
Impairment of the Retinal Endothelial Cell Barrier Induced by Long-Term Treatment with VEGF-A165 No Longer Depends on the Growth Factor’s Presence


Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target Protein | IC50 | Final Concentrations | Reference | Provider (1) | |
|---|---|---|---|---|---|---|
| Nintedanib | VEGFR1 | 34 | nM | 10 or 100 nM | [32] | Selleckchem |
| VEGFR2 | 13 | nM | ||||
| VEGFR3 | 13 | nM | ||||
| FGFR1 | 69 | nM | ||||
| FGFR2 | 37 | nM | ||||
| FGFR3 | 108 | nM | ||||
| PDGFRα | 59 | nM | ||||
| PDGFRβ | 65 | nM | ||||
| Tivozanib | VEGFR1 | 30 | nM | 10 or 100 nM | [31] | Selleckchem |
| VEGFR2 | 6.5 | nM | ||||
| VEGFR3 | 15 | nM | ||||
| PDGFRα | 40 | nM | ||||
| PDGFRβ | 49 | nM | ||||
| c-Kit | 48 | nM | ||||
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Index Measurements
2.3. Preparation of Cell Extracts and Subcellular Fractions and Western Blot Analyses
2.4. Determination of VEGF-A
2.5. Immunofluorescence Stainings
2.6. Statistical Analyses
3. Results
3.1. VEGF-A165-Induced Persistent Impairment of the iBREC Barrier Was Associated with Changes of the Expression Levels of TJ-Protein Claudin-1 and Intgrins α5 and β1
3.2. Binding of VEGF-A Was Not Sufficient to Sustainably Revert Its Detrimental Long-Term Changes
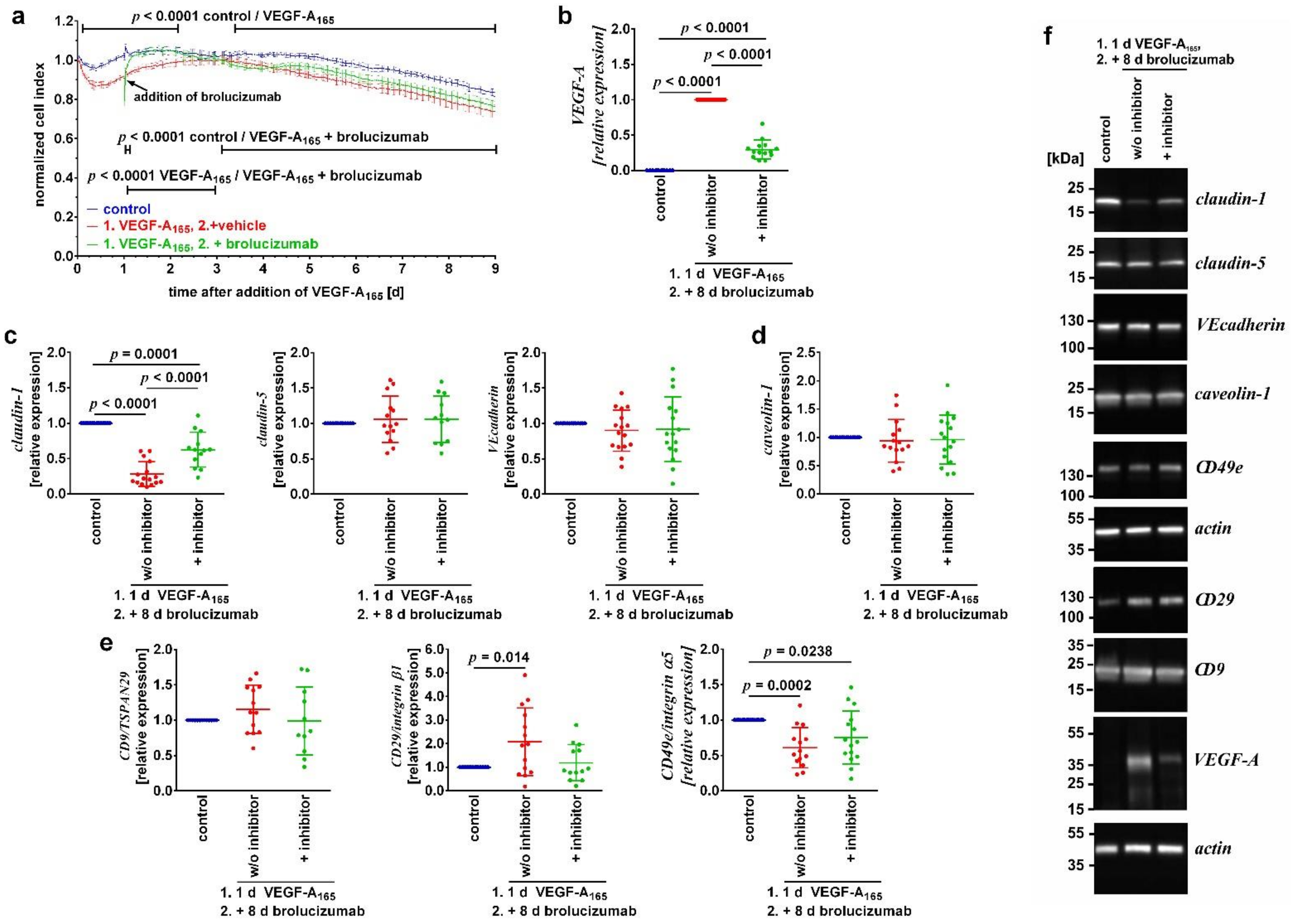
3.3. Inhibitors of VEGFR2 Only Reverted VEGF-A165-Induced Dysfunction When Added during the Early Phase of the Growth Factor’s Action
4. Discussion
| Target | Incubation with VEGF-A165 for | |||
|---|---|---|---|---|
| 1 Day | 2 Days | 6 Days | 9 Days | |
| VEGF-A165 | 36 ± 1.8 ng/mL | 25 ± 3.4 ng/mL [15] | 14 ± 1.2 ng/mL [15] | 6 ± 0.9 ng/mL |
| Target | Incubation with VEGF-A165 for (1) | |||
|---|---|---|---|---|
| 1 Day | 2 Days | 6 Days | 9 Days | |
| Regulation of paracellular and transcellular flow: | ||||
| Claudin-1 | ↓↓ [35] | ↓↓ [15,35] | ↓↓ [15] | ↓↓ |
| Claudin-5 | Unchanged [35] | Unchanged [15,35] | ↑ [15] | Unchanged |
| VEcadherin | Unchanged [35] | Unchanged [15,35] | Unchanged [15] | Unchanged |
| Caveolin-1 | Unchanged [35] | Unchanged [15,35] | Unchanged [15] | Unchanged |
| PLVAP | Not detected | ↑↑ [15] | ↑↑↑ [15] | Not detected |
| Adhesion to extracellular matrix: | ||||
| CD9/TSPAN29 | Unchanged | Unchanged [15] | Unchanged [15] | Unchanged |
| CD29/integrin β1 | ↑ | ↑ [15] | ↑ [15] | ↑ |
| CD49e/integrin α5 | ↑ | ↑ [15] | Unchanged [15] | ↓ |
| WNT-pathway: | ||||
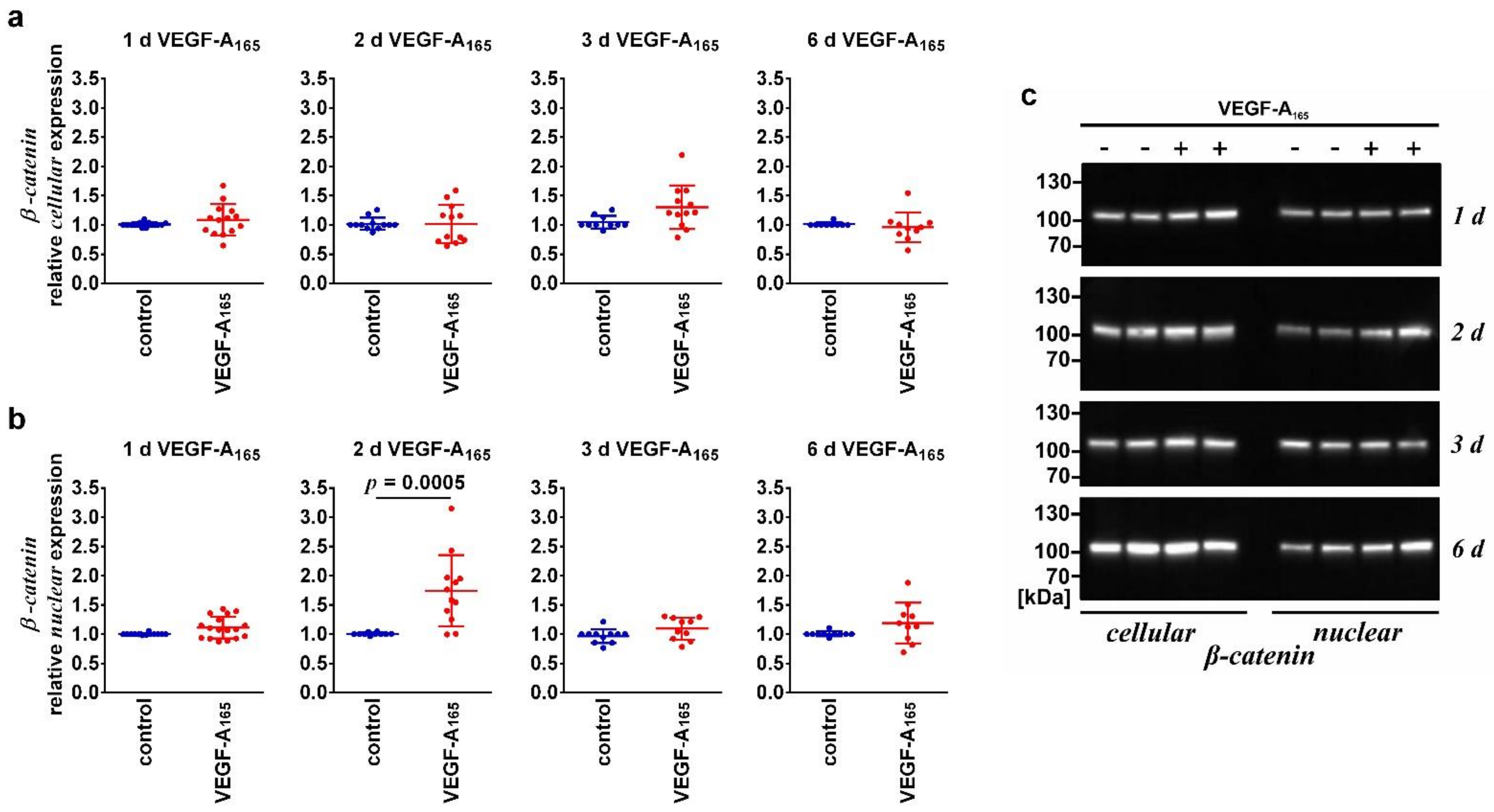
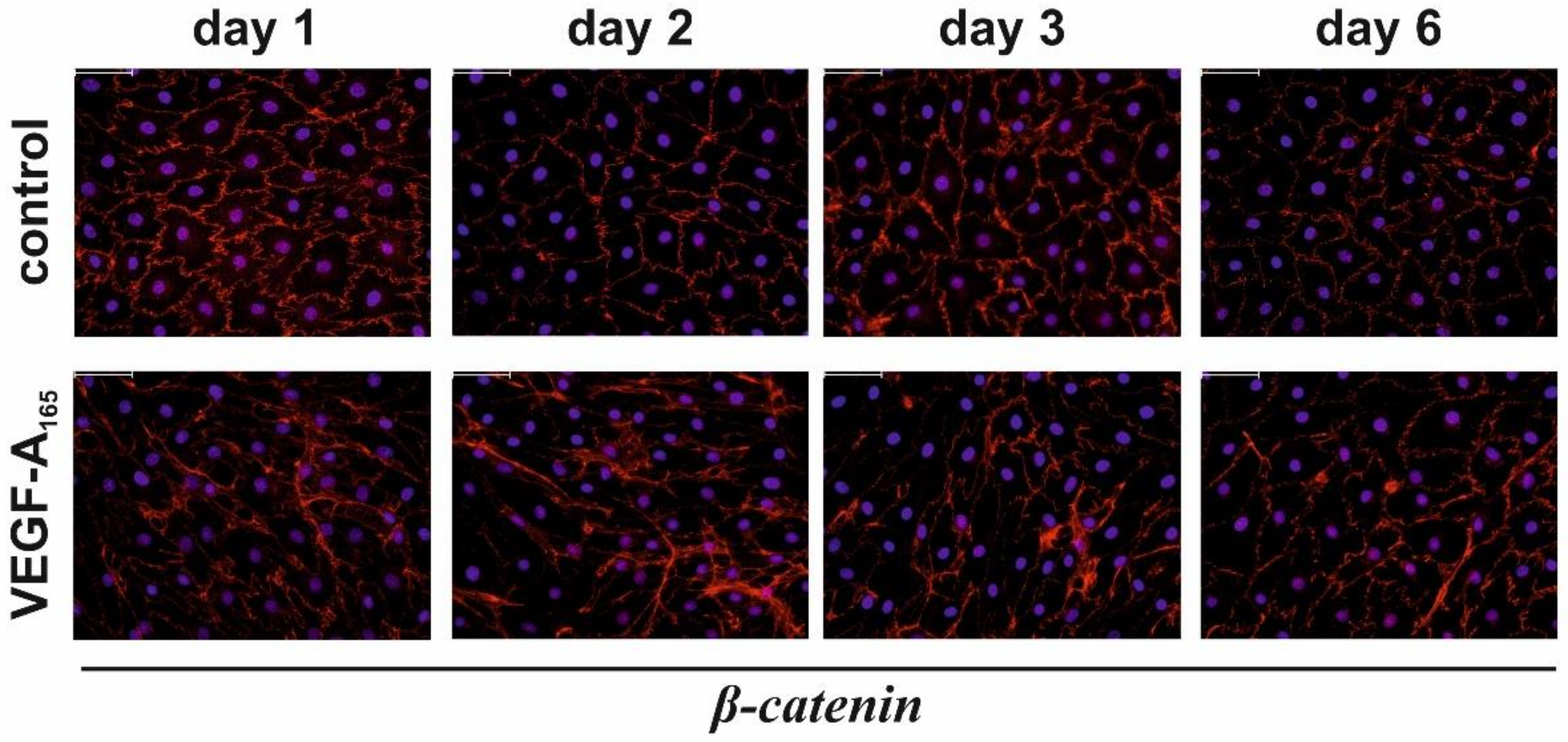
| β-Catenin (nuclear) | Unchanged | ↑ | Unchanged | |
| β-Catenin (cellular) | Unchanged | Unchanged | Unchanged | |
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E.; et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Retinal and choroidal neovascularization. J. Cell Physiol. 2000, 184, 301–310. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Ocular neovascularization. J. Mol. Med. 2013, 91, 311–321. [Google Scholar] [CrossRef]
- Uemura, A.; Fruttiger, M.; D’Amore, P.A.; De Falco, S.; Joussen, A.M.; Sennlaub, F.; Brunck, L.R.; Johnson, K.T.; Lambrou, G.N.; Rittenhouse, K.D.; et al. VEGFR1 signaling in retinal angiogenesis and microinflammation. Prog. Retin. Eye Res. 2021, 84, 100954. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Barber, A.J.; Hollinger, L.A.; Wolpert, E.B.; Gardner, T.W. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occludens 1. J. Biol. Chem. 1999, 274, 23463–23467. [Google Scholar] [CrossRef] [Green Version]
- Deissler, H.; Deissler, H.; Lang, G.E. Inhibition of VEGF is sufficient to completely restore barrier malfunction induced by growth factors in microvascular retinal endothelial cells. Br. J. Ophthalmol. 2011, 95, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Deissler, H.L.; Deissler, H.; Lang, G.K.; Lang, G.E. VEGF but not PlGF disturbs the barrier of retinal endothelial cells. Exp. Eye Res. 2013, 115, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, S.; McCollum, G.W.; Bretz, C.A.; Yang, R.; Capozzi, M.E.; Penn, J.S. Modulation of VEGF-induced retinal vascular permeability by peroxisome proliferator-activated receptor-β/δ. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8232–8240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’Angelo, R.; Sidoti, A. Oxidative Stress and the Neurovascular Unit. Life 2021, 11, 767. [Google Scholar] [CrossRef] [PubMed]
- Qaum, T.; Xu, Q.; Joussen, A.M.; Clemens, M.W.; Qin, W.; Miyamoto, K.; Hassessian, H.; Wiegand, S.J.; Rudge, J.; Yancopoulos, G.D.; et al. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2408–2413. [Google Scholar]
- Wisniewska-Kruk, J.; van der Wijk, A.E.; van Veen, H.A.; Gorgels, T.G.; Vogels, I.M.; Versteeg, D.; Van Noorden, C.J.; Schlingemann, R.O.; Klaassen, I. Plasmalemma vesicle-associated protein has a key role in blood-retinal barrier loss. Am. J. Pathol. 2016, 186, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Bosma, E.K.; van Noorden, C.J.F.; Schlingemann, R.O.; Klaassen, I. The role of plasmalemma vesicle-associated protein in pathological breakdown of blood–brain and blood–retinal barriers: Potential novel therapeutic target for cerebral edema and diabetic macular edema. Fluids Barriers CNS 2018, 15, 24. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Zhang, H.; Hou, Y.; Wei, T.; Liu, J. Plasmalemma vesicle-associated protein: A crucial component of vascular homeostasis. Exp. Ther. Med. 2016, 12, 1639–1644. [Google Scholar] [CrossRef] [Green Version]
- Deissler, H.L.; Rehak, M.; Busch, C.; Wolf, A. Blocking of VEGF-A is not sufficient to completely revert its long-term effects on the barrier formed by retinal endothelial cells. Exp. Eye Res. 2022, 216, 108945. [Google Scholar] [CrossRef]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev. Cell 2009, 16, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.H.; Nguyen, H.K.; Lee, J.E.; Suh, W. Therapeutic effect of apatinib-loaded nanoparticles on diabetes-induced retinal vascular leakage. Int. J. Nanomed. 2016, 11, 3101–3109. [Google Scholar] [CrossRef] [Green Version]
- Deissler, H.L.; Lang, G.K.; Lang, G.E. Inhibition of single routes of intracellular signaling is not sufficient to neutralize the biphasic disturbance of a retinal endothelial cell barrier induced by VEGF-A165. Cell. Physiol. Biochem. 2017, 42, 1493–1513. [Google Scholar] [CrossRef] [Green Version]
- Bruns, A.F.; Shane, P.H.; Odell, A.F.; Jopling, H.M.; Hooper, N.M.; Zachary, I.C.; Walker, J.H.; Ponnambalam, S. Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic 2010, 11, 161–174. [Google Scholar] [CrossRef]
- Eichmann, A.; Simons, M. VEGF signaling inside vascular endothelial cells and beyond. Curr. Opin. Cell Biol. 2012, 24, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Ewan, L.C.; Jopling, H.M.; Jia, H.; Mittar, S.; Bagherzadeh, A.; Howell, G.J.; Walker, J.J.; Zachary, I.C.; Ponnambalam, S. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic 2006, 7, 1270–1282. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.; Takagi, H.; Otani, A.; Koyama, S.; Kemmochi, S.; Uemura, A.; Honda, Y. Selective induction of neuropilin-1 by vascular endothelial growth factor (VEGF): A mechanism contributing to VEGF-induced angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Chathery, Y.; Wu, Y.; Rathore, N.; Tong, R.K.; Peale, F.; Bagri, A.; Tessier-Lavigne, M.; Koch, A.W.; Watts, R.J. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J. Biol. Chem. 2007, 282, 24049–24056. [Google Scholar] [CrossRef] [Green Version]
- Deissler, H.L.; Stutzer, J.-N.; Lang, G.K.; Grisanti, S.; Lang, G.E.; Ranjbar, M. VEGF receptor 2 inhibitor nintedanib completely reverts VEGF-A165-induced disturbances of barriers formed by retinal endothelial cells or long-term cultivated ARPE-19 cells. Exp. Eye Res. 2020, 194, 108004. [Google Scholar] [CrossRef]
- Li, Y.; Baccouche, B.; Olayinka, O.; Serikbaeva, A.; Kazlauskas, A. The role of the Wnt pathway in VEGF/Anti-VEGFdependent control of the endothelial cell barrier. Investig. Ophthalmol. Vis. Sci. 2021, 62, 17. [Google Scholar] [CrossRef]
- Díaz-Coránguez, M.; Lin, C.-M.; Liebner, S.; Antonetti, D.A. Norrin restores blood-retinal barrier properties after vascular endothelial growth factor-induced permeability. J. Bio. Chem. 2020, 295, 4647–4660. [Google Scholar] [CrossRef] [Green Version]
- Deissler, H.; Deissler, H.; Lang, G.K.; Lang, G.E. Generation and characterization of iBREC: Novel hTERT-immortalized bovine retinal endothelial cells. Int. J. Mol. Med. 2005, 15, 65–70. [Google Scholar] [CrossRef]
- Deissler, H.; Kuhn, E.M.; Lang, G.E.; Deissler, H. Tetraspanin CD9 is involved in the migration of retinal microvascular endothelial cells. Int. J. Mol. Med. 2007, 20, 643–652. [Google Scholar] [CrossRef]
- Jäckle, A.; Ziemssen, F.; Kuhn, E.M.; Kampmeier, J.; Lang, G.K.; Lang, G.E.; Deissler, H.; Deissler, H.L. Sitagliptin and the blood-retina barrier: Effects on retinal endothelial cells manifested only after prolonged exposure. J. Diab. Res. 2020, 2020, 2450781. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; Das, A.; Do, D.V.; Dugel, P.U.; Gomes, A.; Holz, F.G.; Koh, A.; Pan, C.K.; Sepah, Y.J.; Patel, N.; et al. Brolucizumab: Evolution through Preclinical and Clinical Studies and the Implications for the Management of Neovascular Age-Related Macular Degeneration. Ophthalmology 2020, 127, 963–976. [Google Scholar] [CrossRef]
- Nakamura, K.; Taguchi, E.; Miura, T.; Yamamoto, A.; Takahashi, K.; Bichat, F.; Guilbaud, N.; Hasegawa, K.; Kubo, K.; Fujiwara, Y.; et al. KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res. 2006, 66, 9134–9142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, C.; Rehak, M.; Hollborn, M.; Wiedemann, P.; Lang, G.K.; Lang, G.E.; Wolf, A.; Deissler, H.L. Type of culture medium determines properties of cultivated retinal endothelial cells: Induction of substantial phenotypic conversion by standard DMEM. Heliyon 2021, 7, e06037. [Google Scholar] [CrossRef] [PubMed]
- Dugel, P.U.; Koh, A.; Ogura, Y.; Jaffe, G.J.; Schmidt-Erfurth, U.; Brown, D.M.; Gomes, A.V.; Warburton, J.; Weichselberger, A.; Holz, F.G.; et al. HAWK and HARRIER: Phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology 2020, 127, 72–84. [Google Scholar] [CrossRef]
- Deissler, H.L.; Sommer, K.; Lang, G.K.; Lang, G.E. Transport and fate of aflibercept in VEGF-A165-challenged retinal endothelial cells. Exp. Eye Res. 2020, 198, 108156. [Google Scholar] [CrossRef]
- Sun, M.; Fu, H.; Cheng, H.; Cao, Q.; Zhao, Y.; Mou, X.; Zhang, X.; Liu, X.; Ke, Y. A dynamic real-time method for monitoring epithelial barrier function in vitro. Anal. Biochem. 2012, 425, 96–103. [Google Scholar] [CrossRef]
- Muellerleile, L.M.; Buxbaum, B.; Nell, B.; Fux, D.A. In-vitro binding analysis of anti-human vascular endothelial growth factor antibodies bevacizumab and aflibercept with canine, feline, and equine vascular endothelial growth factor. Res. Vet. Sci. 2019, 124, 233–238. [Google Scholar] [CrossRef]
- Walz, J.M.; Boehringer, D.; Deissler, H.L.; Faerber, L.; Goepfer, J.; Heiduschka, P.; Kleeberger, S.M.; Klettner, A.; Krohne, T.U.; Schneiderhan-Marra, N.; et al. Pre-Analytical Parameters Affecting Vascular Endothelial Growth Factor Measurement in Plasma: Identifying Confounders. PLoS ONE 2016, 11, e0145375, eCollection 2016. [Google Scholar] [CrossRef]
- Deissler, H.L.; Lang, G.K.; Lang, G.E. Fate of the Fc fusion protein aflibercept in retinal endothelial cells: Competition of recycling and degradation. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.Z.; Zhu, L.; Jackson, J.A.; Gabos, S.; Sun, X.-J.; Wang, X.-B.; Xu, X. Dynamic monitoring of cytotoxicity on microelectronic sensors. Chem. Res. Toxicol. 2005, 18, 154–161. [Google Scholar] [CrossRef]
- Strohl, L.L.; Zang, J.B.; Ding, W.; Manni, M.; Zhou, X.K.; Granstein, R.D. Norepinephrine and adenosine-5′-triphosphate synergize in inducing IL-6 production by human dermal microvascular endothelial cells. Cytokine 2013, 64, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Valle, M.L.; Dworshak, J.; Sharma, A.; Ibrahim, A.S.; Al-Shabrawey, M.; Sharma, S. Inhibition of interleukin-6 trans-signaling prevents inflammation and endothelial barrier disruption in retinal endothelial cells. Exp. Eye Res. 2019, 178, 27–36. [Google Scholar] [CrossRef]
- Yu, H.; Huang, X.; Ma, Y.; Gao, M.; Wang, O.; Gao, T.; Shen, Y.; Liu, X. Interleukin-8 regulates endothelial permeability by down-regulation of tight junction but not dependent on integrins induced focal adhesions. Int. J. Biol. Sci. 2013, 9, 966–979. [Google Scholar] [CrossRef]
- Aveleira, C.A.; Lin, C.M.; Abcouwer, S.F.; Ambrósio, A.F.; Antonetti, D.A. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 2010, 59, 2782–2882. [Google Scholar] [CrossRef] [Green Version]
- Deissler, H.L.; Lang, G.K.; Lang, G.E. Capacity of aflibercept to counteract VEGF-stimulated abnormal behavior of retinal microvascular endothelial cells. Exp. Eye Res. 2014, 122, 20–31. [Google Scholar] [CrossRef] [Green Version]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef] [Green Version]
- Im, J.-E.; Song, S.-H.; Suh, W. Src tyrosine kinase regulates the stem cell factor–induced breakdown of the blood–retinal barrier. Mol. Vis. 2016, 22, 1213–1220. Available online: http://www.molvis.org/molvis/v22/1213 (accessed on 8 March 2022).
- Dejana, E.; Giampietro, C. Vascular endothelial-cadherin and vascular stability. Curr. Opin. Hematol. 2012, 19, 218–223. [Google Scholar] [CrossRef]
- Banan, A.; Zhang, L.J.; Shaikh, M.; Fields, J.Z.; Choudhary, S.; Forsyth, C.B.; Farhadi, A.; Keshavarzian, A. θ isoform of protein kinase C alters barrier function in intestinal epithelium through modulation of distinct claudin isotypes: A novel mechanism for regulation of permeability. J. Pharmacol. Exp. Ther. 2005, 313, 962–982. [Google Scholar] [CrossRef]
- Kluger, M.S.; Clark, P.R.; Tellides, G.; Gerke, V.; Pober, J.S. Claudin-5 controls intercellular barriers of human dermal microvascular but not human umbilical vein endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2014, 33, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Wisniewska-Kruk, J.; Hoeben, K.A.; Vogels, I.M.; Gaillard, P.J.; Van Noorden, C.J.; Schlingemann, R.O.; Klaassen, I. A novel co-culture model of the blood-retinal barrier based on primary retinal endothelial cells, pericytes and astrocytes. Exp. Eye Res. 2012, 96, 181–190. [Google Scholar] [CrossRef]
- Busch, C.; Zur, D.; Fraser-Bell, S.; Laíns, I.; Santos, A.R.; Lupidi, M.; Cagini, C.; Gabrielle, P.H.; Couturier, A.; Mané-Tauty, V.; et al. Shall we stay, or shall we switch? Continued anti-VEGF therapy versus early switch to dexamethasone implant in refractory diabetic macular edema. Acta Diabetol. 2018, 55, 789–796. [Google Scholar] [CrossRef]
- Hussain, R.M.; Ciulla, T.A. Treatment strategies for refractory diabetic macular edema: Switching anti-VEGF treatments, adopting corticosteroid-based treatments, and combination therapy. Expert Opin. Biol. Ther. 2016, 16, 365–374. [Google Scholar] [CrossRef] [Green Version]




| Target | Host, Type or Conjugate | Source (1) | Working Concentrations |
|---|---|---|---|
| Actin | mouse, monoclonal | clone 5J11: Abcam, #ab190301 or bio-techne, #NBP2-25142 | 700 ng/mL |
| β-Catenin | rabbit, monoclonal | clone 6B3: Cell Signaling Technology B.V., #9582S | 1:100 |
| Caveolin-1 | rabbit, polyclonal | Abcam, #ab2910 | 20 ng/mL |
| CD9 | mouse, monoclonal | clone IVA: Exbio | 40 ng/mL |
| CD29 | mouse, monoclonal | clone TS2/16: Thermo Fisher Scientific, #14-0299-82 | 170 ng/mL |
| Claudin-1 | rabbit, polyclonal | Thermo Fisher Scientific, #51-9000 | 0.25 µg/ml |
| Claudin-5 | rabbit, polyclonal | Thermo Fisher Scientific, #34-1600 | 100 ng/mL |
| PLVAP | rabbit, polyclonal | Thermo Fisher Scientific, #PA5-110183 | 6 µg/ml |
| VEcadherin | rabbit, polyclonal | Cell Signaling Technology B.V., #2158S | 1:1000 |
| VEGF-A | goat, polyclonal | bio-techne, #AF1603 | 1:2000 |
| Whole IgG, rabbit | goat, polyclonal, coupled to HRP (2) | Biorad, #170-5046 | 1:30,000 |
| Whole IgG, mouse | goat, polyclonal, coupled to HRP (2) | Biorad, #170-5047 | 1:30,000 |
| IgG, H + L chains, goat | donkey, polyclonal, coupled to HRP (2) | bio-techne, #HAF109 | 1:4000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deissler, H.L.; Rehak, M.; Wolf, A. Impairment of the Retinal Endothelial Cell Barrier Induced by Long-Term Treatment with VEGF-A165 No Longer Depends on the Growth Factor’s Presence. Biomolecules 2022, 12, 734. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12050734
Deissler HL, Rehak M, Wolf A. Impairment of the Retinal Endothelial Cell Barrier Induced by Long-Term Treatment with VEGF-A165 No Longer Depends on the Growth Factor’s Presence. Biomolecules. 2022; 12(5):734. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12050734
Chicago/Turabian StyleDeissler, Heidrun L., Matus Rehak, and Armin Wolf. 2022. "Impairment of the Retinal Endothelial Cell Barrier Induced by Long-Term Treatment with VEGF-A165 No Longer Depends on the Growth Factor’s Presence" Biomolecules 12, no. 5: 734. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12050734