
Transgenic Mouse Overexpressing Spermine Oxidase in Cerebrocortical Neurons: Astrocyte Dysfunction and Susceptibility to Epileptic Seizures

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
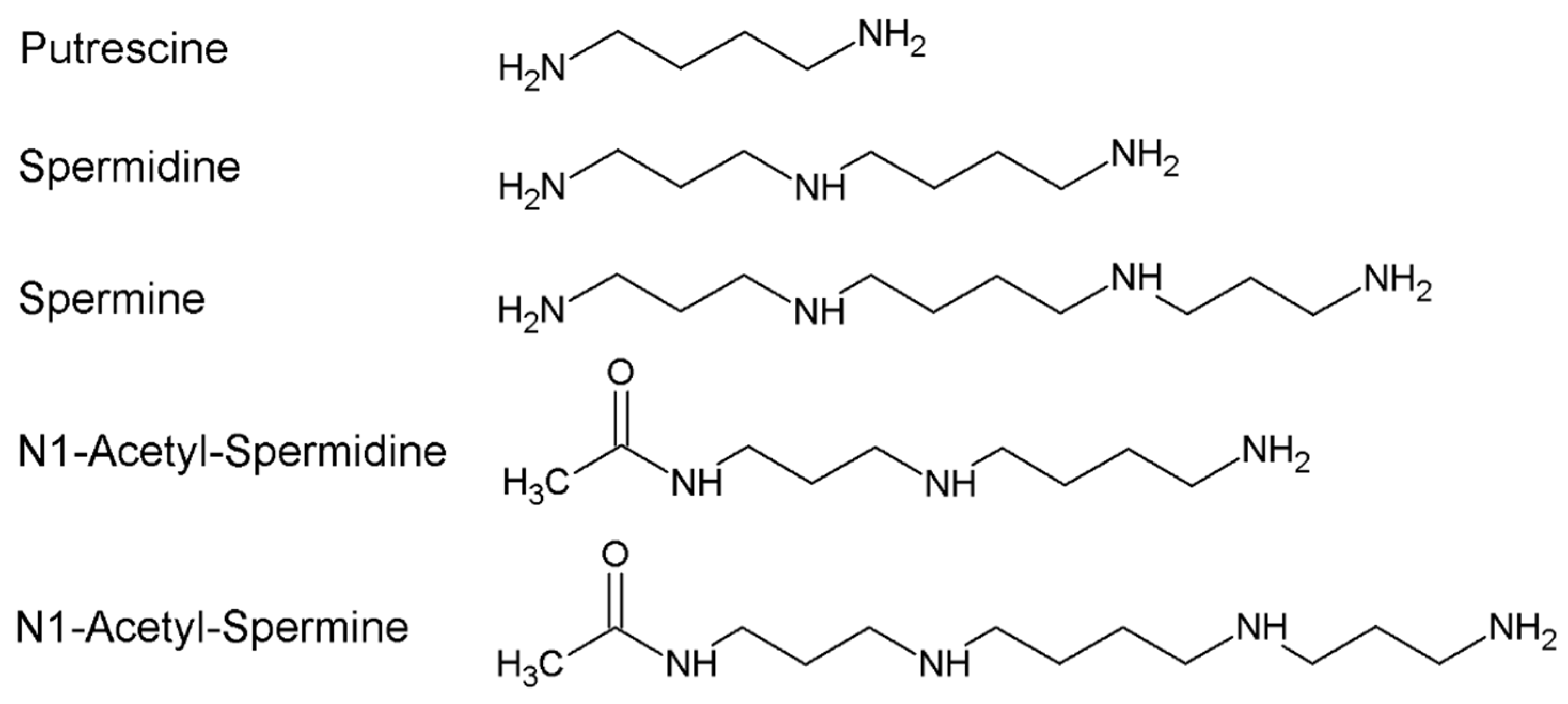
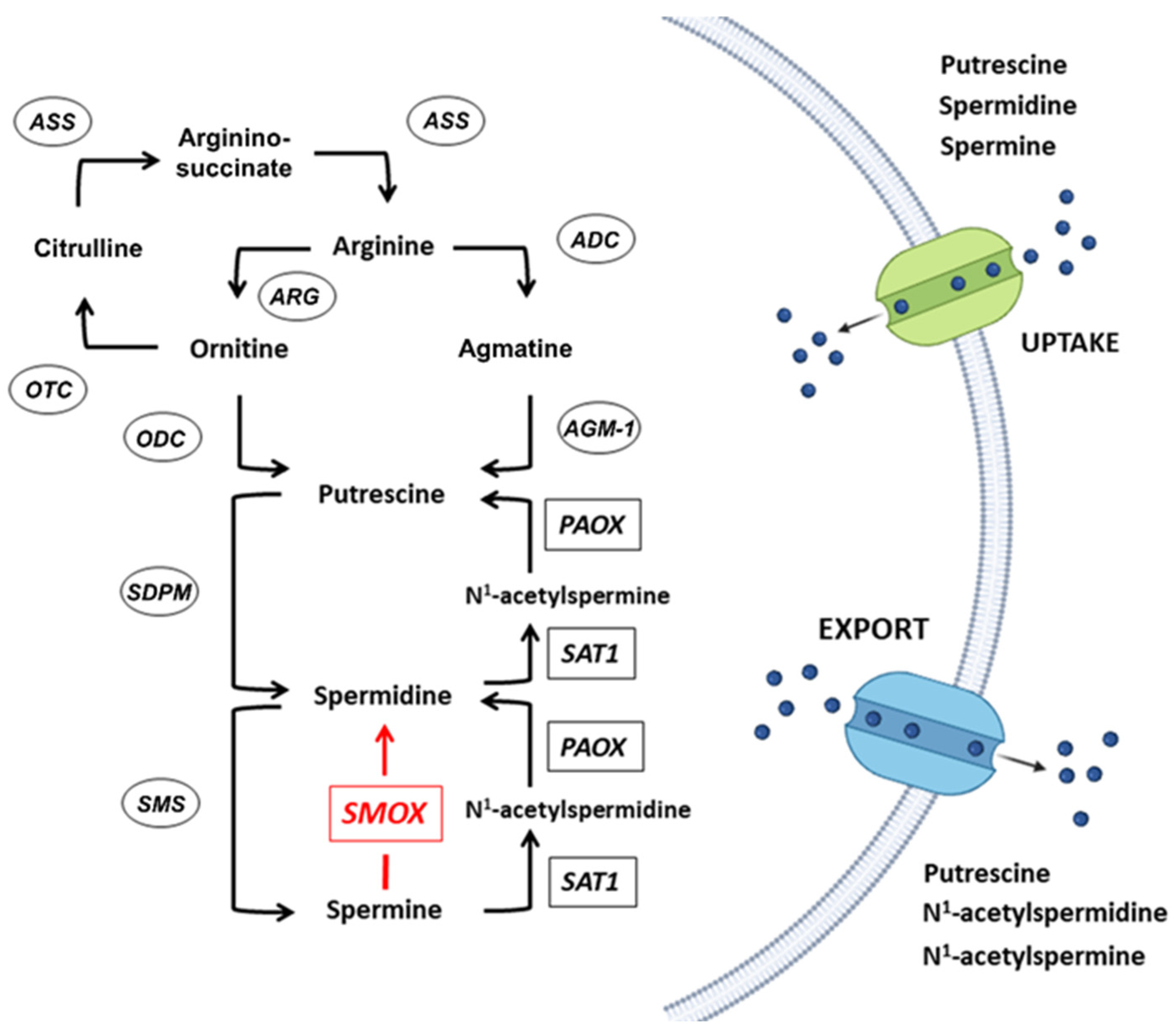
:1. Polyamines Metabolism
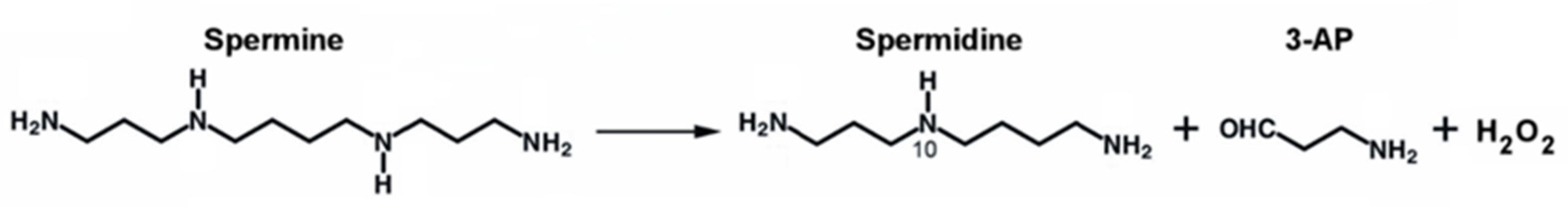
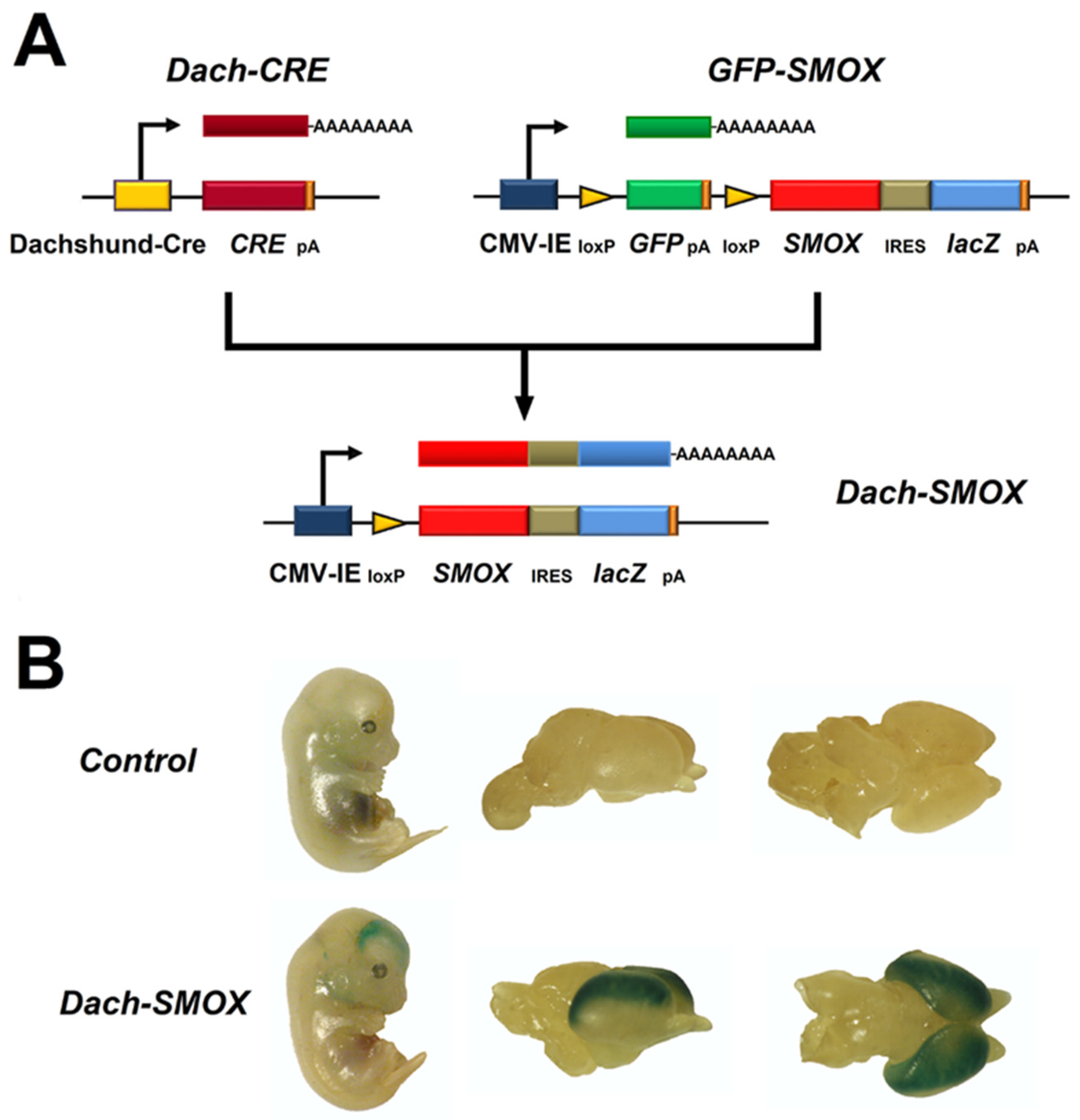
2. The SMOX Overexpressing Mouse: An Animal Model of Chronic Spm Catabolism Activation
3. Reactive Astrocytosis
4. Reduced Spm Content in Reactive Astrocyte Processes
5. Expression of AMPA GluA2-Lacking Receptors linked to Ca2+ Entry and Activation of Glutamate Release from Reactive Astrocyte Processes
6. Increased xc− Transporter and Increased Glutamate In-Out Transport from Reactive Astrocyte Processes
7. Reduced Expression of EAAT1 and EAAT2 in Reactive Astrocyte Processes
8. Neuron Loss
9. Chronic Oxidative Stress
10. Chronic Excitotoxic Stress
11. Susceptibility to Seizure
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, S.; Cleveland, J.L. Polyamine Homeostasis in Development and Disease. Med. Sci. 2021, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Sagar, N.A.; Tarafdar, S.; Agarwal, S.; Tarafdar, A.; Sharma, S. Polyamines: Functions, Metabolism, and Role in Human Disease Management. Med. Sci. 2021, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Cason, A.L.; Ikeguchi, Y.; Skinner, C.; Wood, T.C.; Holden, K.R.; Lubs, H.A.; Martinez, F.; Simensen, R.J.; Stevenson, R.E.; Pegg, A.E.; et al. X-linked spermine synthase gene (SMS) defect: The first polyamine deficiency syndrome. Eur. J. Hum. Genet. EJHG 2003, 11, 937–944. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, A.; Gwadry, F.G.; Ffrench-Mullen, J.M.; Canetti, L.; Gingras, Y.; Casero, R.A., Jr.; Rouleau, G.; Benkelfat, C.; Turecki, G. Implication of SSAT by gene expression and genetic variation in suicide and major depression. Arch. Gen. Psychiatry 2006, 63, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Ikeguchi, Y.; Bewley, M.C.; Pegg, A.E. Aminopropyltransferases: Function, structure and genetics. J. Biochem. 2006, 139, 161–169. [Google Scholar] [CrossRef]
- Cervelli, M.; Bellini, A.; Bianchi, M.; Marcocci, L.; Nocera, S.; Polticelli, F.; Federico, R.; Amendola, R.; Mariottini, P. Mouse spermine oxidase gene splice variants. Nuclear subcellular localization of a novel active isoform. Eur. J. Biochem. 2004, 271, 760–770. [Google Scholar] [CrossRef]
- Wallace, H.M. The physiological role of the polyamines. Eur. J. Clin. Investig. 2000, 30, 1–3. [Google Scholar] [CrossRef]
- Vujcic, S.; Diegelman, P.; Bacchi, C.J.; Kramer, D.L.; Porter, C.W. Identification and characterization of a novel flavin-containing spermine oxidase of mammalian cell origin. Biochem. J. 2002, 367, 665–675. [Google Scholar] [CrossRef] [Green Version]
- Cervelli, M.; Polticelli, F.; Federico, R.; Mariottini, P. Heterologous expression and characterization of mouse spermine oxidase. J. Biol. Chem. 2003, 278, 5271–5276. [Google Scholar] [CrossRef] [Green Version]
- Cervelli, M.; Amendola, R.; Polticelli, F.; Mariottini, P. Spermine oxidase: Ten years after. Amino Acids 2012, 42, 441–450. [Google Scholar] [CrossRef]
- Cervelli, M.; Bellavia, G.; D’Amelio, M.; Cavallucci, V.; Moreno, S.; Berger, J.; Nardacci, R.; Marcoli, M.; Maura, G.; Piacentini, M.; et al. A New Transgenic Mouse Model for Studying the Neurotoxicity of Spermine Oxidase Dosage in the Response to Excitotoxic Injury. PLoS ONE 2013, 8, e64810. [Google Scholar] [CrossRef]
- Cervelli, M.; Fratini, E.; Amendola, R.; Bianchi, M.; Signori, E.; Ferraro, E.; Lisi, A.; Federico, R.; Marcocci, L.; Mariottini, P. Increased spermine oxidase (SMO) activity as a novel differentiation marker of myogenic C2C12 cells. Int. J. Biochem. Cell Biol. 2009, 41, 934–944. [Google Scholar] [CrossRef]
- Polticelli, F.; Salvi, D.; Mariottini, P.; Amendola, R.; Cervelli, M. Molecular evolution of the polyamine oxidase gene family in Metazoa. BMC Evol. Biol. 2012, 12, 90. [Google Scholar] [CrossRef] [Green Version]
- Ceci, R.; Duranti, G.; Leonetti, A.; Pietropaoli, S.; Spinozzi, F.; Marcocci, L.; Amendola, R.; Cecconi, F.; Sabatini, S.; Mariottini, P.; et al. Adaptive responses of heart and skeletal muscle to spermine oxidase overexpression: Evaluation of a new transgenic mouse model. Free. Radic. Biol. Med. 2017, 103, 216–225. [Google Scholar] [CrossRef]
- Masuko, T.; Kusama-Eguchi, K.; Sakata, K.; Kusama, T.; Chaki, S.; Okuyama, S.; Williams, K.; Kashiwagi, K.; Igarashi, K. Polyamine transport, accumulation, and release in brain. J. Neurochem. 2003, 84, 610–617. [Google Scholar] [CrossRef]
- Williams, K. Interactions of polyamines with ion channels. Biochem. J. 1997, 325 Pt 2, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Tomitori, H.; Mizuno, S.; Higashi, K.; Füll, C.; Fukiwake, T.; Terui, Y.; Leewanich, P.; Nishimura, K.; Toida, T.; et al. Binding of spermine and ifenprodil to a purified, soluble regulatory domain of the N-methyl-D-aspartate receptor. J. Neurochem. 2008, 107, 1566–1577. [Google Scholar] [CrossRef] [Green Version]
- Donevan, S.D.; Rogawski, M.A. Intracellular polyamines mediate inward rectification of Ca(2+)-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Proc. Natl. Acad. Sci. USA 1995, 92, 9298–9302. [Google Scholar] [CrossRef] [Green Version]
- Kamboj, S.K.; Swanson, G.T.; Cull-Candy, S.G. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J. Physiol. 1995, 486 Pt 2, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Bhukel, A.; Madeo, F.; Sigrist, S.J. Spermidine boosts autophagy to protect from synapse aging. Autophagy 2017, 13, 444–445. [Google Scholar] [CrossRef] [Green Version]
- Hofer, S.J.; Liang, Y.; Zimmermann, A.; Schroeder, S.; Dengjel, J.; Kroemer, G.; Eisenberg, T.; Sigrist, S.J.; Madeo, F. Spermidine-induced hypusination preserves mitochondrial and cognitive function during aging. Autophagy 2021, 17, 2037–2039. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Madeo, F.; Eisenberg, T.; Büttner, S.; Ruckenstuhl, C.; Kroemer, G. Spermidine: A novel autophagy inducer and longevity elixir. Autophagy 2010, 6, 160–162. [Google Scholar] [CrossRef]
- de Cabo, R.; Carmona-Gutierrez, D.; Bernier, M.; Hall, M.N.; Madeo, F. The search for antiaging interventions: From elixirs to fasting regimens. Cell 2014, 157, 1515–1526. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.K.; Pech, U.; Bhukel, A.; Fulterer, A.; Ender, A.; Mauermann, S.F.; Andlauer, T.F.; Antwi-Adjei, E.; Beuschel, C.; Thriene, K.; et al. Spermidine Suppresses Age-Associated Memory Impairment by Preventing Adverse Increase of Presynaptic Active Zone Size and Release. PLoS Biol. 2016, 14, e1002563. [Google Scholar] [CrossRef]
- Davies, D.A.; Adlimoghaddam, A.; Albensi, B.C. Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease. Cells 2021, 10, 1884. [Google Scholar] [CrossRef]
- Yoshida, M.; Higashi, K.; Jin, L.; Machi, Y.; Suzuki, T.; Masuda, A.; Dohmae, N.; Suganami, A.; Tamura, Y.; Nishimura, K.; et al. Identification of acrolein-conjugated protein in plasma of patients with brain infarction. Biochem. Biophys. Res. Commun. 2010, 391, 1234–1239. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Modulation of cellular function by polyamines. Int. J. Biochem. Cell Biol. 2010, 42, 39–51. [Google Scholar] [CrossRef]
- Ivanova, S.; Botchkina, G.I.; Al-Abed, Y.; Meistrell, M., 3rd; Batliwalla, F.; Dubinsky, J.M.; Iadecola, C.; Wang, H.; Gregersen, P.K.; Eaton, J.W.; et al. Cerebral ischemia enhances polyamine oxidation: Identification of enzymatically formed 3-aminopropanal as an endogenous mediator of neuronal and glial cell death. J. Exp. Med. 1998, 188, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, S.; Batliwalla, F.; Mocco, J.; Kiss, S.; Huang, J.; Mack, W.; Coon, A.; Eaton, J.W.; Al-Abed, Y.; Gregersen, P.K.; et al. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. Proc. Natl. Acad. Sci. USA 2002, 99, 5579–5584. [Google Scholar] [CrossRef] [Green Version]
- Tomitori, H.; Usui, T.; Saeki, N.; Ueda, S.; Kase, H.; Nishimura, K.; Kashiwagi, K.; Igarashi, K. Polyamine oxidase and acrolein as novel biochemical markers for diagnosis of cerebral stroke. Stroke 2005, 36, 2609–2613. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.; Nishimura, K.; Ishii, I.; Omura, T.; Okuyama, S.; Kashiwagi, K.; Igarashi, K. Intense correlation between brain infarction and protein-conjugated acrolein. Stroke 2009, 40, 3356–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiki, R.; Park, H.; Ishii, I.; Yoshida, M.; Nishimura, K.; Toida, T.; Tatsukawa, H.; Kojima, S.; Ikeguchi, Y.; Pegg, A.E.; et al. Brain infarction correlates more closely with acrolein than with reactive oxygen species. Biochem. Biophys. Res. Commun. 2011, 404, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Cervelli, M.; Angelucci, E.; Germani, F.; Amendola, R.; Mariottini, P. Inflammation, carcinogenesis and neurodegeneration studies in transgenic animal models for polyamine research. Amino Acids 2014, 46, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Cervetto, C.; Vergani, L.; Passalacqua, M.; Ragazzoni, M.; Venturini, A.; Cecconi, F.; Berretta, N.; Mercuri, N.; D’Amelio, M.; Maura, G.; et al. Astrocyte-Dependent Vulnerability to Excitotoxicity in Spermine Oxidase-Overexpressing Mouse. Neuromolecular Med. 2016, 18, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Pietropaoli, S.; Leonetti, A.; Cervetto, C.; Venturini, A.; Mastrantonio, R.; Baroli, G.; Persichini, T.; Colasanti, M.; Maura, G.; Marcoli, M.; et al. Glutamate Excitotoxicity Linked to Spermine Oxidase Overexpression. Mol. Neurobiol. 2018, 55, 7259–7270. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Baroli, G.; Fratini, E.; Pietropaoli, S.; Marcoli, M.; Mariottini, P.; Cervelli, M. Epileptic seizures and oxidative stress in a mouse model over-expressing spermine oxidase. Amino Acids 2020, 52, 129–139. [Google Scholar] [CrossRef]
- Wang, D.D.; Bordey, A. The astrocyte odyssey. Prog. Neurobiol. 2008, 86, 342–367. [Google Scholar] [CrossRef]
- Parpura, V.; Heneka, M.T.; Montana, V.; Oliet, S.H.R.; Schousboe, A.; Haydon, P.G.; Stout, R.F., Jr.; Spray, D.C.; Reichenbach, A.; Pannicke, T.; et al. Glial cells in (patho)physiology. J. Neurochem. 2012, 121, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Meldolesi, J. Astrocytes: News about Brain Health and Diseases. Biomedicines 2020, 8, 394. [Google Scholar] [CrossRef]
- Cervetto, C.; Venturini, A.; Passalacqua, M.; Guidolin, D.; Genedani, S.; Fuxe, K.; Borroto-Esquela, D.O.; Cortelli, P.; Woods, A.; Maura, G.; et al. A2A-D2 receptor-receptor interaction modulates gliotransmitter release from striatal astrocyte processes. J. Neurochem. 2017, 140, 268–279. [Google Scholar] [CrossRef]
- Cervetto, C.; Averna, M.; Vergani, L.; Pedrazzi, M.; Amato, S.; Pelassa, S.; Giuliani, S.; Baldini, F.; Maura, G.; Mariottini, P.; et al. Reactive Astrocytosis in a Mouse Model of Chronic Polyamine Catabolism Activation. Biomolecules 2021, 11, 1274. [Google Scholar] [CrossRef]
- Skatchkov, S.N.; Antonov, S.M.; Eaton, M.J. Glia and glial polyamines. Role in brain function in health and disease. Biochem. Suppl. Ser. A Membr. Cell Biol. 2016, 10, 73–98. [Google Scholar] [CrossRef]
- Skatchkov, S.N.; Woodbury-Fariña, M.A.; Eaton, M. The role of glia in stress: Polyamines and brain disorders. Psychiatr. Clin. N. Am. 2014, 37, 653–678. [Google Scholar] [CrossRef] [Green Version]
- Fage, D.; Voltz, C.; Scatton, B.; Carter, C. Selective release of spermine and spermidine from the rat striatum by N-methyl-D-aspartate receptor activation in vivo. J. Neurochem. 1992, 58, 2170–2175. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Kofuji, P.; Araque, A. G-Protein-Coupled Receptors in Astrocyte-Neuron Communication. Neuroscience 2021, 456, 71–84. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Sidoryk-Wegrzynowicz, M.; Wegrzynowicz, M.; Lee, E.; Bowman, A.B.; Aschner, M. Role of astrocytes in brain function and disease. Toxicol. Pathol. 2011, 39, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eulenburg, V.; Gomeza, J. Neurotransmitter transporters expressed in glial cells as regulators of synapse function. Brain Res. Rev. 2010, 63, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Moussawi, K.; Riegel, A.; Nair, S.; Kalivas, P.W. Extracellular glutamate: Functional compartments operate in different concentration ranges. Front. Syst. Neurosci. 2011, 5, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatkowski, M.; Barbour, B.; Attwell, D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 1990, 348, 443–446. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Perea, G.; Araque, A. GLIA modulates synaptic transmission. Brain Res. Rev. 2010, 63, 93–102. [Google Scholar] [CrossRef]
- Parpura, V.; Zorec, R. Gliotransmission: Exocytotic release from astrocytes. Brain Res. Rev. 2010, 63, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Santello, M.; Calì, C.; Bezzi, P. Gliotransmission and the tripartite synapse. Adv. Exp. Med. Biol. 2012, 970, 307–331. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef]
- Cervetto, C.; Frattaroli, D.; Venturini, A.; Passalacqua, M.; Nobile, M.; Alloisio, S.; Tacchetti, C.; Maura, G.; Agnati, L.F.; Marcoli, M. Calcium-permeable AMPA receptors trigger vesicular glutamate release from Bergmann gliosomes. Neuropharmacology 2015, 99, 396–407. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Semyanov, A.; Zorec, R. Physiology of Astroglial Excitability. Function 2020, 1, zqaa016. [Google Scholar] [CrossRef]
- Bernardinelli, Y.; Muller, D.; Nikonenko, I. Astrocyte-synapse structural plasticity. Neural Plast. 2014, 2014, 232105. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Pedrazzi, M.; Patrone, M.; Passalacqua, M.; Ranzato, E.; Colamassaro, D.; Sparatore, B.; Pontremoli, S.; Melloni, E. Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J. Immunol. 2007, 179, 8525–8532. [Google Scholar] [CrossRef] [Green Version]
- Schiweck, J.; Eickholt, B.J.; Murk, K. Important Shapeshifter: Mechanisms Allowing Astrocytes to Respond to the Changing Nervous System During Development, Injury and Disease. Front. Cell. Neurosci. 2018, 12, 261. [Google Scholar] [CrossRef] [Green Version]
- Derouiche, A.; Geiger, K.D. Perspectives for Ezrin and Radixin in Astrocytes: Kinases, Functions and Pathology. Int. J. Mol. Sci. 2019, 20, 3776. [Google Scholar] [CrossRef] [Green Version]
- Lavialle, M.; Aumann, G.; Anlauf, E.; Pröls, F.; Arpin, M.; Derouiche, A. Structural plasticity of perisynaptic astrocyte processes involves ezrin and metabotropic glutamate receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 12915–12919. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.S.; Hu, S.; Feng, A.; Rock, R.B. Reactive oxygen species from human astrocytes induced functional impairment and oxidative damage. Neurochem. Res. 2013, 38, 2148–2159. [Google Scholar] [CrossRef] [Green Version]
- Malpica-Nieves, C.J.; Rivera, Y.; Rivera-Aponte, D.E.; Phanstiel, O.; Veh, R.W.; Eaton, M.J.; Skatchkov, S.N. Uptake of Biotinylated Spermine in Astrocytes: Effect of Cx43 siRNA, HIV-Tat Protein and Polyamine Transport Inhibitor on Polyamine Uptake. Biomolecules 2021, 11, 1187. [Google Scholar] [CrossRef]
- Malpica-Nieves, C.J.; Rivera-Aponte, D.E.; Tejeda-Bayron, F.A.; Mayor, A.M.; Phanstiel, O.; Veh, R.W.; Eaton, M.J.; Skatchkov, S.N. The involvement of polyamine uptake and synthesis pathways in the proliferation of neonatal astrocytes. Amino Acids 2020, 52, 1169–1180. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar]
- Kucheryavykh, Y.V.; Pearson, W.L.; Kurata, H.T.; Eaton, M.J.; Skatchkov, S.N.; Nichols, C.G. Polyamine permeation and rectification of Kir4.1 channels. Channels 2007, 1, 172–178. [Google Scholar] [CrossRef]
- Nichols, C.G.; Lee, S.J. Polyamines and potassium channels: A 25-year romance. J. Biol. Chem. 2018, 293, 18779–18788. [Google Scholar] [CrossRef] [Green Version]
- Song, I.; Huganir, R.L. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002, 25, 578–588. [Google Scholar] [CrossRef]
- Rakhade, S.N.; Fitzgerald, E.F.; Klein, P.M.; Zhou, C.; Sun, H.; Huganir, R.L.; Jensen, F.E. Glutamate Receptor 1 Phosphorylation at Serine 831 and 845 Modulates Seizure Susceptibility and Hippocampal Hyperexcitability after Early Life Seizures. J. Neurosci. 2013, 33, 4623. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Z.; Chung, J.M.; Chung, K.; Kang, M.G. Reactive oxygen species (ROS) modulate AMPA receptor phosphorylation and cell-surface localization in concert with pain-related behavior. Pain 2012, 153, 1905–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhade, S.N.; Zhou, C.; Aujla, P.K.; Fishman, R.; Sucher, N.J.; Jensen, F.E. Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 7979–7990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, J.; Basedow, F.J.; Dekker, A.D.; Papantoniou, C. Phosphorylation of AMPA-type glutamate receptors: The trigger of epileptogenesis? J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 5879–5880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lia, A.; Henriques, V.J.; Zonta, M.; Chiavegato, A.; Carmignoto, G.; Gómez-Gonzalo, M.; Losi, G. Calcium Signals in Astrocyte Microdomains, a Decade of Great Advances. Front. Cell. Neurosci. 2021, 15, 673433. [Google Scholar] [CrossRef]
- Ahmadpour, N.; Kantroo, M.; Stobart, J.L. Extracellular Calcium Influx Pathways in Astrocyte Calcium Microdomain Physiology. Biomolecules 2021, 11, 1467. [Google Scholar] [CrossRef]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef]
- Bridges, R.; Lutgen, V.; Lobner, D.; Baker, D.A. Thinking outside the cleft to understand synaptic activity: Contribution of the cystine-glutamate antiporter (System xc−) to normal and pathological glutamatergic signaling. Pharmacol. Rev. 2012, 64, 780–802. [Google Scholar] [CrossRef] [Green Version]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc⁻ cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Lehre, K.P.; Danbolt, N.C. The number of glutamate transporter subtype molecules at glutamatergic synapses: Chemical and stereological quantification in young adult rat brain. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 8751–8757. [Google Scholar] [CrossRef] [Green Version]
- Volterra, A.; Trotti, D.; Tromba, C.; Floridi, S.; Racagni, G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 1994, 14, 2924–2932. [Google Scholar] [CrossRef]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 1998, 19, 328–334. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A.M. ROS and brain diseases: The good, the bad, and the ugly. Oxidative Med. Cell. Longev. 2013, 2013, 963520. [Google Scholar] [CrossRef]
- Vergani, L.; Cristina, L.; Paola, R.; Luisa, A.M.; Shyti, G.; Edvige, V.; Giuseppe, M.; Elena, G.; Laura, C.; Adriana, V. Metals, metallothioneins and oxidative stress in blood of autistic children. Res. Autism Spectr. Disord. 2011, 5, 286–293. [Google Scholar] [CrossRef]
- Juárez-Rebollar, D.; Rios, C.; Nava-Ruíz, C.; Méndez-Armenta, M. Metallothionein in Brain Disorders. Oxidative Med. Cell. Longev. 2017, 2017, 5828056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, J.; Aschner, M.; Zatta, P.; Vasák, M. Roles of the metallothionein family of proteins in the central nervous system. Brain Res. Bull. 2001, 55, 133–145. [Google Scholar] [CrossRef]
- Aschner, M.; Syversen, T.; Souza, D.O.; Rocha, J.B. Metallothioneins: Mercury species-specific induction and their potential role in attenuating neurotoxicity. Exp. Biol. Med. 2006, 231, 1468–1473. [Google Scholar] [CrossRef]
- Cabungcal, J.H.; Steullet, P.; Kraftsik, R.; Cuenod, M.; Do, K.Q. Early-life insults impair parvalbumin interneurons via oxidative stress: Reversal by N-acetylcysteine. Biol. Psychiatry 2013, 73, 574–582. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free. Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef]
- Penkowa, M. Metallothioneins are multipurpose neuroprotectants during brain pathology. FEBS J. 2006, 273, 1857–1870. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the balance between ROS and antioxidants: When to use the synthetic antioxidants. Oxidative Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Khan, M.A.; Moskal, J.R.; Todd, K.G.; Tanay, V.A.; Baker, G. Aldehyde load in ischemia-reperfusion brain injury: Neuroprotection by neutralization of reactive aldehydes with phenelzine. Brain Res. 2006, 1122, 184–190. [Google Scholar] [CrossRef]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxidative Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Guo, C.; Ma, Y.Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef]
- Mattson, M.P. Chapter 11—Excitotoxicity. In Stress: Physiology, Biochemistry, and Pathology; Fink, G., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 125–134. [Google Scholar]
- Najm, I.; el-Skaf, G.; Tocco, G.; Vanderklish, P.; Lynch, G.; Baudry, M. Seizure activity-induced changes in polyamine metabolism and neuronal pathology during the postnatal period in rat brain. Brain Research. Dev. Brain Res. 1992, 69, 11–21. [Google Scholar] [CrossRef]
- Baudry, M.; Najm, I. Kainate-induced seizure activity stimulates the polyamine interconversion pathway in rat brain. Neurosci. Lett. 1994, 171, 151–154. [Google Scholar] [CrossRef]
- Hayashi, Y.; Baudry, M. Effect of kainate-induced seizure activity on the polyamine interconversion pathway in juvenile rat brain. Brain Res. Dev. Brain Res. 1995, 87, 96–99. [Google Scholar] [CrossRef]
- Hayashi, Y.; Morizumi, Y.; Hattori, Y.; Tanaka, J. Pentylenetetrazol-induced kindling stimulates the polyamine interconversion pathway in rat brain. Brain Res. 1999, 828, 184–188. [Google Scholar] [CrossRef]
- Shin, E.J.; Jeong, J.H.; Chung, Y.H.; Kim, W.K.; Ko, K.H.; Bach, J.H.; Hong, J.S.; Yoneda, Y.; Kim, H.C. Role of oxidative stress in epileptic seizures. Neurochem. Int. 2011, 59, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, C.C.; Almeida, A.B.; Araújo, P.V.; de Abreu, R.N.; Chaves, E.M.; do Vale, O.C.; Macêdo, D.S.; Woods, D.J.; Fonteles, M.M.; Vasconcelos, S.M. Oxidative stress and epilepsy: Literature review. Oxidative Med. Cell. Longev. 2012, 2012, 795259. [Google Scholar] [CrossRef] [Green Version]
- Barker-Haliski, M.; White, H.S. Glutamatergic Mechanisms Associated with Seizures and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022863. [Google Scholar] [CrossRef] [Green Version]
- Wetherington, J.; Serrano, G.; Dingledine, R. Astrocytes in the epileptic brain. Neuron 2008, 58, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Kardos, J.; Szabó, Z.; Héja, L. Framing Neuro-Glia Coupling in Antiepileptic Drug Design. J. Med. Chem. 2016, 59, 777–787. [Google Scholar] [CrossRef]
- Heuser, K.; de Curtis, M.; Steinhäuser, C. Editorial: Glial Dysfunction in Epileptogenesis. Front. Neurol. 2021, 12, 716308. [Google Scholar] [CrossRef]
- Verhoog, Q.P.; Holtman, L.; Aronica, E.; van Vliet, E.A. Astrocytes as Guardians of Neuronal Excitability: Mechanisms Underlying Epileptogenesis. Front. Neurol. 2020, 11, 591690. [Google Scholar] [CrossRef]
- Stanfield, P.R.; Sutcliffe, M.J. Spermine is fit to block inward rectifier (Kir) channels. J. Gen. Physiol. 2003, 122, 481–484. [Google Scholar] [CrossRef]
- Fleidervish, I.A.; Libman, L.; Katz, E.; Gutnick, M.J. Endogenous polyamines regulate cortical neuronal excitability by blocking voltage-gated Na+ channels. Proc. Natl. Acad. Sci. USA 2008, 105, 18994–18999. [Google Scholar] [CrossRef] [Green Version]
- Pál, B. Involvement of extrasynaptic glutamate in physiological and pathophysiological changes of neuronal excitability. Cell. Mol. Life Sci. CMLS 2018, 75, 2917–2949. [Google Scholar] [CrossRef]
- Lopatin, A.N.; Makhina, E.N.; Nichols, C.G. The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. J. Gen. Physiol. 1995, 106, 923–955. [Google Scholar] [CrossRef] [Green Version]
- Bellot-Saez, A.; Kékesi, O.; Morley, J.W.; Buskila, Y. Astrocytic modulation of neuronal excitability through K(+) spatial buffering. Neurosci. Biobehav. Rev. 2017, 77, 87–97. [Google Scholar] [CrossRef]
- Djukic, B.; Casper, K.B.; Philpot, B.D.; Chin, L.S.; McCarthy, K.D. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 11354–11365. [Google Scholar] [CrossRef]
- Haj-Yasein, N.N.; Jensen, V.; Vindedal, G.F.; Gundersen, G.A.; Klungland, A.; Ottersen, O.P.; Hvalby, O.; Nagelhus, E.A. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia 2011, 59, 1635–1642. [Google Scholar] [CrossRef] [Green Version]
- David, Y.; Cacheaux, L.P.; Ivens, S.; Lapilover, E.; Heinemann, U.; Kaufer, D.; Friedman, A. Astrocytic dysfunction in epileptogenesis: Consequence of altered potassium and glutamate homeostasis? J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 10588–10599. [Google Scholar] [CrossRef] [Green Version]
- Kinboshi, M.; Ikeda, A.; Ohno, Y. Role of Astrocytic Inwardly Rectifying Potassium (Kir) 4.1 Channels in Epileptogenesis. Front. Neurol. 2020, 11, 626658. [Google Scholar] [CrossRef]
- Wang, F.; Qi, X.; Zhang, J.; Huang, J.H. Astrocytic modulation of potassium under seizures. Neural Regen. Res. 2020, 15, 980–987. [Google Scholar] [CrossRef]
- Galanopoulou, A.S.; Löscher, W.; Lubbers, L.; O’Brien, T.J.; Staley, K.; Vezzani, A.; D’Ambrosio, R.; White, H.S.; Sontheimer, H.; Wolf, J.A.; et al. Antiepileptogenesis and disease modification: Progress, challenges, and the path forward-Report of the Preclinical Working Group of the 2018 NINDS-sponsored antiepileptogenesis and disease modification workshop. Epilepsia Open 2021, 6, 276–296. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 2011, 20, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grone, B.P.; Baraban, S.C. Animal models in epilepsy research: Legacies and new directions. Nat. Neurosci. 2015, 18, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New avenues for anti-epileptic drug discovery and development. Nat. Reviews. Drug Discov. 2013, 12, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.R.; Belarde, J.A.; Johnson, H.F.; Aizenman, C.D. A neuroprotective role for polyamines in a Xenopus tadpole model of epilepsy. Nat. Neurosci. 2011, 14, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Godoy, L.D.; Liberato, J.L.; da Silva Junior, P.I.; dos Santos, W.F. Mygalin: A new anticonvulsant polyamine in acute seizure model and neuroethological schedule. Cent. Nerv. Syst. Agents Med. Chem. 2013, 13, 122–131. [Google Scholar] [CrossRef]
- Ferchmin, P.A.; Pérez, D.; Biello, M. Spermine is neuroprotective against anoxia and N-methyl-D-aspartate in hippocampal slices. Brain Res. 2000, 859, 273–279. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Liu, H.; Pearson, L.; Kapoor, M.; Harrison, J.C.; Sammut, I.A.; Jackson, D.M.; Appleton, I. Neuroprotective effects of spermine following hypoxic-ischemic-induced brain damage: A mechanistic study. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 1114–1116. [Google Scholar] [CrossRef] [Green Version]
- Halonen, T.; Sivenius, J.; Miettinen, R.; Halmekytö, M.; Kauppinen, R.; Sinervirta, R.; Alakuijala, L.; Alhonen, L.; MacDonald, E.; Jänne, J.; et al. Elevated seizure threshold and impaired spatial learning in transgenic mice with putrescine overproduction in the brain. Eur. J. Neurosci. 1993, 5, 1233–1239. [Google Scholar] [CrossRef]
- Hayashi, Y. Effects of intra-amygdaloid injections of alpha-difluoromethylornithine and putrescine on the development of electrical kindling in rats. Brain Res. 1991, 560, 181–185. [Google Scholar] [CrossRef]
- Hayashi, Y.; Hattori, Y.; Hori, Y. Involvement of putrescine in the development of kindled seizure in rats. J. Neurochem. 1992, 58, 562–566. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcoli, M.; Cervetto, C.; Amato, S.; Fiorucci, C.; Maura, G.; Mariottini, P.; Cervelli, M. Transgenic Mouse Overexpressing Spermine Oxidase in Cerebrocortical Neurons: Astrocyte Dysfunction and Susceptibility to Epileptic Seizures. Biomolecules 2022, 12, 204. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12020204
Marcoli M, Cervetto C, Amato S, Fiorucci C, Maura G, Mariottini P, Cervelli M. Transgenic Mouse Overexpressing Spermine Oxidase in Cerebrocortical Neurons: Astrocyte Dysfunction and Susceptibility to Epileptic Seizures. Biomolecules. 2022; 12(2):204. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12020204
Chicago/Turabian StyleMarcoli, Manuela, Chiara Cervetto, Sarah Amato, Cristian Fiorucci, Guido Maura, Paolo Mariottini, and Manuela Cervelli. 2022. "Transgenic Mouse Overexpressing Spermine Oxidase in Cerebrocortical Neurons: Astrocyte Dysfunction and Susceptibility to Epileptic Seizures" Biomolecules 12, no. 2: 204. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12020204