
What Antarctic Plants Can Tell Us about Climate Changes: Temperature as a Driver for Metabolic Reprogramming

 , ,
, ,  , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Colobanthus Quitensis Protein Database Building
2.3. Protein Extraction for Proteomic Analysis
2.4. SDS-PAGE and Mass Spectrometry Analysis
2.5. Protein Identification and Quantification and Statistical Analyses
2.6. Bioinformatic Analysis
2.7. Enzymatic Activity Assays
2.8. Thiobarbituric Acid Reactive Substance Measurement
2.9. RNA Extraction and Quantitative Reverse Transcriptase–PCR Analysis
2.10. Data Analyses
3. Results and Discussion
3.1. De Novo Colobanthus Quitensis Protein Database Building
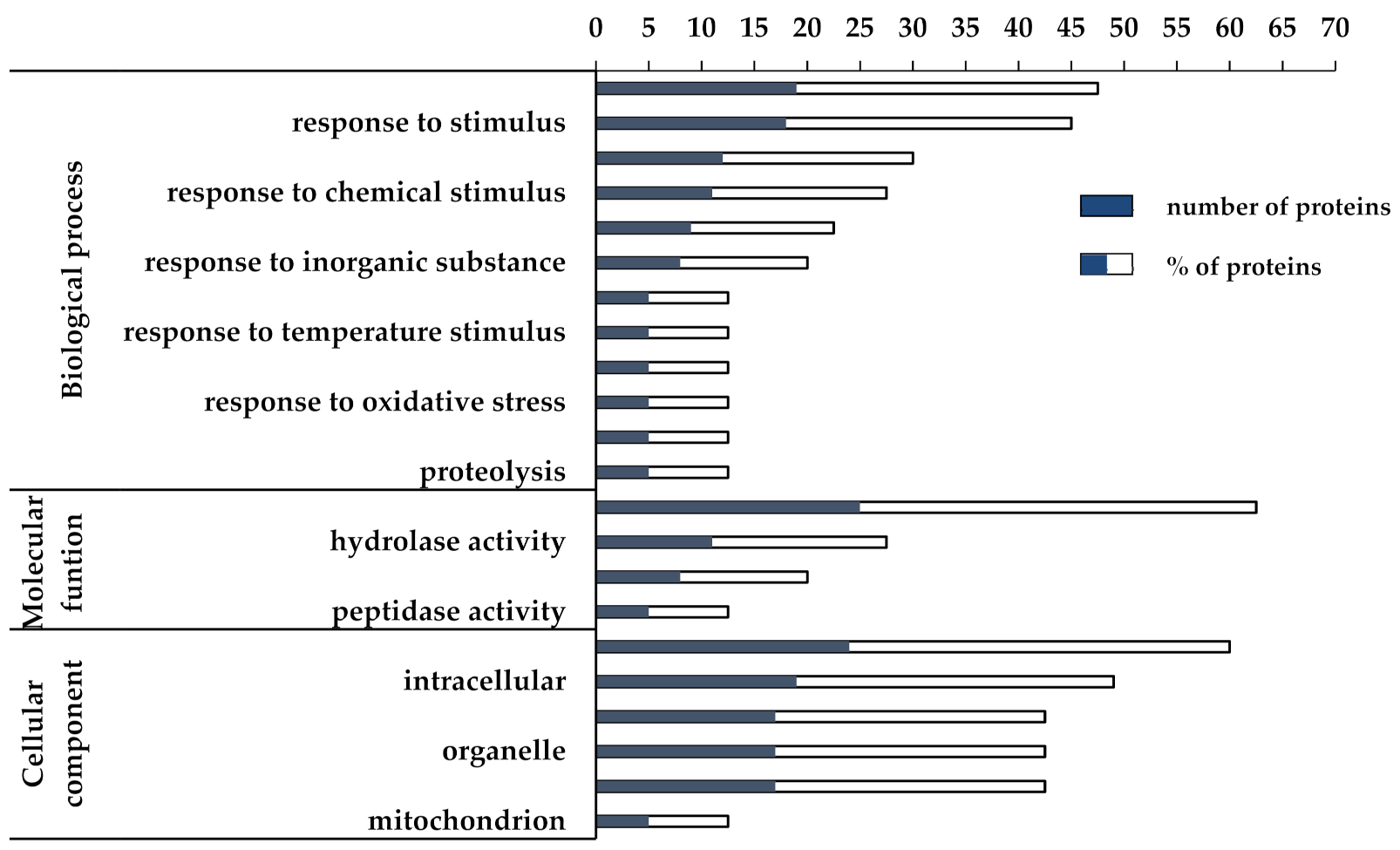
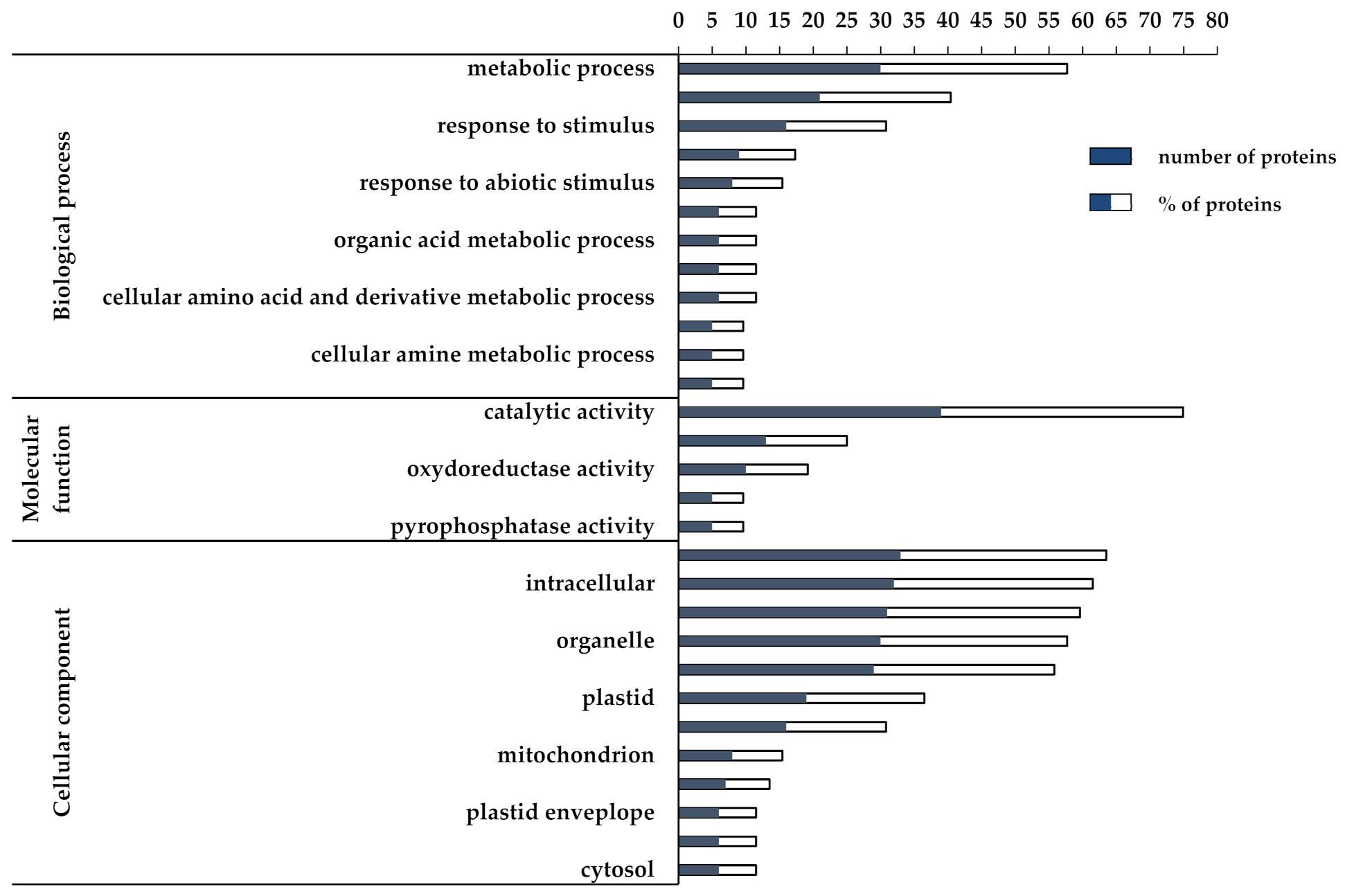
3.2. Proteomic and Functional Enrichment Analysis
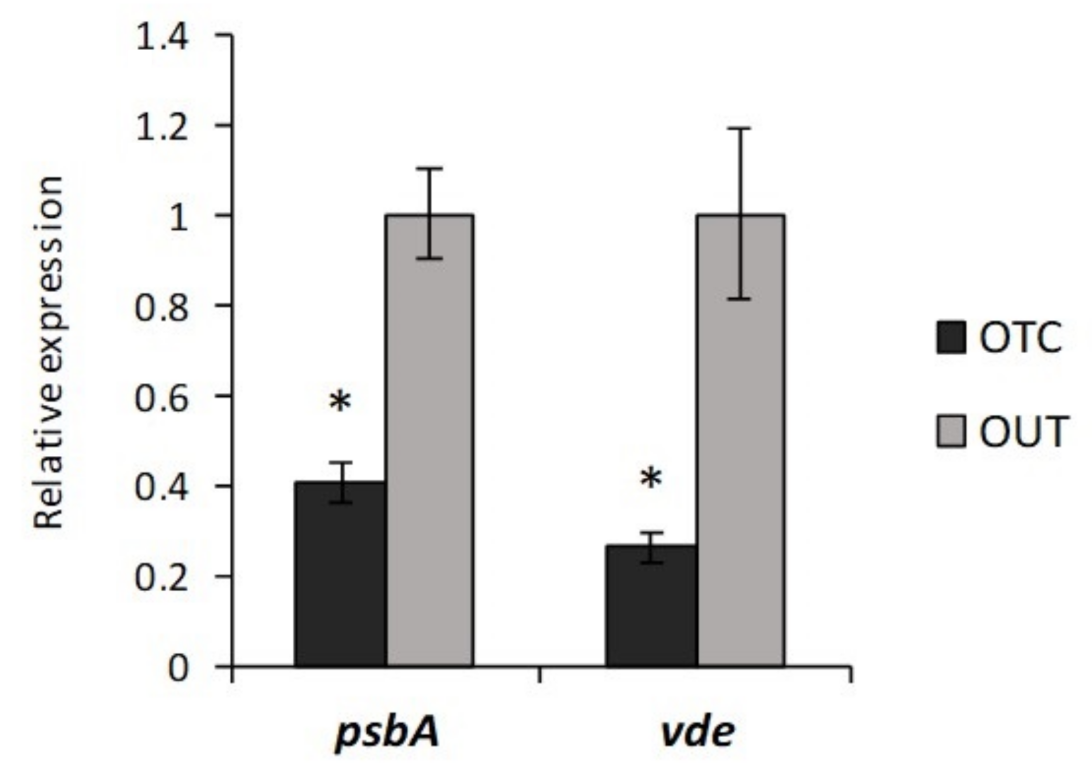
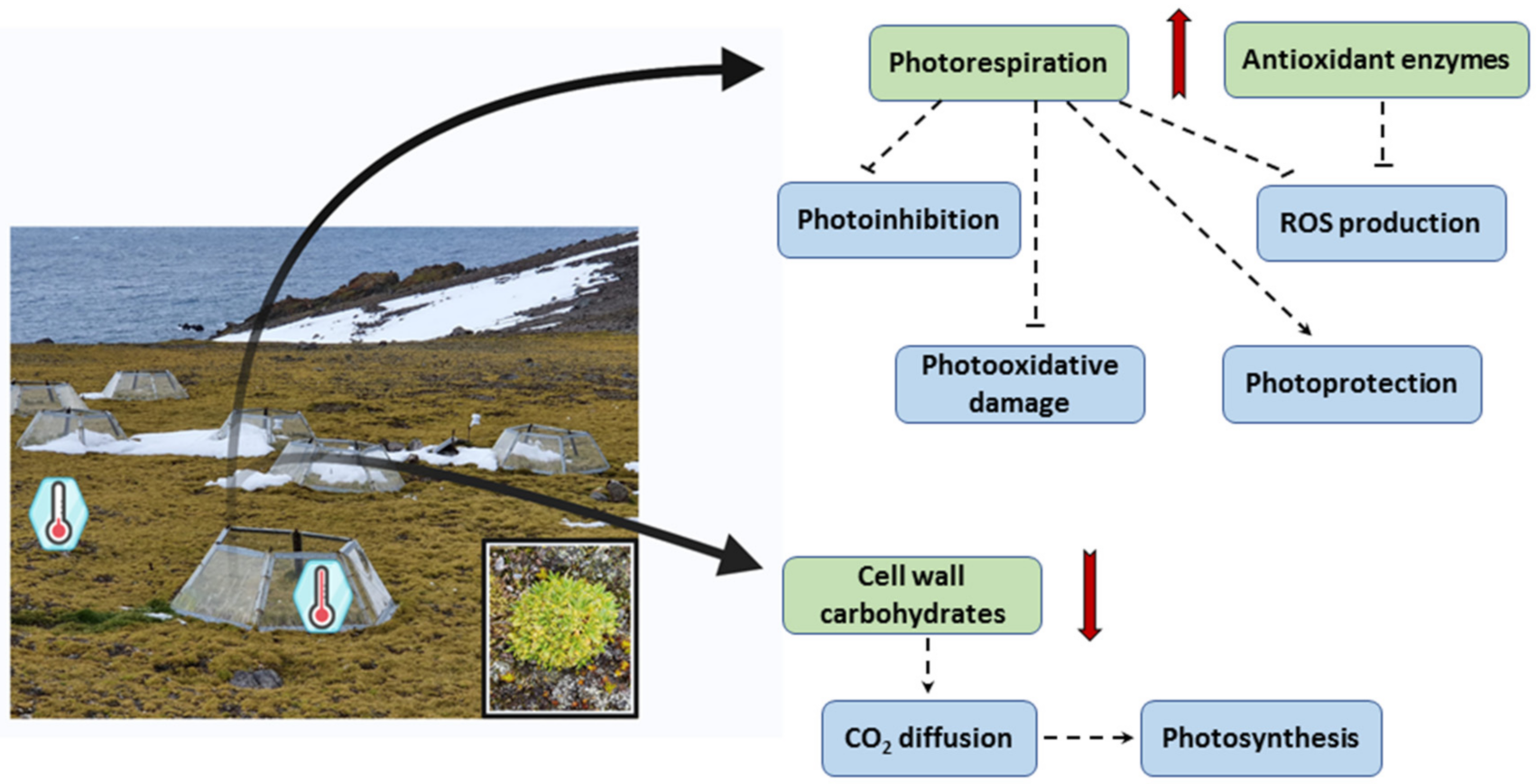
3.3. Photorespiration Protects Photosynthetic Apparatus Counteracting Photooxidation in C. quitensis Plants under Warming Conditions
3.4. Warming Conditions Influence High Irradiance-Induced Photodamage in C. quitensis
3.5. Temperature-Induced Lipocalin (TIL) and HSPs Contribute to Oxidative-Stress Control
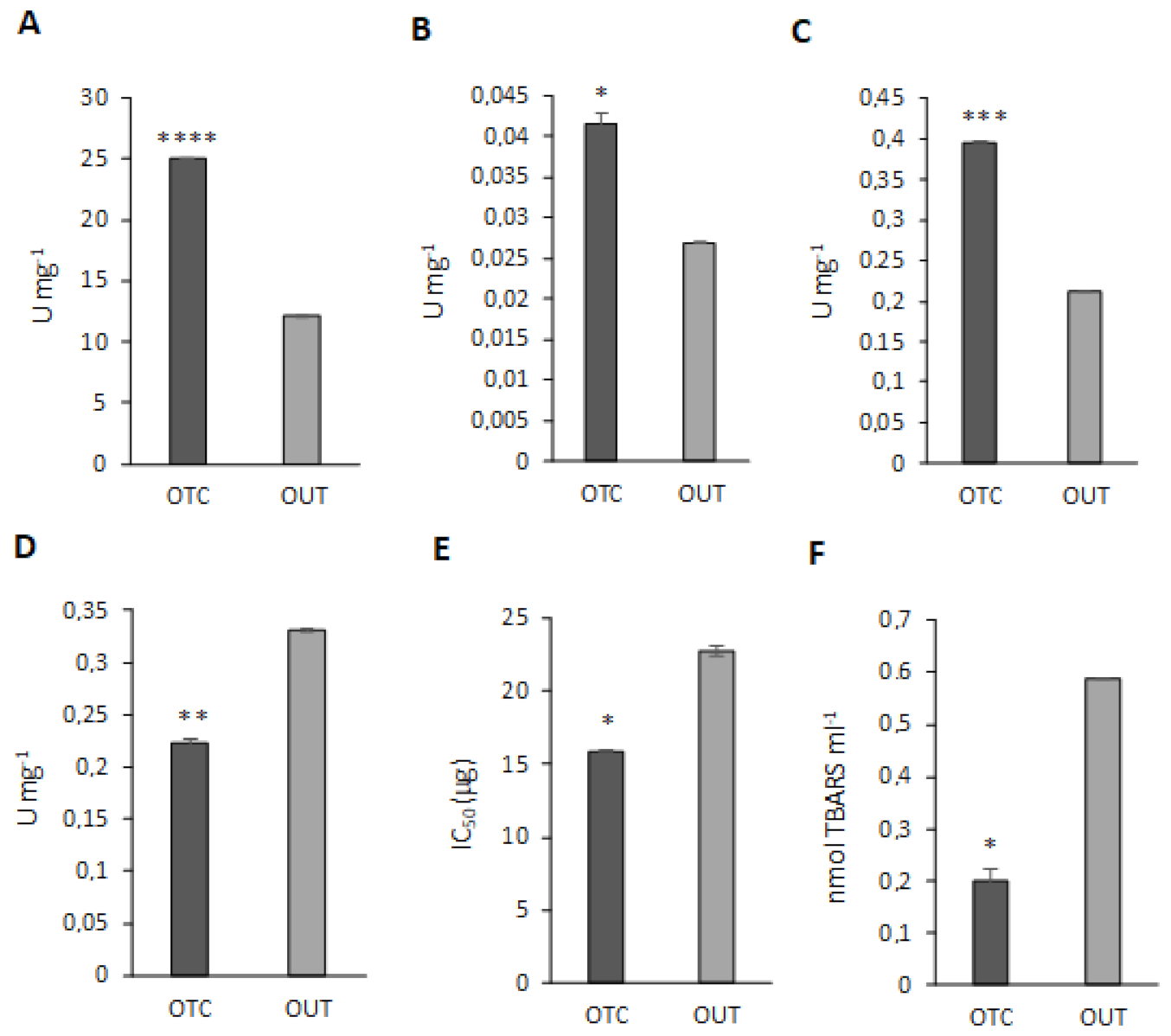
3.6. Antioxidant Enzyme Activity Assays
3.7. Warming Influences Carbohydrates Accumulation in C. quitensis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaughan, D.G.; Marshall, G.J.; Connolley, W.M.; Parkinson, C.; Mulvaney, R.; Hodgson, D.A.; King, J.C.; Pudsey, C.J.; Turner, J. Recent rapid regional climate warming on the Antarctic Peninsula. Clim. Chang. 2003, 60, 243–274. [Google Scholar] [CrossRef]
- Turner, J.; Colwell, S.R.; Marshall, G.J.; Lachlan-Cope, T.A.; Carleton, A.M.; Jones, P.D.; Lagun, V.; Reid, P.A.; Iagovkina, S. Antarctic climate change during the last 50 years. Int. J. Climatol. 2005, 25, 279–294. [Google Scholar] [CrossRef]
- Smith, R.I.L. The enigma of Colobanthus quitensis and Deschampsia antarctica in Antarctica. In Antarctic Biology in a Global Context; Huiskes, A.H.L., Gieskes, W.W.C., Rozema, J., Schorno, R.M.L., van der Vies, S.M., Wolff, W.J., Eds.; Backhuys Publishers: Leiden, The Netherlands, 2003; pp. 234–239. [Google Scholar]
- Sáez, P.L.; Bravo, L.A.; Cavieres, L.A.; Vallejos, V.; Sanhueza, C.; Font-Carrascosa, M.; Gil-Pelegrín, E.; Peguero-Pina, J.J.; Galmés, J. Photosynthetic limitations in Antarctic vascular plants: Importance of the leaf anatomical traits and Rubisco kinetics parameters. J. Exp. Bot. 2017, 68, 2871–2883. [Google Scholar] [CrossRef] [Green Version]
- Cavieres, L.A.; Sáez, P.; Sanhueza, C.; Sierra-Almeida, A.; Rabert, C.; Corcuera, L.J.; Bravo, L.A. Ecophysiological traits of Antarctic vascular plants: Their importance in the responses to climate change. Plant Ecol. 2016, 217, 343–358. [Google Scholar] [CrossRef]
- Torres-Díaz, C.; Gallardo-Cerda, J.; Lavin, P.; Oses, R.; Carrasco-Urra, F.; Atala, C.; Acuña-Rodríguez, I.S.; Convey, P.; Molina-Montenegro, M.A. Biological interactions and simulated climate change modulates the ecophysiological performance of Colobanthus quitensis in the Antarctic ecosystem. PLoS ONE 2016, 11, e0164844. [Google Scholar] [CrossRef] [Green Version]
- Nibert, M.L.; Manny, A.R.; Debat, H.J.; Firth, A.E.; Bertini, L.; Caruso, C. A barnavirus sequence mined from a transcriptome of the Antarctic pearlwort Colobanthus quitensis. Arch. Virol. 2018, 163, 1921–1926. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, G.I.; Torres-Díaz, C.; Bravo, L.A.; Balboa, K.; Caruso, C.; Bertini, L.; Proietti, S.; Molina-Montenegro, M.A. In silico analysis of metatranscriptomic data from the Antarctic vascular plant Colobanthus quitensis: Responses to a global warming scenario through changes in fungal gene expression levels. Fungal. Ecol. 2020, 43, 100873. [Google Scholar] [CrossRef]
- Torres-Mellado, G.A.; Jaña, R.; Casanova-Katny, M.A. Antarctic hairgrass expansion in the South Shetland archipelago and Antarctic Peninsula revisited. Polar Biol. 2011, 34, 1679–1688. [Google Scholar] [CrossRef]
- Cannone, N.; Guglielmin, M.; Convey, P.; Worland, M.R.; Favero Longo, S.E. Vascular plant changes in extreme environments: Effects of multiple drivers. Clim. Chang. 2016, 134, 651–665. [Google Scholar] [CrossRef] [Green Version]
- Day, T.A.; Ruhland, C.T.; Xiong, F.S. Warming increases aboveground plant biomass and C stocks in vascular-plant dominated Antarctic tundra. Glob. Chang. Biol. 2008, 14, 1827–1843. [Google Scholar] [CrossRef]
- Sáez, P.L.; Cavieres, L.A.; Galmés, J.; Gil-Pelegrín, E.; Peguero-Pina, J.J.; Sancho-Knapik, D.; Vivas, M.; Sanhueza, C.; Ramírez, C.F.; Rivera, B.K.; et al. In situ warming in the Antarctic: Effects on growth and photosynthesis in the Antarctic vascular plants. New Phytol. 2018, 218, 1406–1418. [Google Scholar] [CrossRef] [Green Version]
- Sierra-Almeida, A.; Cavieres, L.A.; Bravo, L.A. Warmer Temperatures Affect the in situ Freezing Resistance of the Antarctic Vascular Plants. Front. Plant Sci. 2018, 9, 1456. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Lu, H.; White, I.; King, J.C.; Phillips, T.; Hosking, J.S.; Bracegirdle, T.J.; Marshall, G.J.; Mulvaney, R.; Deb, P. Absence of 21st century warming on Antarctic Peninsula consistent with natural variability. Nature 2016, 535, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente-Moreno, M.J.; Omranian, N.; Sáez, P.; Figueroa, C.M.; Del-Saz, N.; Elso, M.; Poblete, L.; Orf, I.; Cuadros-Inostroza, A.; Cavieres, L.; et al. Cytochrome respiration pathway and sulphur metabolism sustain stress tolerance to low temperature in the Antarctic species Colobanthus quitensis. New Phytol. 2020, 225, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Arthofer, W.; Bertini, L.; Caruso, C.; Cicconardi, F.; Delph, L.F.; Fields, P.D.; Ikeda, M.; Minegishi, Y.; Proietti, S. Transcriptome sequencing of the Antarctic Colobanthus quitensis (Kunth) Bartl (Caryophillaceae). Mol. Ecol. Resour. 2015, 15, 1014–1015. [Google Scholar] [CrossRef]
- Hollister, R.D.; Webber, P.J. Biotic validation of small open-top chambers in a tundra ecosysem. Glob. Chang. Biol. 2000, 6, 835–842. [Google Scholar] [CrossRef]
- Bokhorst, S.; Huiskes, A.; Aerts, R.; Convey, P.; Cooper, E.J.; Dalen, L.; Erschbamer, B.; Gudmundsson, J.; Hofgaard, A.; Hollister, R.D.; et al. Variable temperature effects of Open Top Chambers at polar and alpine sites explained by irradiance and snow depth. Glob. Chang. Biol. 2013, 19, 64–74. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Iaconis, D.; Monti, M.; Renda, M.; van Koppen, A.; Tammaro, R.; Chiaravalli, M.; Cozzolino, F.; Pignata, P.; Crina, C.; Pucci, P.; et al. The centrosomal OFD1 protein interacts with the translation machinery and regulates the synthesis of specific targets. Sci. Rep. 2017, 7, 1224. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, F.; Landolfi, A.; Iacobucci, I.; Monaco, V.; Caterino, M.; Celentano, S.; Zuccato, C.; Cattaneo, E.; Monti, M. New Label-Free Methods for Protein Relative Quantification Applied to the Investigation of an Animal Model of Huntington Disease. PLoS ONE 2020, 15, e0238037. [Google Scholar] [CrossRef]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR Is a Web App for Creating, Exploring, Labeling and Sharing Volcano Plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. AgriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucl. Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Bertini, L.; Focaracci, F.; Proietti, S.; Papetti, P.; Caruso, C. Physiological response of Posidonia oceanica to heavy metal pollution along the Tyrrhenian coast. Funct. Plant Biol. 2019, 46, 933–941. [Google Scholar] [CrossRef]
- Bertini, L.; Proietti, S.; Focaracci, F.; Canini, F.; Bravo, L.A.; Rabert, C.; Caruso, C. Identification and validation of new reference genes for accurate quantitative reverse transcriptase-PCR normalization in the Antarctic plant Colobanthus quitensis under abiotic stress conditions. Polar Biol. 2021, 44, 389–405. [Google Scholar] [CrossRef]
- Laurino, S.; Grossi, G.; Pucci, P.; Flagiello, A.; Bufo, S.A.; Bianco, G.; Salvia, R.; Vinson, B.S.; Vogel, H.; Falabella, P. Identification of major Toxoneuron nigriceps venom proteins using an integrated transcriptomic/proteomic approach. Insect Biochem. Mol. Biol. 2016, 76, 49–61. [Google Scholar] [CrossRef]
- Soto-Suárez, M.; Serrato, A.J.; Rojas-González, J.A.; Bautista, R.; Sahrawy, M. Transcriptomic and proteomic approach to identify differentially expressed genes and proteins in Arabidopsis thaliana mutants lacking chloroplastic 1 and cytosolic FBPases reveals several levels of metabolic regulation. BMC Plant Biol. 2016, 16, 258. [Google Scholar] [CrossRef] [Green Version]
- Scieuzo, C.; Salvia, R.; Franco, A.; Pezzi, M.; Cozzolino, F.; Chicca, M.; Scapoli, C.; Vogel, H.; Monti, M.; Ferracini, C.; et al. An integrated transcriptomic and proteomic approach to identify the main Torymus sinensis venom components. Sci. Rep. 2021, 11, 5032. [Google Scholar] [CrossRef] [PubMed]
- Balbi, V.; Devoto, A. Jasmonate signalling network in Arabidopsis thaliana: Crucial regulatory nodes and new physiological scenarios. New Phytol. 2008, 177, 301–318. [Google Scholar] [CrossRef]
- Convey, P. Antarctic terrestrial ecosystems: Responses to environmental changes. Polarforsch 2006, 75, 101–111. [Google Scholar]
- Xiong, F.S.; Ruhland, T.C.; Day, T.A. Photosynthetic temperature response of the Antarctic vascular plants Colobanthus quitensis and Deschampsia antarctica. Physiol. Plant. 1999, 106, 276–286. [Google Scholar] [CrossRef]
- Derks, A.; Schaven, K.; Bruce, D. Diverse mechanisms for photoprotection in photosynthesis. Dynamic regulation of photosystem II excitation in response to rapid environmental change. Biochim. Biophys. Acta 2015, 1847, 468–485. [Google Scholar] [CrossRef] [Green Version]
- Voss, I.; Sunil, B.; Scheibe, R.; Raghavendra, A.S. Emerging concept for the role of photorespiration as an important part of abiotic stress response. Plant Biol. 2013, 15, 713–722. [Google Scholar] [CrossRef]
- Sunil, B.; Saini, D.; Bapatla, R.B.; Aswani, V.; Raghavendra, A.S. Photorespiration is complemented by cyclic electron flow and the alternative oxidase pathway to optimize photosynthesis and protect against abiotic stress. Photosynth. Res. 2019, 139, 67–79. [Google Scholar] [CrossRef]
- Bauwe, H.; Hagemann, M.; Kern, R.; Timm, S. Photorespiration has a dual origin and manifold links to central metabolism. Curr. Opin. Plant Biol. 2012, 15, 269–275. [Google Scholar] [CrossRef]
- Foyer, C.H.; Bloom, A.J.; Queval, G.; Noctor, G. Photorespiratory metabolism: Genes, mutants, energetics, and redox signaling. Annu. Rev. Plant Biol. 2009, 60, 455–484. [Google Scholar] [CrossRef]
- Walker, B.J.; Strand, D.D.; Kramer, D.M.; Cousins, A.B. The response of cyclic electron flow around photosystem I to changes in photorespiration and nitrate assimilation. Plant Physiol. 2014, 165, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Selinski, J.; Scheibe, R. Malate valves: Old shuttles with new perspectives. Plant Biol. 2019, 21, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Bykova, N.V.; Møller, I.M.; Gardeström, P.; Igamberdiev, A.U. The function of glycine decarboxylase complex is optimized to maintain high photorespiratory flux via buffering of its reaction products. Mitochondrion 2014, 19, 357–364. [Google Scholar] [CrossRef]
- Tomaz, T.; Bagard, M.; Pracharoenwattana, I.; Lindén, P.; Lee, C.P.; Carroll, A.J.; Ströher, E.; Smith, S.M.; Gardeström, P.; Millar, A.H. Mitochondrial malate dehydrogenase lowers leaf respiration and alters photorespiration and plant growth in Arabidopsis. Plant Physiol. 2010, 154, 1143–1157. [Google Scholar] [CrossRef] [Green Version]
- Timm, S.; Florian, A.; Jahnke, K.; Nunes-Nesi, A.; Fernie, A.R.; Bauwe, H. The hydroxypyruvate-reducing system in Arabidopsis: Multiple enzymes for the same end. Plant Physiol. 2011, 155, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Timm, S.; Wittmiß, M.; Gamlien, S.; Ewald, R.; Florian, A.; Frank, M.; Wirtz, M.; Hell, R.; Fernie, A.R.; Bauwe, H. Mitochondrial dihydrolipoyl dehydrogenase activity shapes photosynthesis and photorespiration of Arabidopsis thaliana. Plant Cell 2015, 27, 1968–1984. [Google Scholar] [CrossRef] [Green Version]
- Peterhänsel, C.; Krause, K.; Braun, H.P.; Espie, G.S.; Fernie, A.R.; Hanson, D.T.; Keech, O.; Maurino, V.G.; Mielewczik, M.; Sage, R.F. Engineering photorespiration: Current state and future possibilities. Plant Biol. 2013, 15, 754–758. [Google Scholar] [CrossRef]
- Aliyev, J.A. Photosynthesis, photorespiration and productivity of wheat and soybean genotypes. Physiol. Plant. 2012, 145, 369–383. [Google Scholar] [CrossRef]
- Hagemann, M.; Fernie, A.R.; Espie, G.S.; Kern, R.; Eisenhut, M.; Reumann, S.; Bauwe, H.; Weber, A.P.M. Evolution of the biochemistry of the photorespiratory C2 cycle. Plant Biol. 2013, 15, 639–647. [Google Scholar] [CrossRef]
- Foyer, C.H.; Noctor, G. Oxygen processing in photosynthesis: Regulation and signalling. New Phytol. 2000, 146, 359–388. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Torres, E.; Bravo, L.A.; Corcuera, L.J.; Johnson, G.N. Is electron transport to oxygen an important mechanism in photoprotection? Contrasting responses from Antarctic vascular plants. Physiol. Plant. 2007, 130, 185–194. [Google Scholar] [CrossRef]
- Bascuñán-Godoy, L.; Sanhueza, C.; Cuba, M.; Zuñiga, G.E.; Corcuera, L.J.; Bravo, L.A. Cold-acclimation limits low temperature induced photoinhibition by promoting a higher photochemical quantum yield and a more effective PSII restoration in darkness in the Antarctic rather than the Andean ecotype of Colobanthus quitensis Kunt Bartl (Cariophyllaceae). BMC Plant Biol. 2012, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Xu, D.H.; Kang, H.M.; Chen, K.; Wang, G. Photoprotective function of photorespiration in Reaumuria soongorica during different levels of drought stress in natural high irradiance. Photosynthetica 2008, 46, 232. [Google Scholar] [CrossRef]
- Guan, X.; Gu, S. Photorespiration and photoprotection of grapevine (Vitis vinifera L. cv. Cabernet Sauvignon) under water stress. Photosynthetica 2009, 47, 437. [Google Scholar] [CrossRef]
- Zhang, C.; Zhan, D.-X.; Luo, H.-H.; Zhang, Y.-L.; Zhang, W.-F. Photorespiration and photoinhibition in the bracts of cotton under water stress. Photosynthetica 2016, 54, 12–18. [Google Scholar] [CrossRef]
- Langer, T. AAA proteases: Cellular machines for degrading membrane proteins. Trends Biochem. Sci. 2000, 25, 247–251. [Google Scholar] [CrossRef]
- Sokolenko, A.; Pojidaeva, E.; Zinchenko, V.; Panichkin, V.; Glaser, V.M.; Herrmann, R.G.; Shestakov, S.V. The gene complement for proteolysis in the cyanobacterium Synechocystis sp. PCC 6803 and Arabidopsis thaliana chloroplasts. Curr. Genet. 2002, 41, 291–310. [Google Scholar] [CrossRef]
- Sakamoto, W.; Miura, E.; Kaji, Y.; Okuno, T.; Nishizono, M.; Ogura, T. Allelic characterization of the leaf-variegated mutation var2 identifies the conserved amino acid residues of FtsH that are important for ATP hydrolysis and proteolysis. Plant Mol. Biol. 2004, 56, 705–716. [Google Scholar] [CrossRef]
- Kato, Y.; Miura, E.; Ido, K.; Ifuku, K.; Sakamoto, W. The variegated mutants lacking chloroplastic FtsHs are defective in D1 degradation and accumulate reactive oxygen species. Plant Physiol. 2009, 151, 1790–1801. [Google Scholar] [CrossRef] [Green Version]
- Sinvany-Villalobo, G.; Davydov, O.; Ben-Ari, G.; Zaltsman, A.; Raskind, A.; Adam, Z. Expression in multigene families. Analysis of chloroplast and mitochondrial proteases. Plant Physiol. 2004, 135, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Qi, Y.; Malnoe, A.; Choquet, Y.; Wollman, F.A.; De Vitry, C. The high light response and redox control of thylakoid FtsH protease in Chlamydomonas Reinhardtii. Mol. Plant 2017, 10, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Adamska, I. The Elip family of stress proteins in the thylakoid membranes of pro- and eukaryota. In Advances in Photosynthesis and Respiration-Regulation of Photosynthesis; Aro, E.M., Andersson, B., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; Volume 11, pp. 487–505. [Google Scholar] [CrossRef]
- Andersson, U.; Heddad, M.; Adamska, I. Light stress-induced one-helix protein of the chlorophyll a/b-binding family associated with photosystem I. Plant Physiol. 2003, 132, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Hutin, C.; Nussaume, L.; Moise, N.; Moya, I.; Kloppstech, K.; Havaux, M. Early light-induced proteins protect Arabidopsis from photooxidative stress. Proc. Natl. Acad. Sci. USA 2003, 100, 4921–4926. [Google Scholar] [CrossRef] [Green Version]
- Zarter, C.R.; Adams, W.W., 3rd; Ebbert, V.; Adamska, I.; Jansson, S.; Demmig-Adams, B. Winter acclimation of PsbS and related proteins in the evergreen Arctostaphylos uva-ursi as influenced by altitude and light environment. Plant Cell Environ. 2006, 29, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Jin, E.S.; Polle, J.E.W.; Melis, A. Involvement of zeaxanthin and of the Cbr protein in the repair of photosystem II from photoinhibition in the green alga Dunaliella salina. Biochim. Biophys. Acta 2001, 1506, 244–259. [Google Scholar] [CrossRef] [Green Version]
- Jin, E.S.; Yokthongwattana, K.; Polle, J.E.W.; Melis, A. Role of the reversible xanthophyll cycle in the photosystem II damage and repair cycle in Dunaliella salina. Plant Physiol. 2003, 132, 352–364. [Google Scholar] [CrossRef] [Green Version]
- Niyogi, K.K. Safety valves for photosynthesis. Curr. Opin. Plant Biol. 2000, 3, 455–466. [Google Scholar] [CrossRef]
- Akerstrom, B.; Flower, D.R.; Salier, J.-P. Lipocalins: Unity in diversity. BBA Protein Struct M 2000, 1482, 1–8. [Google Scholar] [CrossRef]
- Charron, J.B.; Ouellet, F.; Pelletier, H.; Danyluk, J.; Chauve, C.; Sarhan, F. Identification, expression, and evolutionary analyses of plant lipocalins. Plant Physiol. 2005, 139, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Boca, S.; Koestler, F.; Ksas, B.; Chevalier, A.; Leymarie, J.; Fekete, A.; Mueller, M.J.; Havaux, M. Arabidopsis lipocalins AtCHL and AtTIL have distinct but overlapping functions essential for lipid protection and seed longevity. Plant Cell Environ. 2014, 37, 368–381. [Google Scholar] [CrossRef]
- Chi, W.T.; Fung, R.W.; Liu, H.C.; Hsu, C.C.; Chamg, Y.Y. Temperature-induced lipocalin is required for basal and acquired thermotolerance in Arabidopsis. Plant Cell Environ. 2009, 32, 917–927. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Greensmith, L. Induction of heat shock proteins for protection against oxidative stress. Adv. Drug Deliv. Rev. 2009, 61, 310–318. [Google Scholar] [CrossRef]
- Larkindale, J.; Mishkind, M.; Vierling, E. Plant responses to high temperature. In Plant Abiotic Stress; Jenks, M.A., Hasegawa, P.M., Eds.; Blackwell Publishing: Hoboken, NY, USA, 2005; pp. 100–144. [Google Scholar] [CrossRef]
- Obara, K.; Sumi, K.; Fukuda, H. The use of multiple transcription starts causes the dual targeting of Arabidopsis putative monodehydroascorbate reductase to both mitochondria and chloroplasts. Plant Cell Physiol. 2002, 43, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Dixon, D.P.; Davis, B.G.; Edwards, R. Functional divergence in the glutathione transferase superfamily in plants: Identification of two classes with putative functions in redox homeostasis in Arabidopsis thaliana. J. Biol. Chem. 2002, 277, 30859–30869. [Google Scholar] [CrossRef] [Green Version]
- Johnston, E.J.; Rylott, E.L.; Beynon, E.; Lorenz, A.; Chechik, V.; Bruce, N.C. Monodehydroascorbate reductase mediates TNT toxicity in plants. Science 2015, 349, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Rahantaniaina, M.S.; Li, S.; Chatel-Innocenti, G.; Tuzet, A.; Issakidis-Bourguet, E.; Mhamdi, A.; Noctor, G. Cytosolic and Chloroplastic DHARs Cooperate in Oxidative Stress-Driven Activation of the Salicylic Acid Pathway. Plant Physiol. 2017, 174, 956–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifacio, A.; Martins, M.O.; Ribeiro, C.W.; Fontenele, A.V.; Carvalho, F.E.L.; Margis-Pinheiro, M.; Silveira, J.A.G. Role of peroxidases in the compensation of cytosolic ascorbate peroxidase knockdown in rice plants under abiotic stress. Plant Cell Environ. 2011, 34, 1705–1722. [Google Scholar] [CrossRef] [PubMed]
- Xalxo, R.; Yadu, B.; Chandra, J.; Chandrakar, V.; Keshavkant, S. Alteration in carbohydrate metabolism modulates thermotolerance of plant under heat stress. In Heat Stress Tolerance in Plants: Physiological, Molecular and Genetic Perspectives, 1st ed.; Wani, S.H., Kumar, V., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2020. [Google Scholar] [CrossRef]
- Somerville, C.R.; Bauer, S.; Brininstool, G.; Facette, M.; Hamann, T.; Milne, J.; Osborne, E.; Paredez, A.; Persson, S.; Raab, T.; et al. Toward a systems approach to understanding plant cell walls. Science 2004, 306, 2206–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zúñiga-Feest, A.; Bascuñán-Godoy, L.; Reyes-Diaz, M.; Bravo, L.A.; Corcuera, L.J. Is survival after ice encasement related with sugar distribution in organs of the Antarctic plants Deschampsia antarctica Desv. (Poaceae) and Colobanthus quitensis (Kunth) Bartl. (Caryophyllaceae)? Polar Biol. 2009, 32, 583–591. [Google Scholar] [CrossRef]
- Bravo, L.A.; Ulloa, N.; Zúñiga, G.E.; Casanova, A.; Corcuera, L.J.; Alberdi, M. Cold resistance in Antarctic angiosperms. Physiol. Plant. 2001, 111, 55–65. [Google Scholar] [CrossRef]
- Rösti, J.; Barton, C.J.; Albrecht, S.; Dupree, P.; Pauly, M.; Findlay, K.; Roberts, K.; Seiferta, G.J. UDP-Glucose 4-Epimerase Isoforms UGE2 and UGE4 Cooperate in Providing UDP-Galactose for Cell Wall Biosynthesis and Growth of Arabidopsis thaliana. Plant Cell 2007, 19, 1565–1579. [Google Scholar] [CrossRef] [Green Version]
- Bokhorst, S.; Bjerke, J.W.; Davey, M.P.; Taulavuori, K.; Taulavuori, E.; Laine, K.; Callaghan, T.V.; Gareth, K.P. Impacts of extreme winter warming events on plant physiology in a sub-Arctic heath community. Physiol. Plant. 2010, 140, 128–140. [Google Scholar] [CrossRef]
- Pagter, M.; Andersen, U.B.; Andersen, L. Winter warming delays dormancy release, advances budburst, alters carbohydrate metabolism and reduces yield in a temperate shrub. AoB Plants 2015, 7, plv024. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TAIR Code | TAIR Description | p-Value | log2FC OTC/OUT |
|---|---|---|---|
| AT2G28490 | RmlC-like cupins superfamily protein | 5.29 × 10−4 | 4.81 |
| AT1G54870 | NAD(P)-binding Rossmann-fold superfamily protein | 5.49 × 10−3 | 4.78 |
| AT5G60160 | Zn-dependent exopeptidases superfamily protein | 2.95 × 10−3 | 4.64 |
| AT5G44120 | ATCRA1, CRA1, CRU1, RmlC-like cupins superfamily protein | 4.18 × 10−5 | 4.48 |
| AT5G44120 | ATCRA1, CRA1, CRU1, RmlC-like cupins superfamily protein | 3.14 × 10−3 | 4.07 |
| AT3G54470 | Uridine 5’-monophosphate synthase/UMP synthase (PYRE-F) (UMPS) | 1.54 × 10−4 | 3.80 |
| AT3G51640 | Stress response NST1-like protein | 1.36 × 10−3 | 3.65 |
| AT1G48030 | mtLPD1, mitochondrial lipoamide dehydrogenase 1 | 5.23 × 10−3 | 3.53 |
| AT1G04580 | AAO4, AO4, ATAO-4, ATAO2, aldehyde oxidase 4 | 5.10 × 10−4 | 3.44 |
| AT1G79530 | GAPCP-1, glyceraldehyde-3-phosphate dehydrogenase of plastid 1 | 2.51 × 10−3 | 3.35 |
| AT2G15220 | Plant basic secretory protein (BSP) family protein | 2.51 × 10−3 | 3.35 |
| AT3G22640 | PAP85, cupin family protein | 7.96 × 10−3 | 3.23 |
| AT2G24530 | Transcriptional regulator of RNA poIII, SAGA | 9.63 × 10−3 | 3.03 |
| AT3G09540 | Pectin lyase-like superfamily protein | 2.25 × 10−3 | 2.95 |
| AT1G43670 | Inositol monophosphatase family protein (CYFBP, Fructose-1,6-bisphosphatase, cytosolic) | 7.04 × 10−3 | 2.71 |
| AT4G25150 | HAD superfamily, subfamily IIIB acid phosphatase | 4.62 × 10−3 | 2.63 |
| AT5G19660 | ATS1P, ATSBT6.1, S1P, SITE-1 protease | 9.39 × 10−3 | 2.34 |
| AT1G13750 | Purple acid phosphatases superfamily protein | 7.29 × 10−3 | 2.05 |
| AT2G05840 | PAA2, 20S proteasome subunit PAA2 | 2.86 × 10−3 | 1.93 |
| AT5G67360 | ARA12, subtilase family protein | 1.42 × 10−3 | 1.76 |
| AT4G35090 | CAT2, catalase 2 | 7.64 × 10−4 | 1.61 |
| AT2G24200 | Cytosol aminopeptidase family protein (ATLAP1, LAP1, LEUCYL AMINOPEPTIDASE 1) | 9.74 × 10−4 | 1.60 |
| AT5G58070 | ATTIL, TIL, temperature-induced lipocalin | 5.70 × 10−3 | 1.40 |
| AT1G03090 | MCCA, methylcrotonyl-CoA carboxylase alpha chain, mitochondrial/3-methylcrotonyl-CoA carboxylase 1 (MCCA) | 7.70 × 10−3 | 1.28 |
| AT1G54050 | HSP20-like chaperones superfamily protein | 2.25 × 10−3 | 0.94 |
| AT2G22780 | PMDH1, peroxisomal NAD-malate dehydrogenase 1 | 6.05 × 10−3 | 0.57 |
| AT1G31190 | IMPL1, myo-inositol monophosphatase like 1 | 5.98 × 10−3 | 0.57 |
| AT1G59730 | ATH7, TH7, thioredoxin H-type 7 | 5.98 × 10−3 | 0.57 |
| AT1G71695 | Peroxidase superfamily protein | 5.98 × 10−3 | 0.57 |
| AT1G74310 | ATHSP101, HOT1, HSP101, heat shock protein 101 | 5.98 × 10−3 | 0.57 |
| AT2G05030 | Transposable element gene | 5.98 × 10−3 | 0.57 |
| AT2G05710 | ACO3, aconitase 3 | 5.98 × 10−3 | 0.57 |
| AT2G25890 | Oleosin family protein | 5.98 × 10−3 | 0.57 |
| AT2G29570 | ATPCNA2, PCNA2, proliferating cell nuclear antigen 2 | 5.98 × 10−3 | 0.57 |
| AT2G44050 | COS1, COS1, 6,7-dimethyl-8-ribityllumazine synthase/DMRL synthase/lumazine synthase/riboflavin synthase | 5.98 × 10−3 | 0.57 |
| AT3G01570 | Oleosin family protein | 5.98 × 10−3 | 0.57 |
| AT3G11930 | Adenine nucleotide alpha hydrolases-like superfamily protein | 5.98 × 10−3 | 0.57 |
| AT3G23490 | CYN, cyanase | 5.98 × 10−3 | 0.57 |
| AT3G55620 | emb1624, translation initiation factor IF6 | 5.98 × 10−3 | 0.57 |
| AT4G16260 | Glycosyl hydrolase superfamily protein | 5.98 × 10−3 | 0.57 |
| AT1G53240 | mMDH1, lactate/malate dehydrogenase family protein | 1.06 × 10−3 | 0.41 |
| TAIR Code | TAIR Description | p-Value | log2FC OTC/OUT |
|---|---|---|---|
| AT5G05200 | Protein kinase superfamily protein | 3.33 × 10−3 | −4.39 |
| AT1G77510 | RPT3, regulatory particle triple-A ATPase 3 | 1.03 × 10−4 | −4.33 |
| AT5G58290 | RPT3, regulatory particle triple-A ATPase 3 | 7.35 × 10−4 | −4.23 |
| AT2G40840 | DPE2, disproportionating enzyme 2 | 7.56 × 10−3 | −4.11 |
| AT2G20420 | ATP citrate lyase (ACL) family protein | 5.65 × 10−3 | −3.88 |
| AT3G23570 | Alpha/beta-Hydrolases superfamily protein | 4.85 × 10−3 | −3.76 |
| AT1G79870 | D-isomer specific 2-hydroxyacid dehydrogenase family protein | 5.97 × 10−3 | −3.61 |
| AT1G63940 | MDAR6, monodehydroascorbate reductase 6 | 3.78 × 10−4 | −3.52 |
| AT5G17380 | Thiamine pyrophosphate dependent pyruvate decarboxylase family protein | 5.08 × 10−3 | −3.49 |
| AT1G11860 | Glycine cleavage T-protein family | 7.50 × 10−3 | −3.40 |
| AT1G75270 | DHAR2, dehydroascorbate reductase 2 | 3.35 × 10−3 | −3.32 |
| AT5G48540 | Receptor-like protein kinase-related family protein | 7.88 × 10−3 | −3.30 |
| AT3G55270 | ATMKP1, MKP1, mitogen-activated protein kinase phosphatase 1 | 2.18 × 10−4 | −3.12 |
| AT4G18810 | NAD(P)-binding Rossmann-fold superfamily protein | 1.80 × 10−3 | −3.09 |
| AT4G10960 | UGE5, UDP-D-glucose/UDP-D-galactose 4-epimerase 5 | 1.66 × 10−3 | −3.01 |
| AT4G11150 | emb2448, TUF, TUFF, VHA-E1, vacuolar ATP synthase subunit E1 | 6.54 × 10−3 | −2.99 |
| AT3G03250 | AtUGP1, UGP, UGP1, UDP-GLUCOSE PYROPHOSPHORYLASE 1 | 4.73 × 10−3 | −2.87 |
| AT1G74910 | ADP-glucose pyrophosphorylase family protein | 9.88 × 10−3 | −2.84 |
| AT1G48830 | Ribosomal protein S7e family protein | 5.25 × 10−3 | −2.81 |
| AT1G53450 | Epstein-barr nuclear antigen | 2.81 × 10−3 | −2.74 |
| AT1G72680 | ATCAD1, CAD1, cinnamyl-alcohol dehydrogenase | 3.17 × 10−3 | −2.64 |
| AT5G26780 | SHM2, serine hydroxymethyltransferase 2 | 9.12 × 10−4 | −2.62 |
| AT4G11600 | ATGPX6, GPX6, LSC803, PHGPX, glutathione peroxidase 6 | 3.98 × 10−3 | −2.58 |
| AT2G45300 | RNA 3’-terminal phosphate cyclase/enolpyruvate transferase, alpha/beta | 2.76 × 10−3 | −2.55 |
| ATCG01280 | YCF2.2, chloroplast Ycf2;ATPase, AAA type, core | 1.03 × 10−3 | −2.49 |
| AT1G19920 | APS2, ASA1, pseudouridine synthase/archaeosine transglycosylase-like family protein | 5.55 × 10−3 | −2.42 |
| AT1G09310 | Protein of unknown function, DUF538 | 9.05 × 10−3 | −2.39 |
| AT5G46290 | KAS I, KAS1, 3-ketoacyl-acyl carrier protein synthase I | 2.42 × 10−3 | −2.33 |
| AT3G53580 | Diaminopimelate epimerase family protein | 2.09 × 10−3 | −2.08 |
| AT2G30110 | ATUBA1, MOS5, UBA1, ubiquitin-activating enzyme 1 | 9.46 × 10−3 | −1.99 |
| AT2G37770 | NAD(P)-linked oxidoreductase superfamily protein (AKR4C9, ALDO-KETO REDUCTASE FAMILY 4 MEMBER C9, CHLAKR, CHLOROPLASTIC ALDO-KETO REDUCTASE) | 4.95 × 10−4 | −1.99 |
| AT5G17770 | ATCBR, CBR, CBR1, NADH:cytochrome B5 reductase 1 | 6.18 × 10−3 | −1.92 |
| AT1G10810 | NAD(P)-linked oxidoreductase superfamily protein | 2.89 × 10−3 | −1.90 |
| AT1G09130 | ATP-dependent caseinolytic (Clp) protease/crotonase family protein | 5.48 × 10−3 | −1.89 |
| AT5G61510 | GroES-like zinc-binding alcohol dehydrogenase family protein | 4.33 × 10−3 | −1.87 |
| AT2G42590 | GF14 MU, GRF9, general regulatory factor 9 | 6.54 × 10−3 | −1.86 |
| AT3G49830 | P-loop containing nucleoside triphosphate hydrolases superfamily protein | 6.41 × 10−4 | −1.75 |
| AT2G22910 | NAGS1, N-acetyl-l-glutamate synthase 1 | 2.32 × 10−3 | −1.69 |
| AT4G17170 | AT-RAB2, ATRAB-B1B, ATRAB2A, ATRABB1C, RAB-B1B, RAB2A, RABB1C, RAB GTPase homolog B1C | 9.62 × 10−3 | −1.51 |
| AT2G22780 | PMDH1, peroxisomal NAD-malate dehydrogenase 1 | 6.68 × 10−3 | −1.50 |
| AT3G51800 | ATEBP1, ATG2, EBP1, metallopeptidase M24 family protein | 7.91 × 10−3 | −1.50 |
| AT2G27680 | NAD(P)-linked oxidoreductase superfamily protein | 1.01 × 10−3 | −1.38 |
| AT2G30950 | FTSH2, VAR2, FtsH extracellular protease family | 1.02 × 10−3 | −1.38 |
| AT3G22840 | ELIP, ELIP1, chlorophyll A-B binding family protein | 1.02 × 10−3 | −1.38 |
| AT1G62780 | Dimethylallyl, adenosine tRNA methylthiotransferase | 1.38 × 10−3 | −1.20 |
| AT4G24280 | cpHsc70-1, chloroplast heat shock protein 70-1 | 8.01 × 10−3 | −1.09 |
| AT2G21530 | SMAD/FHA domain-containing protein | 5.64 × 10−3 | −1.06 |
| AT5G17920 | ATCIMS, ATMETS, ATMS1, cobalamin-independent synthase family protein | 1.89 × 10−3 | −1.01 |
| AT1G70730 | Phosphoglucomutase/phosphomannomutase family protein (PGM) | 1.22 × 10−3 | −0.94 |
| AT3G04770 | RPSAb, 40s ribosomal protein SA B | 4.08 × 10−4 | −0.90 |
| AT4G35090 | CAT2, catalase 2 | 1.85 × 10−3 | −0.76 |
| AT1G80380 | P-loop containing nucleoside triphosphate hydrolases superfamily protein | 5.56 × 10−3 | −0.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertini, L.; Cozzolino, F.; Proietti, S.; Falconieri, G.S.; Iacobucci, I.; Salvia, R.; Falabella, P.; Monti, M.; Caruso, C. What Antarctic Plants Can Tell Us about Climate Changes: Temperature as a Driver for Metabolic Reprogramming. Biomolecules 2021, 11, 1094. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11081094
Bertini L, Cozzolino F, Proietti S, Falconieri GS, Iacobucci I, Salvia R, Falabella P, Monti M, Caruso C. What Antarctic Plants Can Tell Us about Climate Changes: Temperature as a Driver for Metabolic Reprogramming. Biomolecules. 2021; 11(8):1094. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11081094
Chicago/Turabian StyleBertini, Laura, Flora Cozzolino, Silvia Proietti, Gaia Salvatore Falconieri, Ilaria Iacobucci, Rosanna Salvia, Patrizia Falabella, Maria Monti, and Carla Caruso. 2021. "What Antarctic Plants Can Tell Us about Climate Changes: Temperature as a Driver for Metabolic Reprogramming" Biomolecules 11, no. 8: 1094. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11081094