
Interaction Mode of the Novel Monobactam AIC499 Targeting Penicillin Binding Protein 3 of Gram-Negative Bacteria

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
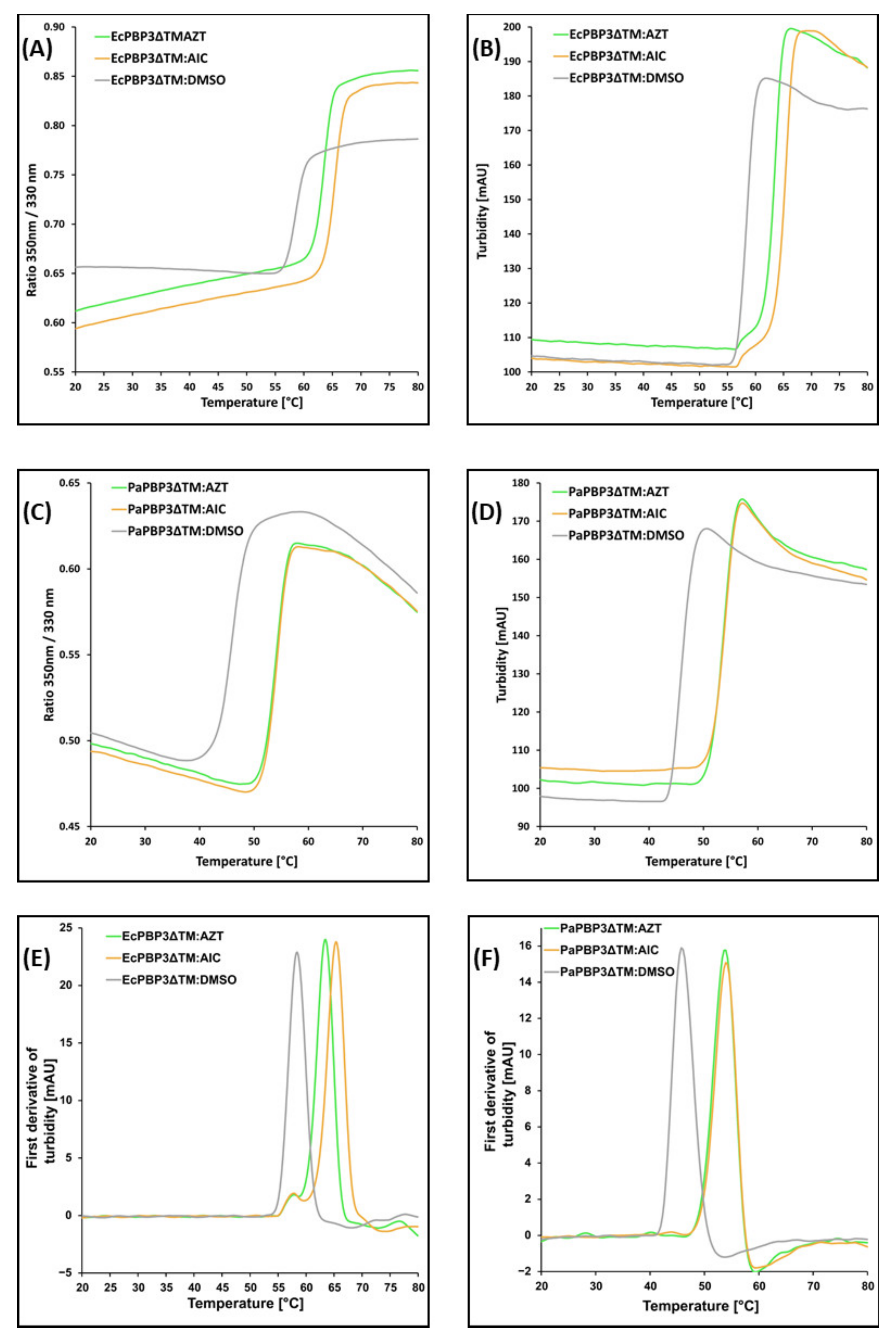
3. Results
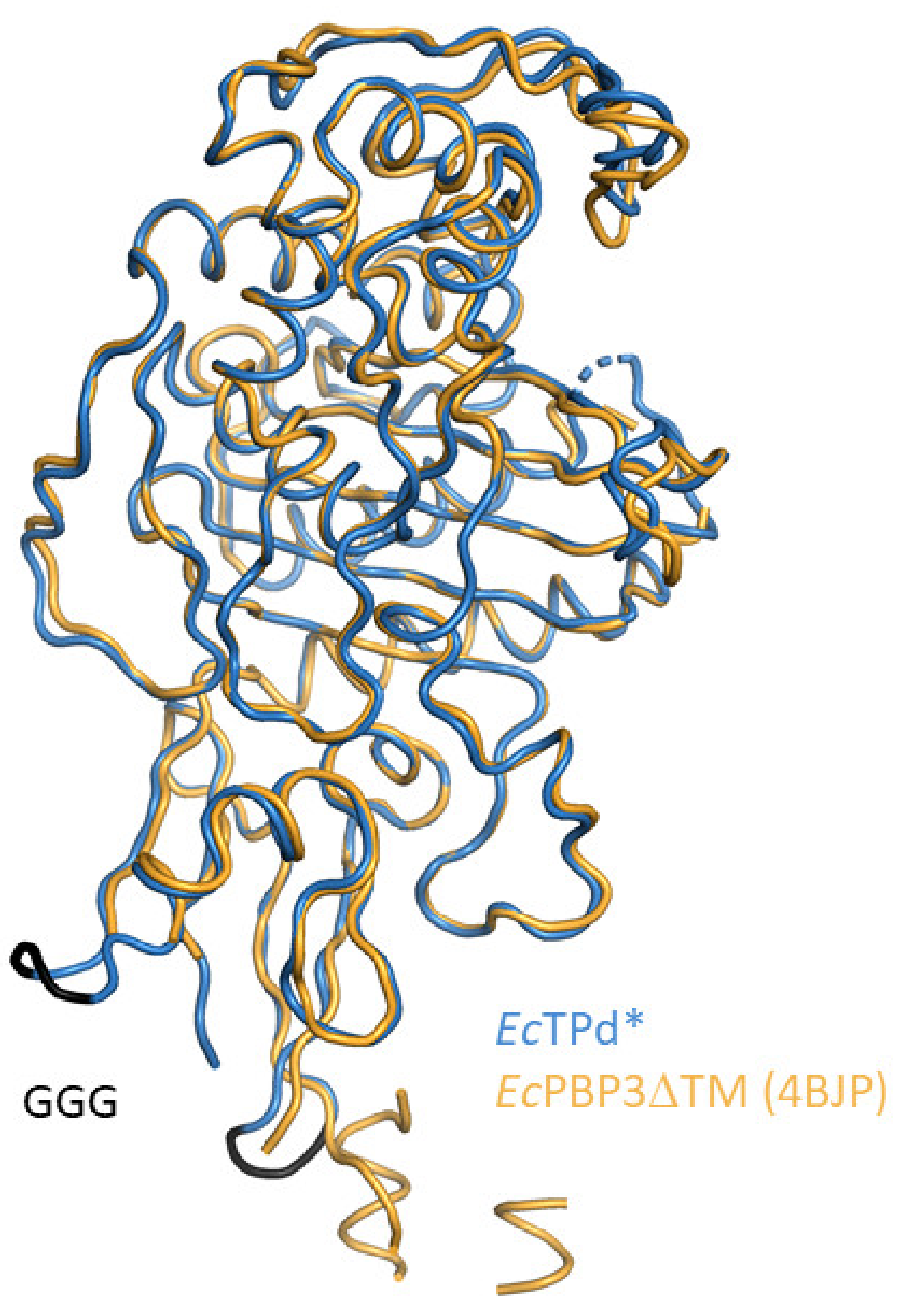
3.1. Structures of E. coli PBP3
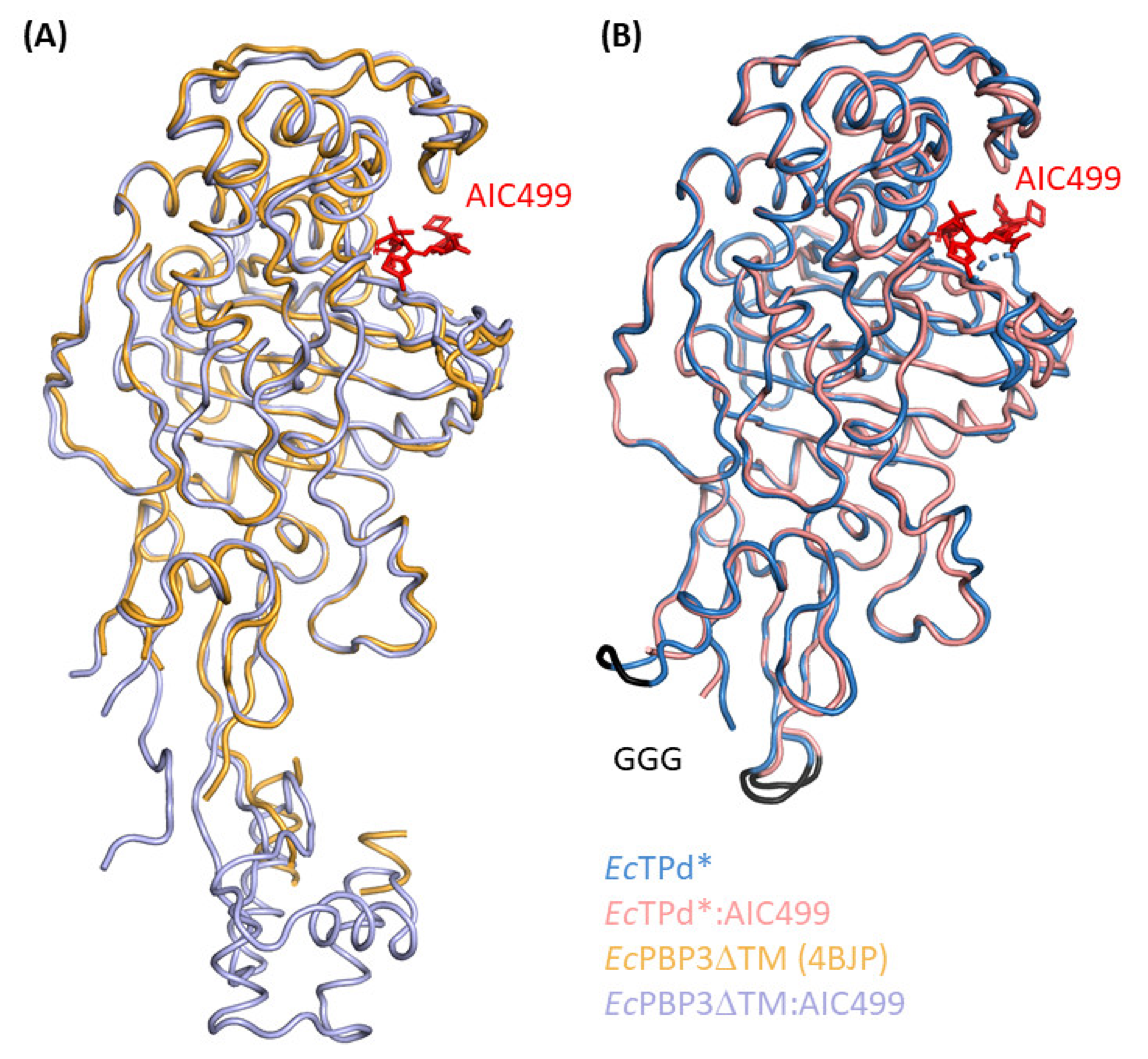
3.1.1. E. coli PBP3 Apo Protein

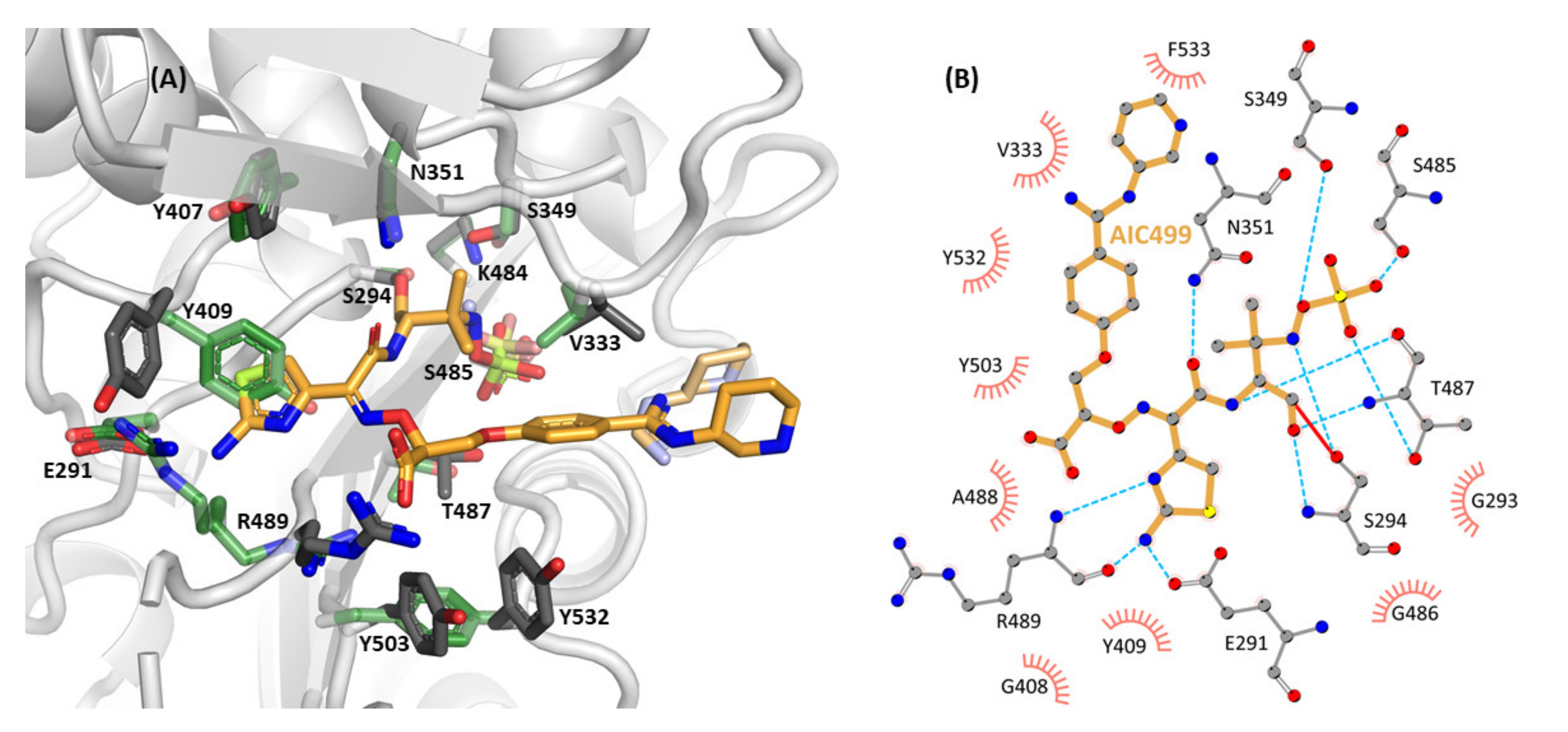
3.1.2. Effect of AIC499 Binding on E. coli PBP3
3.2. Structures of P. aeruginosa PBP3
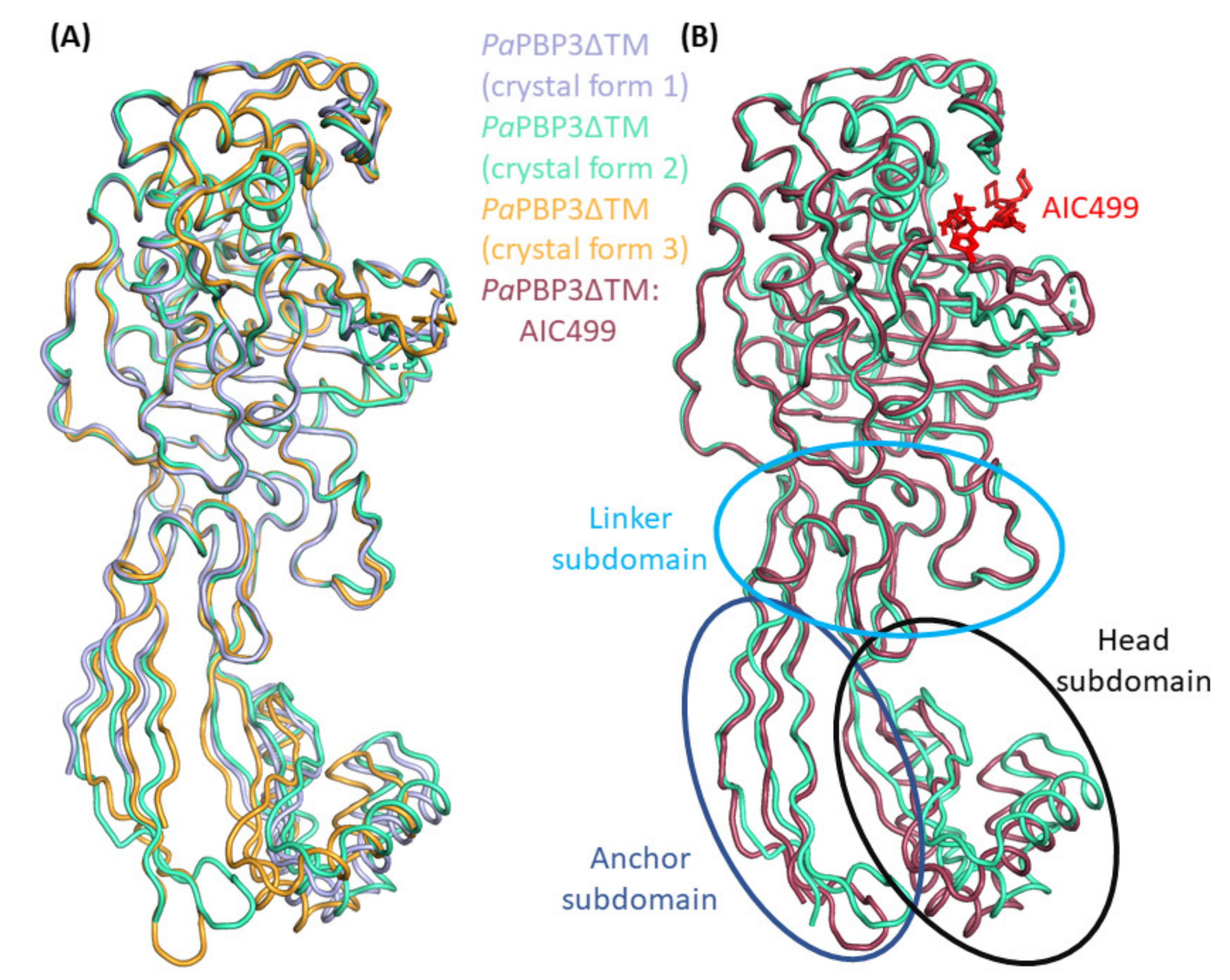
3.2.1. P. aeruginosa PBP3 Apo Protein
3.2.2. Effect of AIC499 Binding on P. aeruginosa PBP3
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

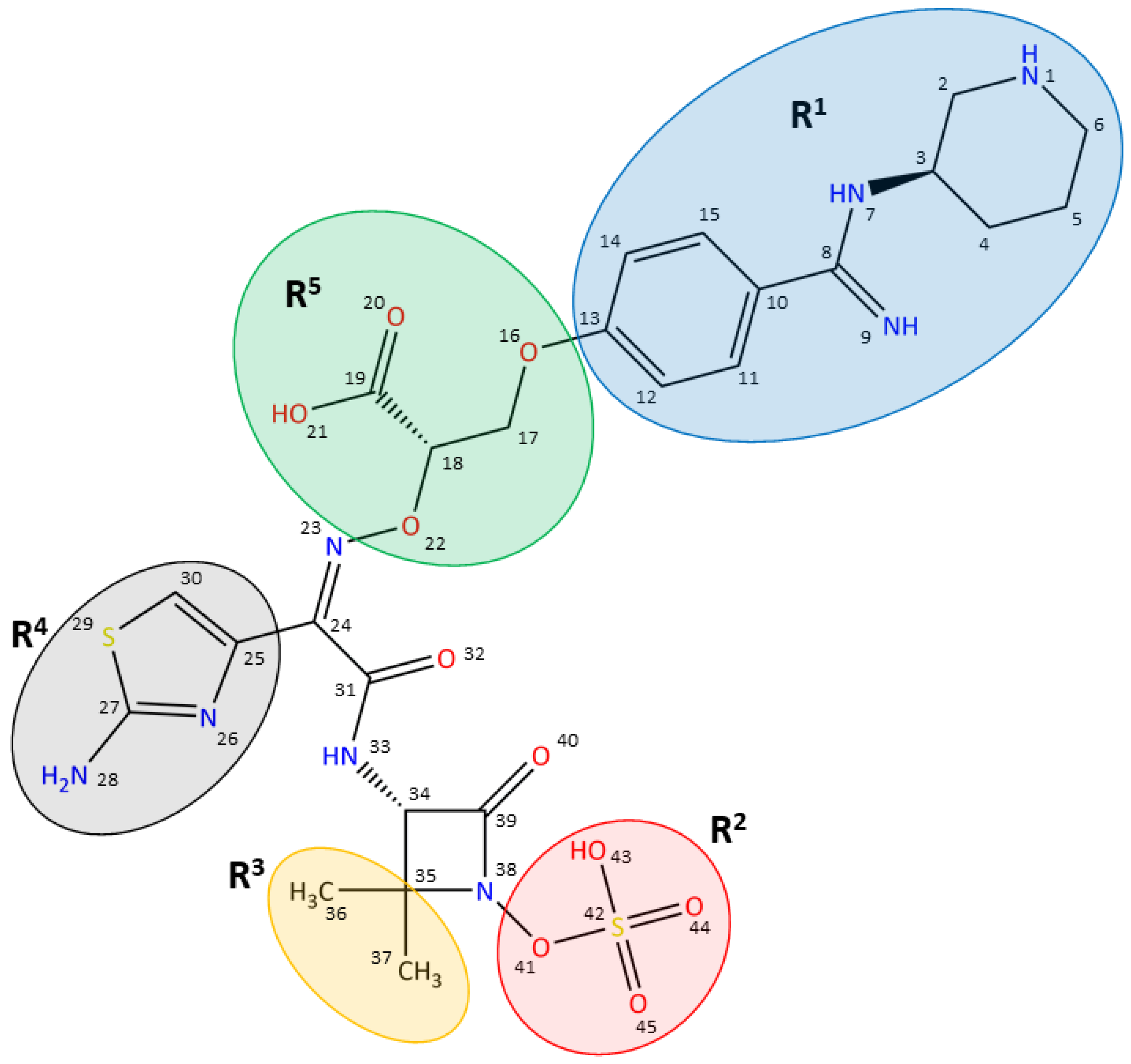{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PaPBP3ΔTM | AIC499 | EcTPd* | ||
|---|---|---|---|---|
| Atom | Distance [Å] | Atom | Distance [Å] | Atom |
| / | / | C2 (R1) | 3.6 (hydrophobic) | Y541 Cε2 |
| V333 Cγ1 | 3.5 (hydrophobic) | C4 (R1) | / | / |
| V333 Cγ2 | 3.6 (hydrophobic) | C11 (R1) | 3.7 (hydrophobic) | V344 Cγ1 |
| Y532 Cε2 | 3.8 (aromatic) | C13 (R1) | 4.2 (aromatic) b | Y540 Cε2 |
| T487 Cβ | 3.5 (hydrophobic) | C14 (R1) | 4.2 (hydrophobic) | T497 Cβ |
| F533 Cβ | 3.7 (aromatic) a | C15 (R1) | 4.0 (aromatic) | Y541 Cδ1 |
| T487 Cβ | 4.3 (hydrophobic) | C18 (R5) | / | / |
| / | / | C19 (R5) | 4.1 (hydrophobic) | Y511 Cε2 |
| R489 Nη2 | 3.0 (H-bond) | O20 (R5) | / | / |
| T487 O | 3.2 (H-bond) | N23 | / | / |
| R489 N | 2.8 (H-bond) | N26 (R4) | 3.0 (H-bond) | K499 N |
| Y409 Cδ1 | 3.8 (aromatic) | C27 (R4) | 3.6 (aromatic) | Y419 Cε1 |
| A488 Cα | 3.8 (hydrophobic) | C27 (R4) | 3.6 (hydrophobic) | A498 Cα |
| E291 Oε1 | 2.5 (H-bond) | N28 (R4) | 2.3 (H-bond) | E304 Oε1 |
| R489 O | 2.5 (H-bond) | N28 (R4) | 2.8 (H-bond) | K499 O |
| / | / | N28 (R4) | 3.1 (H-bond) | K499 N |
| / | / | C30 (R4) | 4.4 (aromatic) | F417 Cδ1 |
| G293 Cα | 3.8 (hydrophobic) | C30 (R4) | 3.8 (hydrophobic) | G306 Cα |
| N351 Nδ2 | 2.8 (H-bond) | O32 | 3.0 (H-bond) | N361 Nγ2 |
| T487 O | 2.8 (H-bond) | N33 | 3.2 (H-bond) | T497 O |
| V333 Cγ2 | 3.6 (hydrophobic) | C36 (R3) | / | / |
| T487 Cβ | 3.8 (hydrophobic) | C36 (R3) | / | / |
| V333 Cγ2 | 3.8 (hydrophobic) | C37 (R3) | 3.6 (hydrophobic) | V344 Cγ1 |
| S349 Cβ | 4.3 (hydrophobic) | C37 (R3) | / | / |
| S294 Oγ | 2.7 (H-bond) | N38 (β-lactam ring) | / | / |
| S349 Oγ | 2.8 (H-bond) | N38 (β-lactam ring) | / | / |
| S294 N | 2.7 (H-bond) | O40 | 2.5 (H-bond) | S307 N |
| T487 N | 2.9 (H-bond) | O40 | 3.2 (H-bond) | T497 N |
| S294 Oγ | 3.0 (H-bond) | O41 (R2) | 3.0 (H-bond) | S307 Oγ |
| K484 Nζ | 2.9 (H-bond) | O44 (R2) | 2.7 (H-bond) | T497 Oγ |
| S485 Oγ | 2.4 (H-bond) | O44 (R2) | / | / |
| T487 Oγ | 2.5 (H-bond) | O45 (R2) | 3.1 (H-bond) | K494 Nζ |
| T487 N | 2.9 (H-bond) | O45 (R2) | 3.1 (H-bond) | T495 O |
| PaPBP3 | Distance [Å] | Aztreonam |
|---|---|---|
| Atom | Atom | |
| T487 Cγ2 | 3.6 (hydrophobic) | C28 (C4; R1) c |
| Y532 Cε2 | 4.4 (hydrophobic) | C28 (C13; R1) c |
| Y503 Cε1 | 3.7 (hydrophobic) | C28 (C13; R1) c |
| F533 Cβ | 3.9 (hydrophobic) | C27 (C15; R1) c |
| R489 Nε | 3.1 (H-bond) | O31 (O20; R5) |
| A488 Cα | 4.2 (hydrophobic) | C25 (C25; R4) |
| Y409 Cδ1 | 3.9 (aromatic) | C26 (C27; R4) |
| A488 Cα | 3.9 (hydrophobic) | C26 (C27; R4) |
| E291 Oε1 | 2.9 (H-bond) | N16 (N28; R4) |
| G293 Cα | 4.0 (hydrophobic) | C25 (C30; R4) |
| N351 Nδ2 | 2.9 (H-bond) | O10 (O32) |
| T487 O | 3.0 (H-bond) | N13 (N33) |
| T487 Cβ | 4.3 (hydrophobic) | C18 (C35; R3) |
| S294 Cβ | 4.4 (hydrophobic) | C18 (C35; R3) |
| V333 Cγ1 | 3.6 (hydrophobic) | C7 (C36; R3) |
| S294 Oγ | 3.1 (H-bond) | N12 (N38; β-lactam ring) |
| S349 Oγ | 3.0 (H-bond) | N12 (N38; β-lactam ring) |
| G293 Cα | 4.5 (hydrophobic) | C20 (C39) |
| S294 N | 3.0 (H-bond) | O9 (O40) |
| T487 N | 2.8 (H-bond) | O9 (O40) |
| K484 Nζ | 3.1 (H-bond) | O34 (O44; R2) |
| S485 Oγ | 2.7 (H-bond) | O34 (O44; R2) |
| T487 Oγ | 2.6 (H-bond) | O33 (O45; R2) |
| T487 N | 3.2 (H-bond) | O33 (O45; R2) |


References
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. London: Review on Antimicrobial Resistance. 2014. Available online: https://amr-review.org/Publications.html (accessed on 15 May 2021).
- Zervosen, A.; Sauvage, E.; Frère, J.-M.; Charlier, P.; Luxen, A. Development of new drugs for an old target—The penicillin binding proteins. Molecules 2012, 17, 12478–12505. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, E.; Terrak, M. Glycosyltransferases and transpeptidases/penicillin-binding proteins: Valuable targets for new antibacterials. Antibiotics 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Bertsche, U. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim. Biophys. Acta Biomembr. 2008, 1778, 1714–1734. [Google Scholar] [CrossRef] [Green Version]
- Goffin, C.; Ghuysen, J.-M. Biochemistry and comparative genomics of SxxK superfamily acyltransferases offer a clue to the mycobacterial paradox: Presence of penicillin-susceptible target proteins versus lack of efficiency of penicillin as therapeutic agent. Microbiol. Mol. Biol. Rev. 2002, 66, 702–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, D.S.; Chen, J.C.; Ghigo, J.-M.; Boyd, D.; Beckwith, J. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 1999, 181, 508–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Errington, J.; Daniel, R.A.; Scheffers, D.-J. Cytokinesis in bacteria. Microbiol. Mol. Biol. Rev. 2003, 67, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Wivagg, C.N.; Kapoor, M.; Barry, Z.; Rohs, P.D.; Suh, H.; Marto, J.A.; Garner, E.C.; Bernhardt, T.G. Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat. Microbiol. 2016, 1, 1–8. [Google Scholar] [CrossRef]
- Meeske, A.J.; Riley, E.P.; Robins, W.P.; Uehara, T.; Mekalanos, J.J.; Kahne, D.; Walker, S.; Kruse, A.C.; Bernhardt, T.G.; Rudner, D.Z. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 2016, 537, 634–638. [Google Scholar] [CrossRef] [Green Version]
- Höltje, J.-V. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 1998, 62, 181–203. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Zaniewski, R.P.; Marr, E.S.; Lacey, B.M.; Tomaras, A.P.; Evdokimov, A.; Miller, J.R.; Shanmugasundaram, V. Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2010, 107, 22002–22007. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, S.; Derouaux, A.; Olatunji, S.; Fraipont, C.; Egan, A.J.; Vollmer, W.; Breukink, E.; Terrak, M. Interplay between penicillin-binding proteins and SEDS proteins promotes bacterial cell wall synthesis. Sci. Rep. 2017, 7, 43306. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, E.; Derouaux, A.; Fraipont, C.; Joris, M.; Herman, R.; Rocaboy, M.; Schloesser, M.; Dumas, J.; Kerff, F.; Nguyen-Disteche, M. Crystal structure of penicillin-binding protein 3 (PBP3) from Escherichia coli. PLoS ONE 2014, 9, e98042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K.; Bradford, P.A. β-Lactams and β-lactamase inhibitors: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Y.-M.; Davies, C. Penicillin-binding protein 3 is essential for growth of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01651-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, L.M.; da Silva, B.N.M.; Barbosa, G.; Barreiro, E.J. β-lactam antibiotics: An overview from a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 208, 112829. [Google Scholar] [CrossRef] [PubMed]
- Sykes, R.; Cimarusti, C.; Bonner, D.; Bush, K.; Floyd, D.; Georgopapadakou, N.; Koster, W.; Liu, W.; Parker, W.; Principe, P. Monocyclic β-lactam antibiotics produced by bacteria. Nature 1981, 291, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Sykes, R.; Bonner, D. Aztreonam: The first monobactam. Am. J. Med. 1985, 78, 2–10. [Google Scholar] [CrossRef]
- McPherson, C.J.; Aschenbrenner, L.M.; Lacey, B.M.; Fahnoe, K.C.; Lemmon, M.M.; Finegan, S.M.; Tadakamalla, B.; O’Donnell, J.P.; Mueller, J.P.; Tomaras, A.P. Clinically relevant Gram-negative resistance mechanisms have no effect on the efficacy of MC-1, a novel siderophore-conjugated monocarbam. Antimicrob. Agents Chemother. 2012, 56, 6334. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, M.E.; Brickner, S.J.; Lall, M.; Casavant, J.; Deschenes, L.; Finegan, S.M.; George, D.M.; Granskog, K.; Hardink, J.R.; Huband, M.D. Preparation, Gram-negative antibacterial activity, and hydrolytic stability of novel siderophore-conjugated monocarbam diols. ACS Med. Chem. Lett. 2011, 2, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Blais, J.; Lopez, S.; Li, C.; Ruzin, A.; Ranjitkar, S.; Dean, C.R.; Leeds, J.A.; Casarez, A.; Simmons, R.L.; Reck, F. In vitro activity of LYS228, a novel monobactam antibiotic, against multidrug-resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62, e00552-18. [Google Scholar] [CrossRef] [Green Version]
- Reck, F.; Bermingham, A.; Blais, J.; Capka, V.; Cariaga, T.; Casarez, A.; Colvin, R.; Dean, C.R.; Fekete, A.; Gong, W. Optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae–identification of LYS228. Bioorganic Med. Chem. Lett. 2018, 28, 748–755. [Google Scholar] [CrossRef]
- Klenke, B.; Wiegand, I.; Schiffer, G.; Broetz-Oesterhelt, H.; Maiti, S.N.; Khan, J.; Reddy, A.; Yang, Z.; Hena, M.; Jia, G.; et al. Amidine Substituted Beta-Lactam Compounds, Their Preparation and Use as Antibacterial Agents. WO2013110643, 23 January 2013. [Google Scholar]
- AiCuris, A.G. AiCuris Initiates Clinical Development of AIC499, a Novel Resistance-Breaking Antibiotic against a Broad Range of MDR Gram-Negative Bacteria. Wuppertal: AiCuris Anti-Infective Cures AG. 2017. Available online: https://www.aicuris.com/81n93/AiCuris-Initiates-Clinical-Development-of-AIC499,-a-Novel-Resistance-Breaking-Antibiotic-against-a-Broad-Range-of-MDR-Gram-Negative-Bacteria.htm (accessed on 28 May 2021).
- Sainsbury, S.; Bird, L.; Rao, V.; Shepherd, S.M.; Stuart, D.I.; Hunter, W.N.; Owens, R.J.; Ren, J. Crystal structures of penicillin-binding protein 3 from Pseudomonas aeruginosa: Comparison of native and antibiotic-bound forms. J. Mol. Biol. 2011, 405, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tickle, I.; Flensburg, C.; Keller, P.; Paciorek, W.; Sharff, A.; Vonrhein, C.; Bricogne, G. Staraniso; Global Phasing Ltd.: Cambridge, UK, 2018. [Google Scholar]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.4.0 Schrodinger, LLC. Available online: https://pymol.org/2/support.html?#citing (accessed on 17 June 2021).
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Huang, Y.; Yang, R.; Craigie, R.; Clore, G.M. A simple and robust protocol for high-yield expression of perdeuterated proteins in Escherichia coli grown in shaker flasks. J. Biomol. NMR 2016, 66, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, M.; Brand, T.; Berger, S. A 1H-NMR thermometer suitable for cryoprobes. Magn. Reson. Chem. 2007, 45, 175–178. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins Struct. Funct. Bioinform. 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Rajavel, M.; Kumar, V.; Nguyen, H.; Wyatt, J.; Marshall, S.H.; Papp-Wallace, K.M.; Deshpande, P.; Bhavsar, S.; Yeole, R.; Bhagwat, S. Structural Characterization of Diazabicyclooctane β-Lactam “Enhancers” in Complex with Penicillin-Binding Proteins PBP2 and PBP3 of Pseudomonas aeruginosa. Mbio 2021, 12, e03058-20. [Google Scholar] [CrossRef]
- Bellini, D.; Koekemoer, L.; Newman, H.; Dowson, C.G. Novel and Improved Crystal Structures of H. influenzae, E. coli and P. aeruginosa Penicillin-Binding Protein 3 (PBP3) and N. gonorrhoeae PBP2: Toward a Better Understanding of β-Lactam Target-Mediated Resistance. J. Mol. Biol. 2019, 431, 3501–3519. [Google Scholar] [CrossRef]
- Yang, D.; Kay, L.E. Improved 1HN-detected triple resonance TROSY-based experiments. J. Biomol. NMR 1999, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Bigam, C.G.; Holm, A.; Hodges, R.S.; Sykes, B.D. 1 H, 13 C and 15 N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 1995, 5, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Musafia, B.; Buchner, V.; Arad, D. Complex salt bridges in proteins: Statistical analysis of structure and function. J. Mol. Biol. 1995, 254, 761–770. [Google Scholar] [CrossRef] [PubMed]






| EcTPd* | EcTPd*: AIC499 | EcPBP3 ΔTM: AIC499 | PaPBP3 ΔTM (cf 1) | PaPBP3 ΔTM (cf 2) | PaPBP3 ΔTM (cf 3) | PaPBP3 ΔTM: AIC499 | |
|---|---|---|---|---|---|---|---|
| PDB entry | 7ONO | 7ONN | 7ONW | 7ONX | 7ONY | 7ONZ | 7ONK |
| Data collection | |||||||
| Beamline | DESY P11 | DESY P11 | ESRF ID23 | DESY P11 | EMBL P13 | EMBL P13 | DESY P11 |
| Wavelength [Å] | 1.0332 | 1.0332 | 0.97242 | 1.0332 | 0.9999 | 0.9999 | 1.0332 |
| Space group | P 62 2 2 | P 62 2 2 | P 64 2 2 | C 1 2 1 | C 1 2 1 | C 1 2 1 | P 21 21 21 |
| Cell dimensions | |||||||
| a, b, c [Å] | 109.3, 109.3, 143.2 | 110.4, 110.4, 142.1 | 106.9, 106.9, 285.8 | 110.6, 82.2, 91.4 | 104.1, 125.0, 74.2 | 151.5, 37.5, 82.8 | 81.0, 91.1, 148.4 |
| α, β, γ [°] | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 116.3, 90 | 90, 122.5, 90 | 90, 112.6, 90 | 90, 90, 90 |
| Resolution range [Å] | 47.74–2.30 (2.60–2.30) | 47.80–1.92 (2.22–1.92) | 48.64–2.70 (3.03–2.70) | 44.40–2.16 (2.34–2.16) | 39.55–1.77 (1.97–1.77) | 40.75–1.86 (2.09–1.86) | 46.92–1.73 (1.90–1.73) |
| CC1/2 [%] | 99.9 (84.0) | 99.9 (78.3) | 99.9 (79.3) | 99.6 (47.1) | 99.8 (64.1) | 99.8 (77.2) | 99.9 (62.4) |
| Rmeas [%] | 10.3 (145.6) | 6.6 (160.2) | 10.1 (121.1) | 11.1 (101.5) | 7.0 (115.9) | 7.1 (96.6) | 9.4 (92.5) |
| I/σ | 20.7 (2.4) | 24.2 (2.3) | 15.4 (2.0) | 7.9 (1.4) | 13.9 (1.8) | 12.3 (1.6) | 10.9 (1.6) |
| Completeness [%] a | 40.8 (6.7) | 45.7 (6.5) | 51.1 (9.0) | 66.6 (15.4) | 55.5 (10.4) | 51.8 (8.8) | 74.2 (15.3) |
| Ellips. Compl. [%] b | 93.9 (74.6) | 95.0 (74.3) | 92.2 (76.1) | 91.4 (57.8) | 70.4 (4.4) | 74.8 (3.6) | 95.9 (64.8) |
| Refinement | |||||||
| Resolution range [Å] | 47.74–2.30 | 47.80–1.92 | 48.64–2.70 | 44.40–2.16 | 39.55–1.77 | 40.75–1.86 | 46.92–1.73 |
| No. unique reflections | 9457 | 18221 | 13994 | 26398 | 42915 | 19031 | 85337 |
| No. protein atoms | 3031 | 3022 | 3516 | 3609 | 3841 | 3668 | 7803 |
| No. ligand atoms | 5 | 135 | 71 | 32 | 118 | 12 | 260 |
| No. water molecules | 16 | 60 | 6 | 146 | 274 | 134 | 750 |
| Rwork [%] | 23.00 | 22.33 | 23.35 | 18.17 | 17.98 | 20.91 | 17.34 |
| Rfree [%] | 28.20 | 26.93 | 27.07 | 21.52 | 21.34 | 25.85 | 21.52 |
| RMSD | |||||||
| Bond lengths [Å] | 0.001 | 0.005 | 0.002 | 0.003 | 0.004 | 0.003 | 0.007 |
| Bond angles [°] | 0.403 | 1.031 | 0.672 | 0.579 | 0.666 | 0.601 | 1.037 |
| Mean B factor [Å2] | 50.49 | 41.23 | 64.13 | 42.63 | 34.16 | 29.60 | 23.60 |
| Ramachandran plot | |||||||
| Favored [%] | 95.52 | 93.98 | 95.01 | 95.98 | 96.79 | 96.30 | 97.72 |
| Allowed [%] | 4.48 | 6.02 | 4.99 | 4.02 | 3.21 | 3.70 | 2.28 |
| Outliers [%] | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freischem, S.; Grimm, I.; López-Pérez, A.; Willbold, D.; Klenke, B.; Vuong, C.; Dingley, A.J.; Weiergräber, O.H. Interaction Mode of the Novel Monobactam AIC499 Targeting Penicillin Binding Protein 3 of Gram-Negative Bacteria. Biomolecules 2021, 11, 1057. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11071057
Freischem S, Grimm I, López-Pérez A, Willbold D, Klenke B, Vuong C, Dingley AJ, Weiergräber OH. Interaction Mode of the Novel Monobactam AIC499 Targeting Penicillin Binding Protein 3 of Gram-Negative Bacteria. Biomolecules. 2021; 11(7):1057. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11071057
Chicago/Turabian StyleFreischem, Stefan, Immanuel Grimm, Arancha López-Pérez, Dieter Willbold, Burkhard Klenke, Cuong Vuong, Andrew J. Dingley, and Oliver H. Weiergräber. 2021. "Interaction Mode of the Novel Monobactam AIC499 Targeting Penicillin Binding Protein 3 of Gram-Negative Bacteria" Biomolecules 11, no. 7: 1057. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11071057