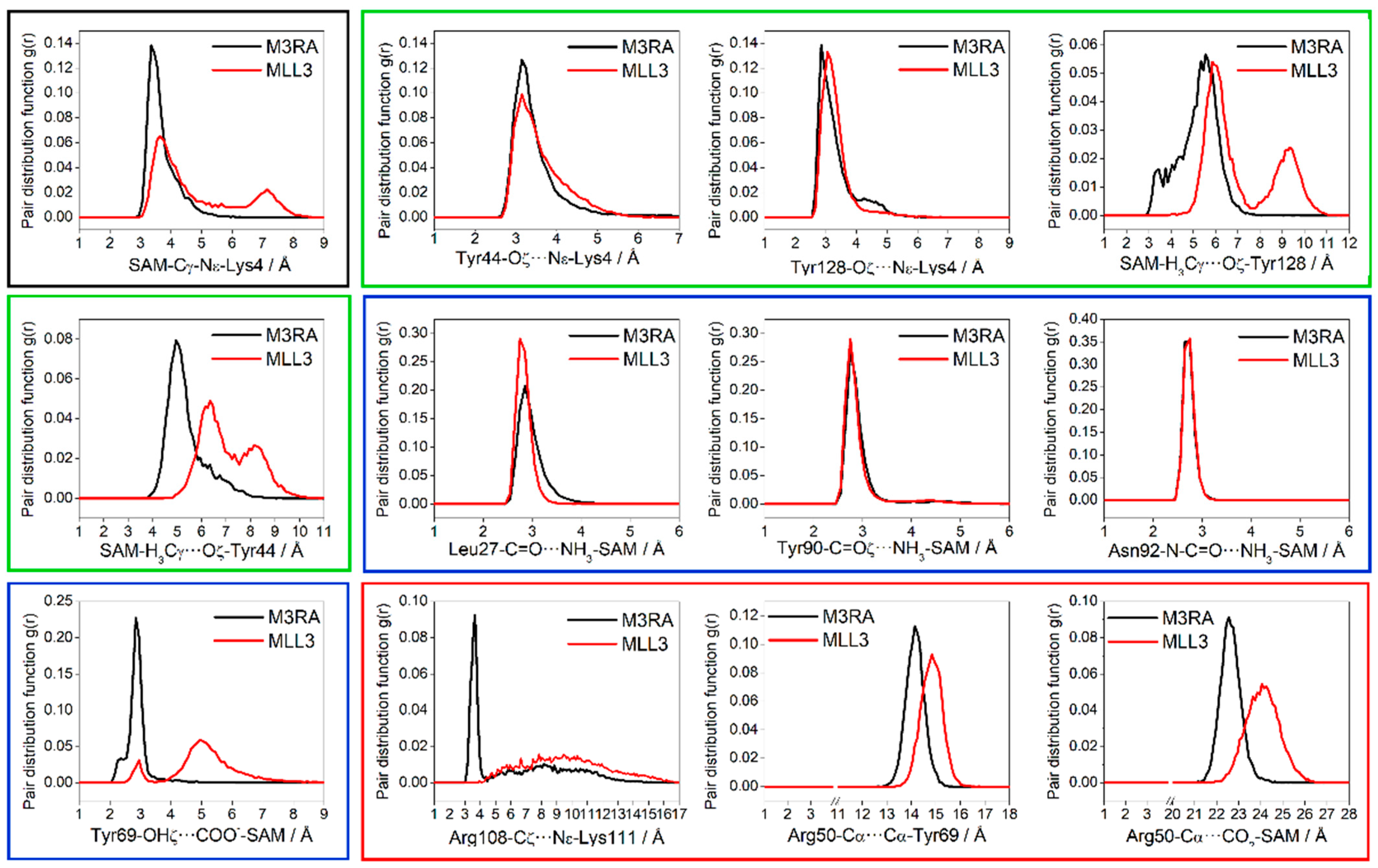
3. Results and Discussion
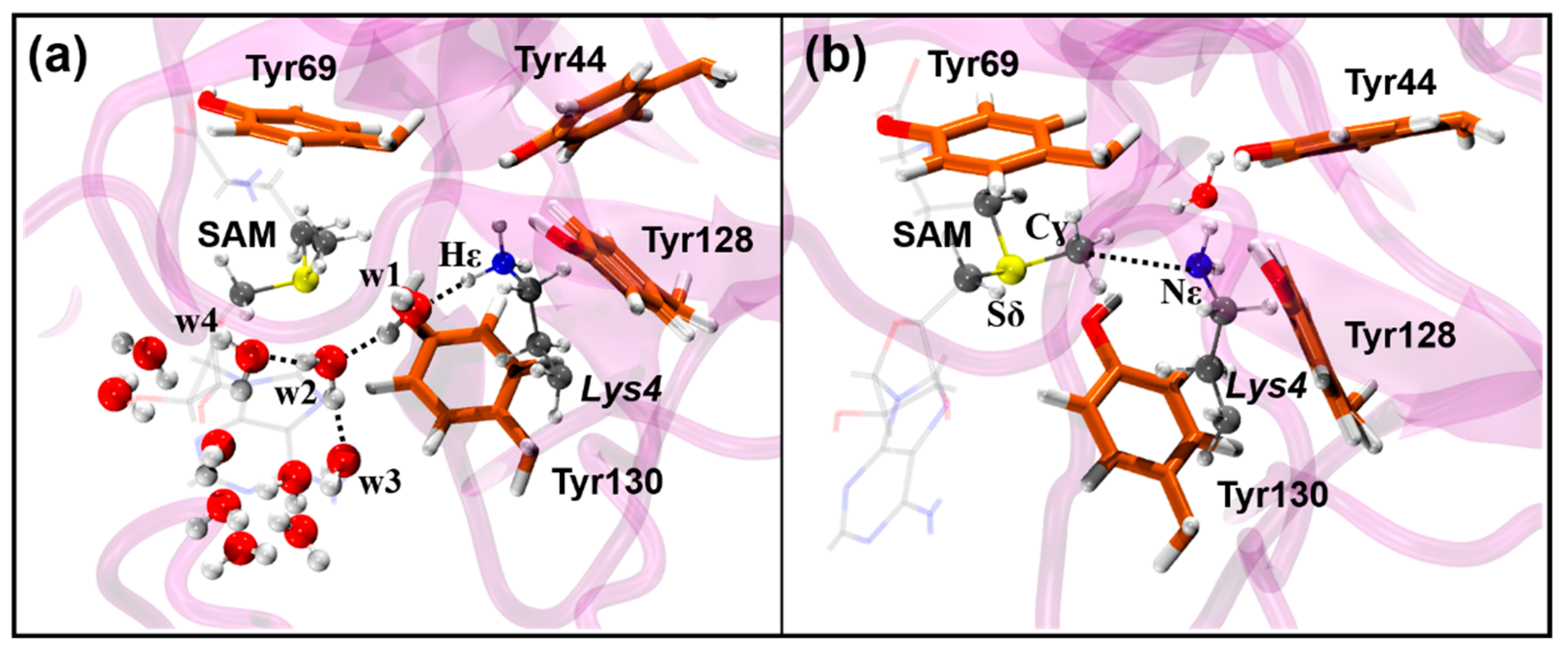
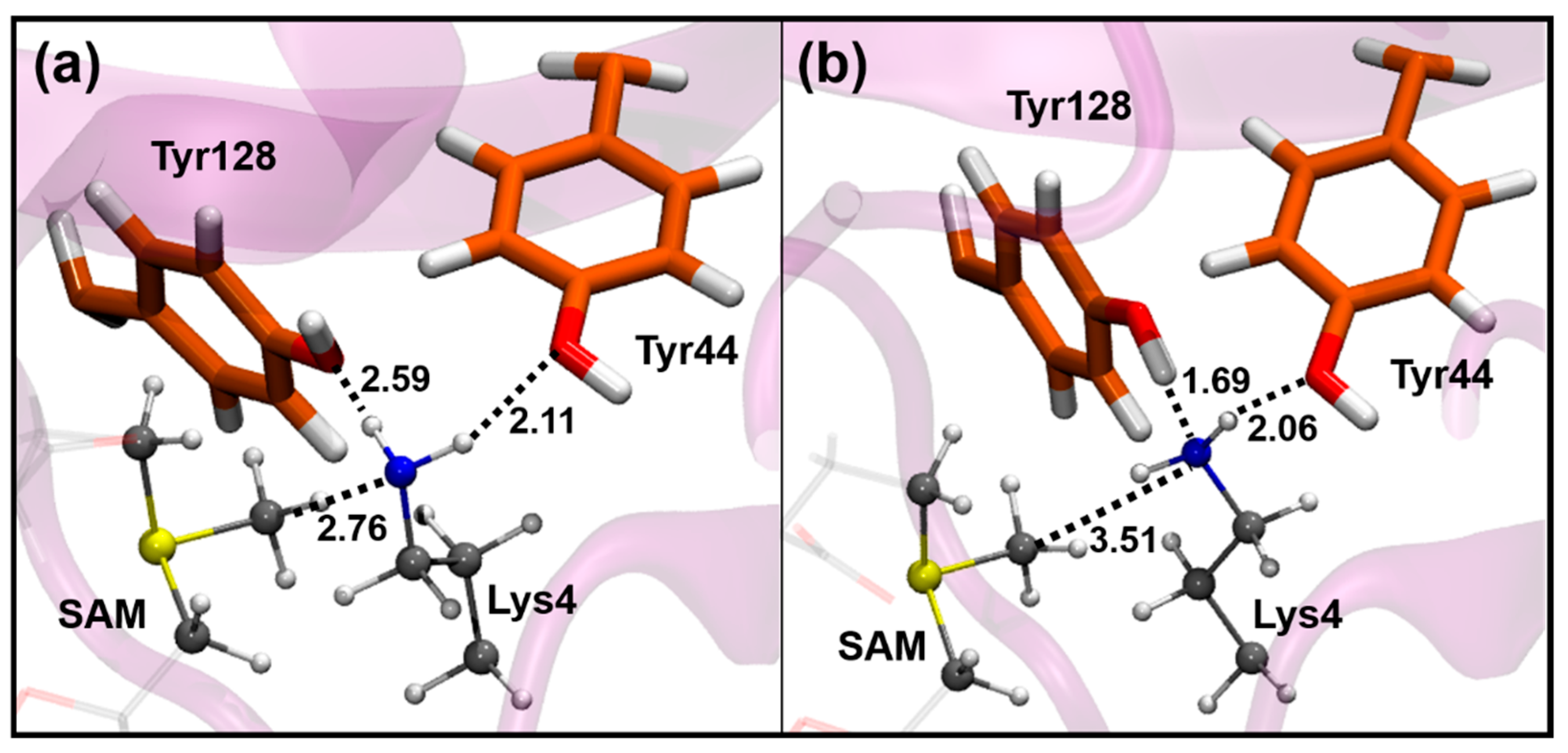
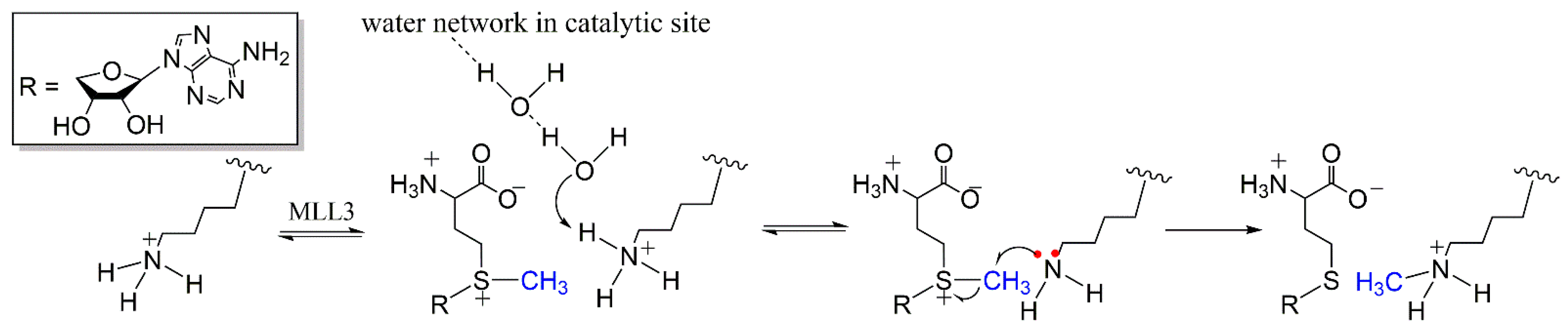
3.1. Activation of the Deprotonation Process
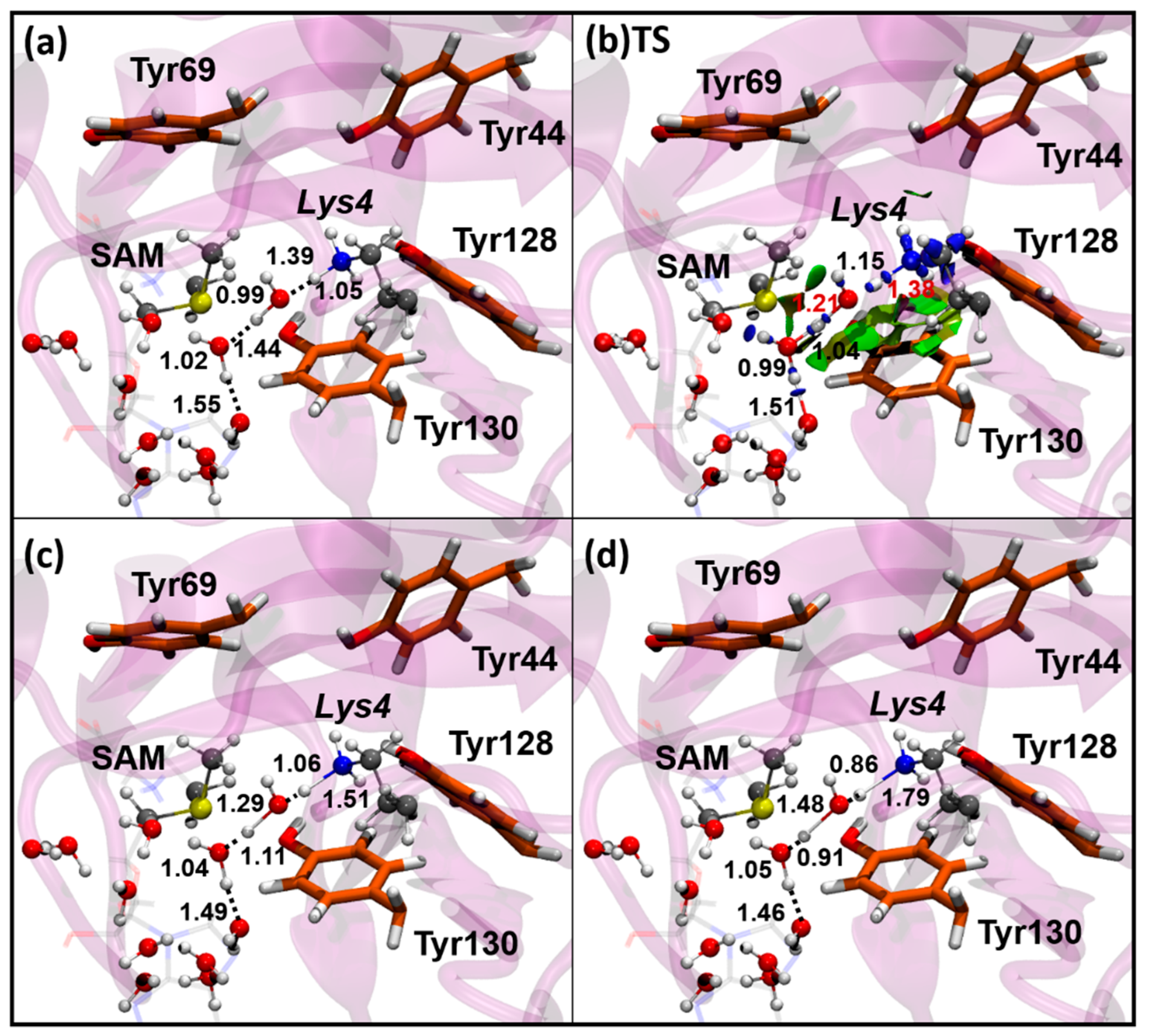
3.2. Methyl-Transfer Reaction Mechanism
3.2.1. Methyl-Transfer Energy Barrier
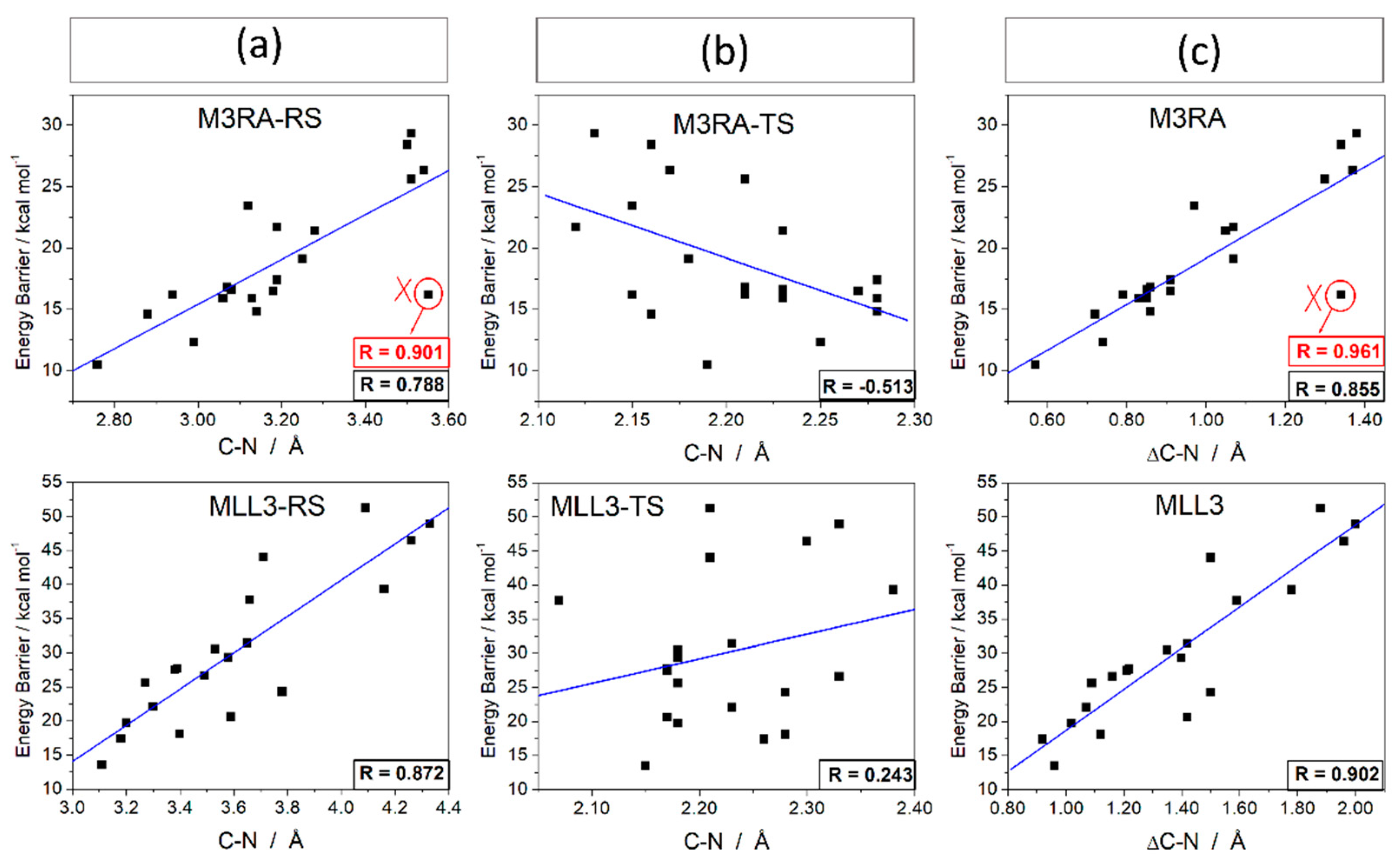
3.2.2. Role of the Donor-Acceptor Distance
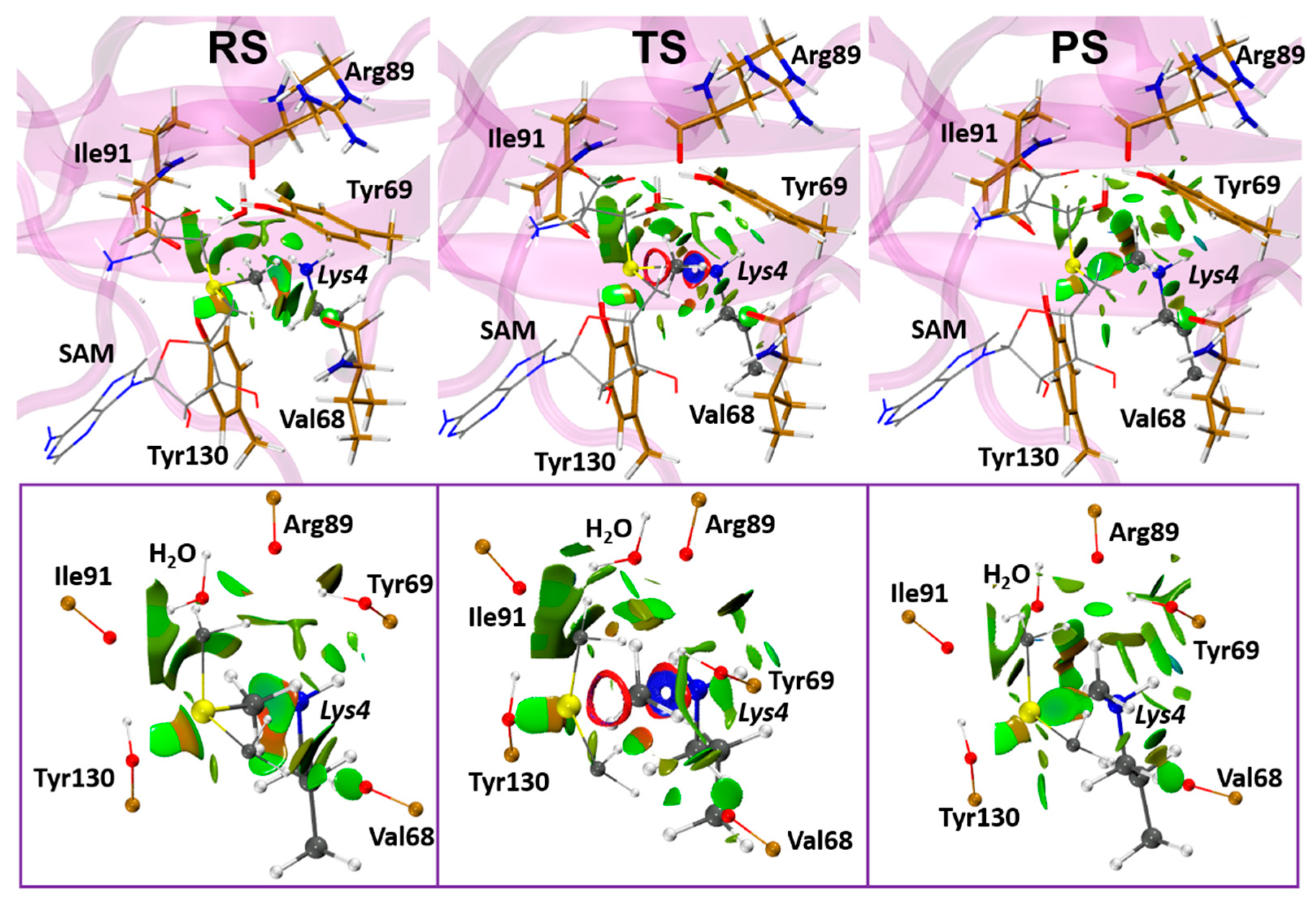
3.2.3. Role of the Enzymatic Environment
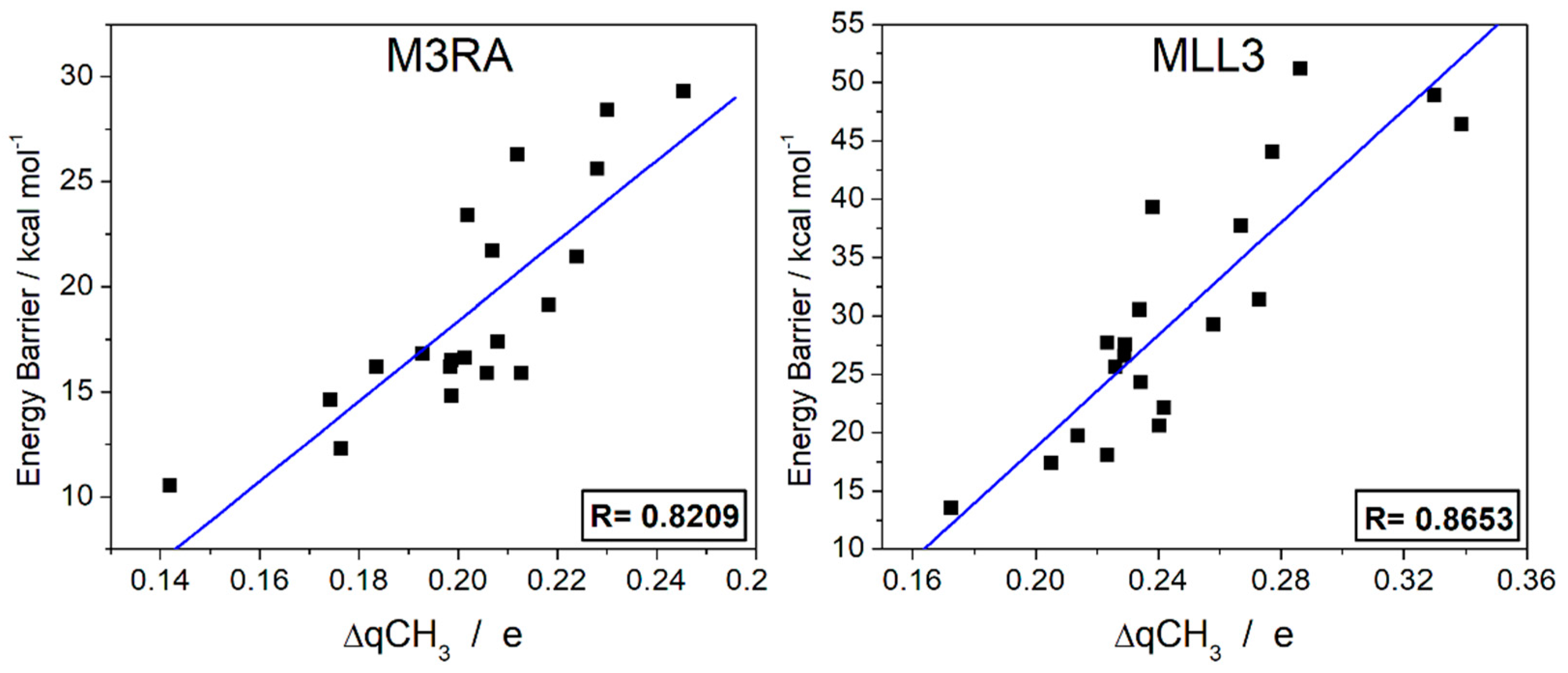
3.2.4. Charge Transfer Analysis
3.2.5. Product Stabilization and Specificity
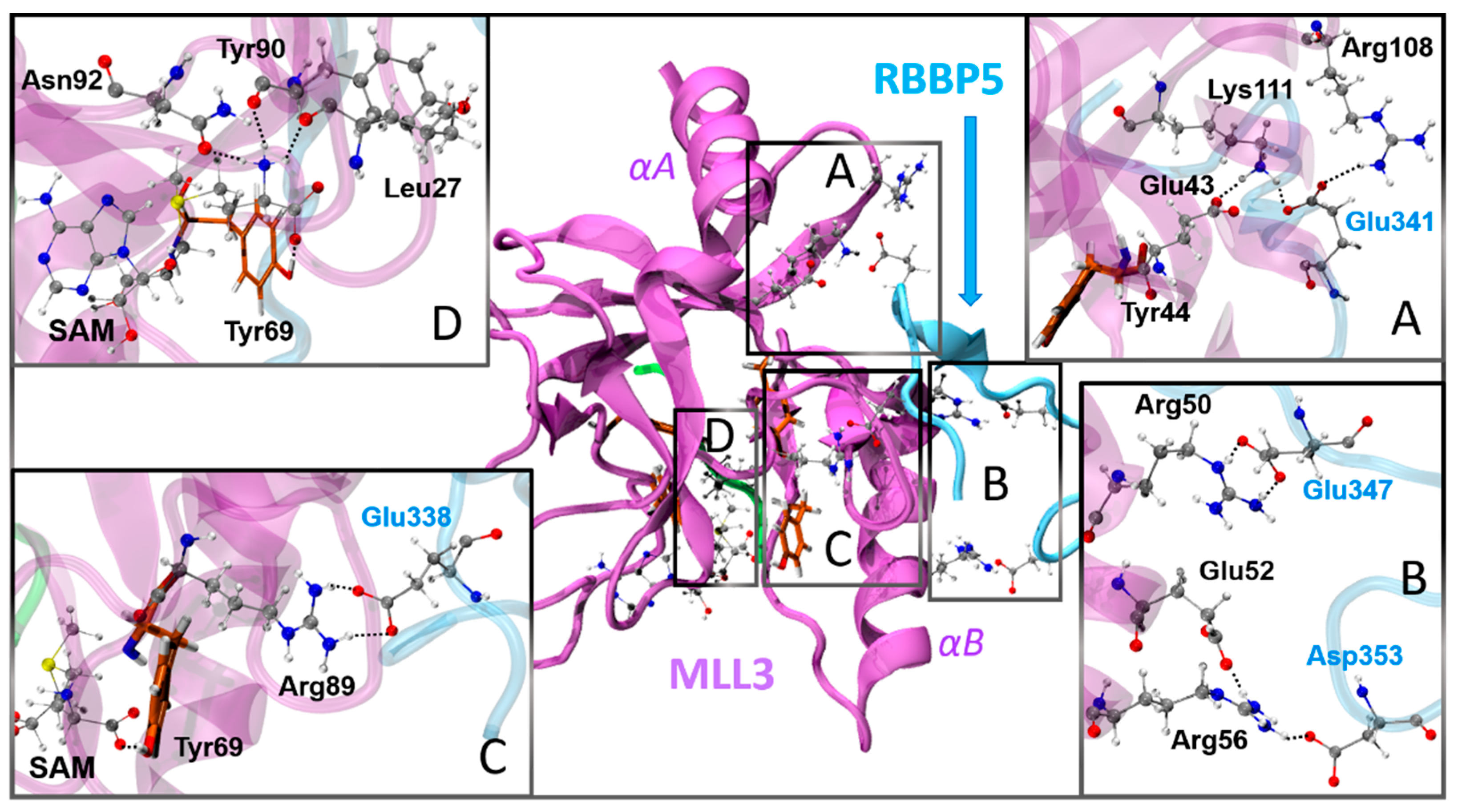
3.3. Activation Mechanism by the Dimer RBBP5-ASH2L Binding
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schneider, R.; Bannister, A.J.; Kouzarides, T. Unsafe SETs: Histone lysine methyltransferases and cancer. Trends Biochem. Sci. 2002, 27, 396–402. [Google Scholar] [CrossRef]
- Wang, X.-X.; Fu, L.; Li, X.; Wu, X.; Zhu, Z.; Fu, L.; Dong, J.-T. Somatic Mutations of the Mixed-Lineage Leukemia 3 (MLL3) Gene in Primary Breast Cancers. Pathol. Oncol. Res. 2011, 17, 429–433. [Google Scholar] [CrossRef]
- Li, B.; Liu, H.Y.; Guo, S.H.; Sun, P.; Gong, F.M.; Jia, B.Q. Association of MLL3 expression with prognosis in gastric cancer. Genet. Mol. Res. 2014, 13, 7513–7518. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudithipudi, S.; Jeltsch, A. Role of somatic cancer mutations in human protein lysine methyltransferases. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Li, M.; Zhang, X.; Jones, S.; Leary, R.J.; Lin, J.C.-H.; Boca, S.M.; Carter, H.; Samayoa, J.; Bettegowda, C.; et al. The Genetic Landscape of the Childhood Cancer Medulloblastoma. Science 2011, 331, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 Is a Haploinsufficient 7q Tumor Suppressor in Acute Myeloid Leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, D.-H.; Lee, S.; Yang, Q.-H.; Lee, D.K.; Lee, S.-K.; Roeder, R.G.; Lee, J.W. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. USA 2009, 106, 8513–8518. [Google Scholar] [CrossRef] [Green Version]
- Kipp, D.R.; Quinn, C.M.; Fortin, P.D. Enzyme-Dependent Lysine Deprotonation in EZH2 Catalysis. Biochemistry 2013, 52, 6866–6878. [Google Scholar] [CrossRef]
- Li, Y.; Han, J.; Zhang, Y.; Cao, F.; Liu, Z.; Li, S.; Wu, J.; Hu, C.; Wang, Y.; Shuai, J.; et al. Structural basis for activity regulation of MLL family methyltransferases. Nature 2016, 530, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Trievel, R.C.; Beach, B.M.; Dirk, L.M.; Houtz, R.L.; Hurley, J.H. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell 2002, 111, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Kwon, T.; Chang, J.H.; Kwak, E.; Lee, C.W.; Joachimiak, A.; Kim, Y.C.; Lee, J.; Cho, Y. Mechanism of histone lysine methyl transfer revealed by the structure of SET7/9-AdoMet. EMBO J. 2003, 22, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Min, J.; Lunin, V.V.; Antoshenko, T.; Dombrovski, L.; Zeng, H.; Allali-Hassani, A.; Campagna-Slater, V.; Vedadi, M.; Arrowsmith, C.H.; et al. Structural Biology of Human H3K9 Methyltransferases. PLoS ONE 2010, 5, e8570. [Google Scholar] [CrossRef] [Green Version]
- Trievel, R.C.; Flynn, E.M.; Houtz, R.L.; Hurley, J.H. Mechanism of multiple lysine methylation by the SET domain enzyme Rubisco LSMT. Nat. Struct. Biol. 2003, 10, 545–552. [Google Scholar] [CrossRef]
- Zhang, X.; Bruice, T.C. Histone Lysine Methyltransferase SET7/9: Formation of a Water Channel Precedes Each Methyl Transfer. Biochemistry 2007, 46, 14838–14844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bruice, T.C. Enzymatic mechanism and product specificity of SET-domain protein lysine methyltransferases. Proc. Natl. Acad. Sci. USA 2008, 105, 5728–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Senn, H.M.; O’Hagan, D.; Thiel, W. Insight into Enzymatic C−F Bond Formation from QM and QM/MM Calculations. J. Am. Chem. Soc. 2005, 127, 13643–13655. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.-B.; Guo, H. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity. Proc. Natl. Acad. Sci. USA 2007, 104, 8797–8802. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, P.; Himo, F. Quantum chemical modeling of enzymatic reactions: The case of histone lysine methyltransferase. J. Comput. Chem. 2010, 31, 1707–1714. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, Y. Catalytic Mechanism and Product Specificity of the Histone Lysine Methyltransferase SET7/9: An ab Initio QM/MM-FE Study with Multiple Initial Structures. J. Am. Chem. Soc. 2006, 128, 1272–1278. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kästner, J.; Sherwood, P. Superlinearly converging dimer method for transition state search. J. Chem. Phys. 2008, 128, 14106. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.; Kästner, J.; Sokol, A.A.; Keal, T.W.; Sherwood, P. ChemShell—a modular software package for QM/MM simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 101–110. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R.; Hättig, C.; Klopper, W.; Sierka, M.; Weigend, F. Turbomole. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 91–100. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- De Silva, P.; Corminboeuf, C. Simultaneous Visualization of Covalent and Noncovalent Interactions Using Regions of Density Overlap. J. Chem. Theory Comput. 2014, 10, 3745–3756. [Google Scholar] [CrossRef]
- Changeux, J.-P. 50 years of allosteric interactions: The twists and turns of the models. Nat. Rev. Mol. Cell Biol. 2013, 14, 819–829. [Google Scholar] [CrossRef]
- Świderek, K.; Tuñón, I.; Williams, I.H.; Moliner, V. Insights on the Origin of Catalysis on Glycine N-Methyltransferase from Computational Modeling. J. Am. Chem. Soc. 2018, 140, 4327–4334. [Google Scholar] [CrossRef]
- Couture, J.-F.; Dirk, L.M.A.; Brunzelle, J.S.; Houtz, R.L.; Trievel, R.C. Structural origins for the product specificity of SET domain protein methyltransferases. Proc. Natl. Acad. Sci. USA 2008, 105, 20659–20664. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | ΔE≠ | ΔE1° | Tyr44-Lys4 a | Tyr128-Lys4 a | Val68-W1 b | Tyr69-SAM c | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| TRIMER | R | TS | R | TS | R | TS | R | TS | ||
| Th-conf-1 | 16.2 | 8.7 | 2.04 | 2.55 | 1.84 | 2.14 | 2.61 | 2.71 | 3.36 | 3.35 |
| Th-conf-2 | 16.5 | 11.7 | 4.66 | 4.77 | 4.18 | 4.30 | 1.68 | 1.75 | 3.28 | 3.27 |
| Th-conf-3 | 10.9 | 6.6 | 2.06 | 3.04 | 1.87 | 2.29 | 1.61 | 1.61 | 3.71 | 3.71 |
| Th-conf-4 | 14.5 | 3.9 | 3.98 | 2.44 | 2.45 | 2.69 | 2.93 | 3.21 | 3.65 | 3.59 |
| Avg. () | 14.5 | 7.7 | 3.19 | 3.45 | 2.59 | 2.86 | 2.63 | 2.58 | 3.50 | 3.48 |
| MONOMER | ||||||||||
| Mh-conf-1 | 33.5 | 10.5 | 5.61 | 5.41 | 4.99 | 4.94 | 3.40 | 3.30 | 5.89 | 5.83 |
| Mh-conf-2 | 19.5 | 17.8 | 4.93 | 5.05 | 4.27 | 4.65 | 1.72 | 1.70 | 3.71 | 3.68 |
| Mh-conf-3 | 32.5 | 26.8 | 3.65 | 3.29 | 3.07 | 2.99 | 3.14 | 3.05 | 3.82 | 3.59 |
| Mh-conf-4 | 27.5 | 18.9 | 2.57 | 3.16 | 3.60 | 3.37 | 3.24 | 2.81 | 4.68 | 4.47 |
| Avg. () | 28.3 | 18.5 | 4.19 | 4.23 | 3.98 | 3.99 | 3.22 | 3.13 | 4.53 | 4.39 |
| Systems | ΔE≠ | ΔE2° | C-N(RS) | C-N(TS) | S-C(RS) | S-C(TS) | S-C-N(RS) | S-C-N(TS) |
|---|---|---|---|---|---|---|---|---|
| T-conf-1 | 21.4 | −11.2 | 3.28 | 2.23 | 1.81 | 2.35 | 143.8 | 171.9 |
| T-conf-2 | 10.5 | −18.9 | 2.76 | 2.19 | 1.82 | 2.19 | 163.6 | 170.3 |
| T-conf-3 | 16.2 | −12.5 | 2.94 | 2.15 | 1.82 | 2.29 | 162.0 | 167.9 |
| T-conf-4 | 14.8 | −20.1 | 3.14 | 2.28 | 1.82 | 2.29 | 153.8 | 174.0 |
| T-conf-5 | 16.5 | −16.6 | 3.18 | 2.27 | 1.81 | 2.30 | 148.2 | 172.8 |
| T-conf-6 | 15.9 | −17.9 | 3.13 | 2.28 | 1.82 | 2.30 | 147.3 | 173.8 |
| T-conf-7 | 16.6 | −12.8 | 3.08 | 2.23 | 1.82 | 2.31 | 159.2 | 175.0 |
| T-conf-8 | 12.3 | −21.1 | 2.99 | 2.25 | 1.82 | 2.25 | 156.9 | 173.2 |
| T-conf-9 | 16.2 | −14.4 | 3.55 | 2.21 | 1.81 | 2.26 | 158.8 | 167.9 |
| T-conf-10 | 15.9 | −14.7 | 3.06 | 2.23 | 1.81 | 2.28 | 133.1 | 168.1 |
| T-conf-11 | 16.8 | −13.2 | 3.07 | 2.21 | 1.81 | 2.29 | 153.6 | 172.0 |
| T-conf-12 | 14.6 | −10.3 | 2.88 | 2.16 | 1.82 | 2.27 | 170.9 | 175.7 |
| T-conf-13 | 25.6 | −2.9 | 3.51 | 2.21 | 1.81 | 2.30 | 146.0 | 174.4 |
| T-conf-14 | 17.4 | −16.1 | 3.19 | 2.28 | 1.82 | 2.31 | 152.0 | 172.4 |
| T-conf-15 | 29.3 | 7.0 | 3.51 | 2.13 | 1.81 | 2.35 | 168.5 | 176.9 |
| T-conf-16 | 23.4 | −9.0 | 3.12 | 2.15 | 1.81 | 2.30 | 121.0 | 158.2 |
| T-conf-17 | 28.4 | −1.7 | 3.50 | 2.16 | 1.81 | 2.29 | 108.2 | 163.5 |
| T-conf-18 | 26.3 | 0.1 | 3.54 | 2.17 | 1.82 | 2.32 | 148.4 | 168.7 |
| T-conf-19 | 19.1 | −3.4 | 3.25 | 2.18 | 1.82 | 2.36 | 169.4 | 174.2 |
| T-conf-20 | 21.7 | −3.5 | 3.19 | 2.12 | 1.81 | 2.35 | 156.0 | 167.2 |
| Avg. () | 18.9 | −10.7 | 3.19 | 2.20 | 1.82 | 2.30 | 151.0 | 170.9 |
| Systems | ΔE≠ | ΔE2° | C-N(RS) | C-N(TS) | S-C(RS) | S-C(TS) | S-C-N(RS) | S-C-N(TS) |
|---|---|---|---|---|---|---|---|---|
| M-conf-1 | 20.6 | −5.6 | 3.59 | 2.17 | 1.81 | 2.29 | 120.2 | 166.2 |
| M-conf-2 | 17.4 | −15.3 | 3.18 | 2.26 | 1.82 | 2.33 | 162.1 | 175.7 |
| M-conf-3 | 22.1 | −3.7 | 3.30 | 2.23 | 1.82 | 2.41 | 157.8 | 177.3 |
| M-conf-4 | 25.6 | −0.7 | 3.27 | 2.18 | 1.81 | 2.38 | 170.1 | 178.9 |
| M-conf-5 | 19.7 | −6.0 | 3.20 | 2.18 | 1.82 | 2.34 | 153.2 | 171.9 |
| M-conf-6 | 48.9 | −2.6 | 4.33 | 2.33 | 1.83 | 2.79 | 151.4 | 164.8 |
| M-conf-7 | 46.4 | 0.5 | 4.26 | 2.30 | 1.82 | 2.73 | 151.8 | 162.6 |
| M-conf-8 | 18.1 | −13.1 | 3.40 | 2.28 | 1.83 | 2.36 | 165.9 | 176.0 |
| M-conf-9 | 39.3 | −7.3 | 4.16 | 2.38 | 1.81 | 2.24 | 111.0 | 156.5 |
| M-conf-10 | 51.2 | −7.2 | 4.09 | 2.21 | 1.81 | 2.49 | 115.8 | 151.8 |
| M-conf-11 | 26.6 | −22.8 | 3.49 | 2.33 | 1.81 | 2.31 | 124.5 | 170.8 |
| M-conf-12 | 44.0 | −4.8 | 3.71 | 2.21 | 1.81 | 2.41 | 108.4 | 149.2 |
| M-conf-13 | 37.7 | −8.5 | 3.66 | 2.07 | 1.81 | 2.44 | 104.8 | 148.9 |
| M-conf-14 | 31.4 | 3.3 | 3.65 | 2.23 | 1.81 | 2.35 | 114.8 | 160.4 |
| M-conf-15 | 27.5 | −0.6 | 3.38 | 2.17 | 1.81 | 2.30 | 119.5 | 163.8 |
| M-conf-16 | 29.3 | −0.2 | 3.58 | 2.18 | 1.81 | 2.34 | 115.6 | 161.6 |
| M-conf-17 | 24.3 | −7.0 | 3.78 | 2.28 | 1.82 | 2.30 | 152.0 | 167.4 |
| M-conf-18 | 27.7 | 0.3 | 3.39 | 2.17 | 1.81 | 2.29 | 114.2 | 166.1 |
| M-conf-19 | 13.5 | −19.3 | 3.11 | 2.15 | 1.81 | 2.24 | 160.3 | 170.3 |
| M-conf-20 | 30.5 | −3.5 | 3.53 | 2.18 | 1.81 | 2.29 | 116.2 | 165.2 |
| Avg. () | 30.1 | −6.2 | 3.60 | 2.22 | 1.81 | 2.38 | 134.5 | 165.3 |
| Systems | M3RA | MLL3 | ||
|---|---|---|---|---|
| R | TS | R | TS | |
| Tyr44-Lys4a | 2.30 | 2.43 | 2.95 | 3.22 |
| Tyr128-Lys4a | 2.55 | 2.48 | 2.28 | 2.54 |
| Val68-Met b | 3.25 | 3.00 | 2.64 | 2.53 |
| Arg89-Met b | 3.20 | 3.22 | 2.85 | 3.30 |
| Ile91-Met b | 3.19 | 3.26 | 3.54 | 3.38 |
| Tyr130-Met c | 3.11 | 2.93 | 3.60 | 2.78 |
| Tyr69-SAM d | 1.57 | 1.57 | 3.01 | 2.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda-Rojas, S.; Blanco-Esperguez, K.; Tuñón, I.; Kästner, J.; Mendizábal, F. Exploration of the Activation Mechanism of the Epigenetic Regulator MLL3: A QM/MM Study. Biomolecules 2021, 11, 1051. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11071051
Miranda-Rojas S, Blanco-Esperguez K, Tuñón I, Kästner J, Mendizábal F. Exploration of the Activation Mechanism of the Epigenetic Regulator MLL3: A QM/MM Study. Biomolecules. 2021; 11(7):1051. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11071051
Chicago/Turabian StyleMiranda-Rojas, Sebastián, Kevin Blanco-Esperguez, Iñaki Tuñón, Johannes Kästner, and Fernando Mendizábal. 2021. "Exploration of the Activation Mechanism of the Epigenetic Regulator MLL3: A QM/MM Study" Biomolecules 11, no. 7: 1051. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11071051