
Functional Role of NBS1 in Radiation Damage Response and Translesion DNA Synthesis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Functional Domains of NBS1

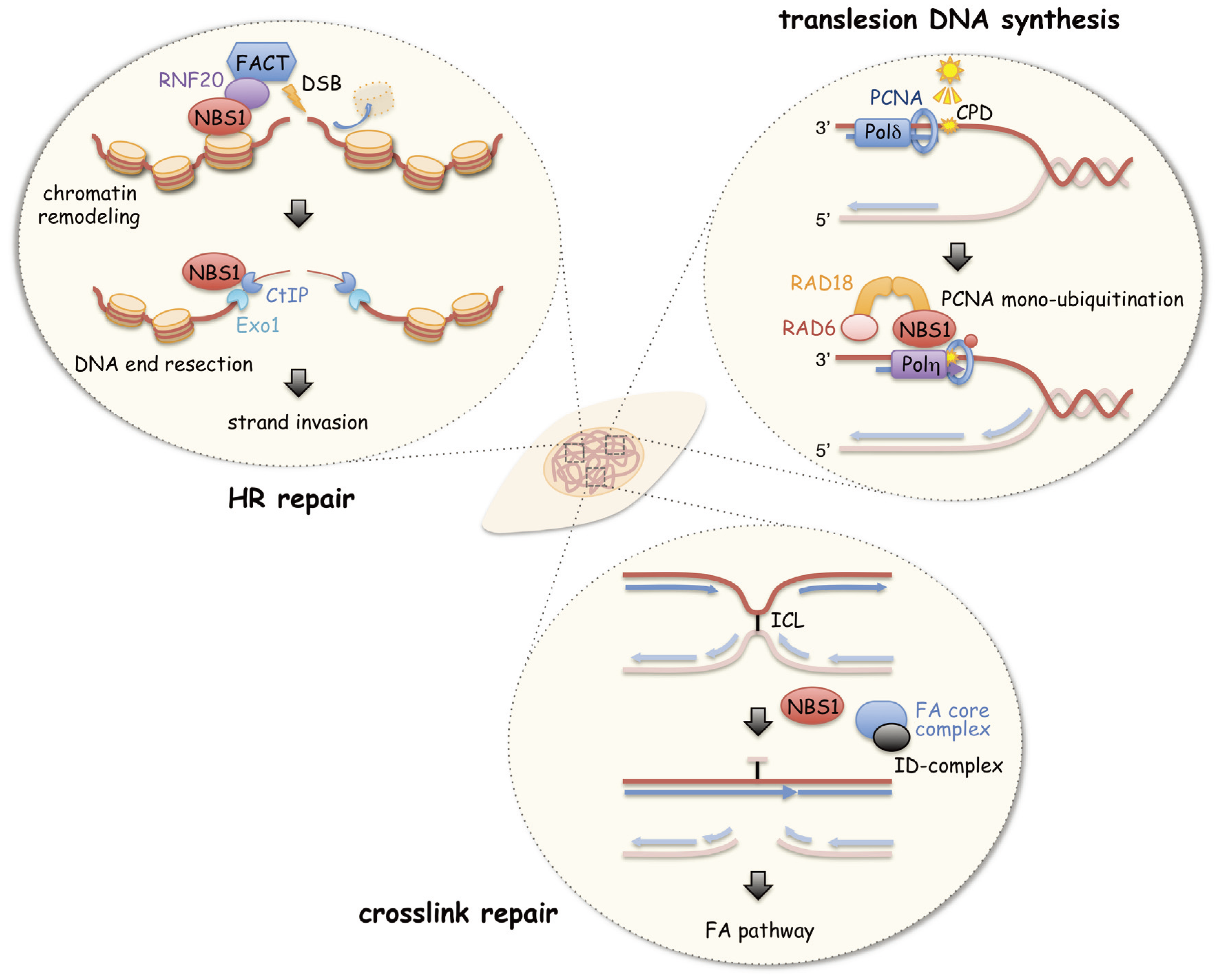
3. A Role of NBS1 in HR Repair
4. A Role of NBS1 in Chromatin Remodeling during HR Repair
5. A Role of NBS1 in Initiation of Translesion DNA Synthesis
6. A Role of NBS1 in ICL Repair
7. Conclusions

Acknowledgements
Author Contributions
Conflicts of Interest
References
- Weemaes, C.M.; Hustinx, T.W.; Scheres, J.M.; van Munster, P.J.; Bakkeren, J.A.; Taalman, R.D. A new chromosomal instability disorder: The Nijmegen breakage syndrome. Acta Paediatr. Scand. 1981, 70, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, H.; Matsuura, S.; Kobayashi, J.; Sakamoto, S.; Komatsu, K. Nijmegen breakage syndrome gene, NBS1, and molecular links to factors for genome stability. Oncogene 2002, 21, 8967–8980. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, S.; Weemaes, C.; Smeets, D.; Takami, H.; Kondo, N.; Sakamoto, S.; Yano, N.; Nakamura, A.; Tauchi, H.; Endo, S.; et al. Genetic Mapping Using Microcell-Mediated Chromosome Transfer Suggests a Locus, f.o.r.Nijmegen Breakage Syndrome at Chromosome 8q21–24. Am. J. Hum. Genet. 1997, 60, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, S.; Tauchi, H.; Nakamura, A.; Kondo, N.; Sakamoto, S.; Endo, S.; Smeets, D.; Solder, B.; Belohradsky, B.H.; der Kaloustian, V.M.; et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nat. Genet. 1998, 19, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; le Beau, M.; Yates, J.R., 3rd; Hays, L.; Morgan, W.F.; Petrini, J.H. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [PubMed]
- Varon, R.; Vissinga, C.; Platzer, M.; Cerosaletti, K.M.; Chrzanowska, K.H.; Saar, K.; Beckmann, G.; Seemanová, E.; Cooper, P.R.; Nowak, N.J.; et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998, 93, 467–476. [Google Scholar] [CrossRef]
- Varon, R.; Dutrannoy, V.; Weikert, G.; Tanzarella, C.; Antoccia, A.; Stockl, L.; Spadoni, E.; Krüger, L.A.; di Masi, A.; Sperling, K.; et al. Mild Nijmegen breakage syndrome phenotype due to alternative splicing. Hum. Mol. Genet. 2006, 15, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, H.; Kobayashi, J.; Tateishi, S.; Kato, A.; Matsuura, S.; Tauchi, H.; Yamada, K.; Takezawa, J.; Sugasawa, K.; Masutani, C.; et al. NBS1 recruits RAD18 via a RAD6-like domain and regulates Pol η-dependent translesion DNA synthesis. Mol. Cell 2011, 43, 788–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, C.B.; Lewis, A.L.; Baldwin, K.K.; Resnick, M.A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. USA 1993, 90, 5613–5617. [Google Scholar] [CrossRef] [PubMed]
- Wyman, C.; Kanaar, R. DNA Double-strand break repair: All’s well that ends well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Iwabuchi, K.; Miyagawa, K.; Sonoda, E.; Suzuki, K.; Takata, M.; Tauchi, H. Current topics in DNA double-strand break repair. J. Radiat. Res. 2008, 49, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, H.; Kobayashi, J.; Morishima, K.; Matsuura, S.; Nakamura, A.; Shiraishi, T.; Ito, E.; Masnada, D.; Delia, D.; Komatsu, K. The forkhead-associated domain of NBS1 is essential for nuclear foci formation after irradiation but not essential for hRAD50-hMRE11-NBS1 complex DNA repair activity. J. Biol. Chem. 2001, 276, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Tauchi, H.; Sakamoto, S.; Nakamura, A.; Morishima, K.-I.; Matsuura, S.; Kobayashi, T.; Tamai, K.; Tanimoto, K.; Komatsu, K. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr. Biol. 2002, 12, 1846–1851. [Google Scholar] [CrossRef]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Fujimoto, H.; Kobayashi, J. Role of NBS1 in DNA damage response and its relationship with cancer development. Transl. Cancer Res. 2013, 2, 178–189. [Google Scholar]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.; Iwasaki, H.; Russell, P.; et al. Nbs1 Flexibly Tethers Ctp1 and Mre11-Rad50 to Coordinate DNA Double-Strand Break Processing and Repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Maraschio, P.; Peretti, D.; Lambiase, S.; Curto Lo, F.; Caufin, D.; Gargantini, L.; Minoli, L.; Zuffardi, O. A new chromosome instability disorder. Clin. Genet. 1986, 30, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, H.; Kobayashi, J.; Morishima, K.-I.; van Gent, D.C.; Shiraishi, T.; Verkaik, N.S.; van Heems, D.; Ito, E.; Nakamura, A.; Sonoda, E.; et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 2002, 420, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Iijima, K.; Mochizuki, D.; Nakamura, K.; Teshigawara, K.; Kobayashi, J.; Matsuura, S.; Tauchi, H.; Komatsu, K. Homologous recombination repair is regulated by domains at the N- and C-terminus of NBS1 and is dissociated with ATM functions. Oncogene 2007, 26, 6002–6009. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Nicolas, A.; Furuse, M.; Nabetani, A.; Ogawa, H.; Shibata, T. Mutations in the MRE11, RAD50, XRS2, and MRE2 genes alter chromatin configuration at meiotic DNA double-stranded break sites in premeiotic and meiotic cells. Proc. Natl. Acad. Sci. USA 1998, 95, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.E. The many interfaces of Mre11. Cell 1998, 95, 583–586. [Google Scholar] [CrossRef]
- Furuse, M.; Nagase, Y.; Tsubouchi, H.; Murakami-Murofushi, K.; Shibata, T.; Ohta, K. Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J. 1998, 17, 6412–6425. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.V.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.V.; Kato, A.; Nakamura, K.; Ikura, T.; Okada, M.; Kobayashi, J.; Yanagihara, H.; Saito, Y.; Tauchi, H.; Komatsu, K. Histone chaperone FACT regulates homologous recombination by chromatin remodeling through interaction with RNF20. J. Cell Sci. 2014, 127, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Demuth, I. An inducible null mutant murine model of Nijmegen breakage syndrome proves the essential function of NBS1 in chromosomal stability and cell viability. Hum. Mol. Genet. 2004, 13, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Iwai, Y.; Sonoda, E.; Sasaki, M.S.; Morrison, C.; Haraguchi, T.; Hiraoka, Y.; Yamashita, Y.M.; Yagi, T.; Takata, M.; Price, C. Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 1999, 18, 6619–6629. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Petersen, S.; Tessarollo, L.; Nussenzweig, A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 2001, 11, 105–109. [Google Scholar] [CrossRef]
- Buerstedde, J.M.; Takeda, S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell 1991, 67, 179–188. [Google Scholar] [CrossRef]
- Johnson, R.D.; Liu, N.; Jasin, M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature 1999, 401, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.-T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J.; et al. Genomic instability in mice lacking histone H2AX. Science 2002, 296, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Chua, K.F.; Sekiguchi, J.; Suh, H.; Whitlow, S.R.; Fleming, J.C.; Monroe, B.C.; Ciccone, D.N.; Yan, C.; Vlasakova, K.; et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. USA 2002, 99, 8173–8178. [Google Scholar] [CrossRef] [PubMed]
- White, C.I.; Haber, J.E. Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. EMBO J. 1990, 9, 663–673. [Google Scholar] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Phelps, S.E.L.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.-M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Dodson, G.E.; Limbo, O.; Nieto, D.; Russell, P. Phosphorylation-regulated binding of Ctp1 to Nbs1 is critical for repair of DNA double-strand breaks. Cell Cycle 2010, 9, 1516–1522. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Forment, J.V.; Coates, J.; Mistrik, M.; Lukas, J.; Bartek, J.; Jackson, S.P. CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 2012, 13, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Kwok, A.; Scully, R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Van Attikum, H.; Gasser, S.M. The histone code at DNA breaks: A guide to repair? Nat. Rev. Mol. Cell Biol. 2005, 6, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.R.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Sung, P. 53BP1, BRCA1 and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar] [CrossRef]
- Van Attikum, H.; Fritsch, O.; Hohn, B.; Gasser, S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004, 119, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Murr, R.; Loizou, J.I.; Yang, Y.-G.; Cuenin, C.; Li, H.; Wang, Z.-Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2005, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Niida, H.; Katsuno, Y.; Sengoku, M.; Shimada, M.; Yukawa, M.; Ikura, M.; Ikura, T.; Kohno, K.; Shima, H.; Suzuki, H.; et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev. 2010, 24, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Tsukuda, T.; Fleming, A.B.; Nickoloff, J.A.; Osley, M.A. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 2005, 438, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Berkovich, E.; Monnat, R.J.; Kastan, M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 2007, 9, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Orphanides, G.; Wu, W.H.; Lane, W.S.; Hampsey, M.; Reinberg, D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 1999, 400, 284–288. [Google Scholar] [PubMed]
- Winkler, D.D.; Luger, K. The histone chaperone FACT: Structural insights and mechanisms for nucleosome reorganization. J. Biol. Chem. 2011, 286, 18369–18374. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Polo, S.E.; Almouzni, G. Prime, repair, restore: The active role of chromatin in the DNA damage response. Mol. Cell 2012, 46, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskaya, R.; Oh, S.; Bondarenko, V.A.; Orphanides, G.; Studitsky, V.M.; Reinberg, D. FACT facilitates transcription-dependent nucleosome alteration. Science 2003, 301, 1090–1093. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.; Kim, H.; Choi, S.H.; Choi, J.; Kim, K.; Gu, J.; Lieber, M.R.; Yang, A.S.; An, W. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol. Cell 2008, 30, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Piro, A.S.; Mayekar, M.K.; Warner, M.H.; Davis, C.P.; Arndt, K.M. Small region of Rtf1 protein can substitute for complete Paf1 complex in facilitating global histone H2B ubiquitylation in yeast. Proc. Natl. Acad. Sci. 2012, 109, 10837–10842. [Google Scholar] [CrossRef] [PubMed]
- Klement, K.; Luijsterburg, M.S.; Pinder, J.B.; Cena, C.S.; del Nero, V.; Wintersinger, C.M.; Dellaire, G.; van Attikum, H.; Goodarzi, A.A. Opposing ISWI- and CHD-class chromatin remodeling activities orchestrate heterochromatic DNA repair. J. Cell Biol. 2014, 207, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Biard, D.S.F. Untangling the relationships between DNA repair pathways by silencing more than 20 DNA repair genes in human stable clones. Nucleic Acids Res. 2007, 35, 3535–3550. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro-Stone, M.; Zaritskaya, L.S.; Price, L.K.; Kaufmann, W.K. Replication fork bypass of a pyrimidine dimer blocking leading strand DNA synthesis. J. Biol. Chem. 1997, 272, 13945–13954. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Boudsocq, F.; Plosky, B.S.; Woodgate, R.; Yang, W. Replication of a cis-syn thymine dimer at atomic resolution. Nature 2003, 424, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R. New functions for Y family polymerases. Mol. Cell 2006, 24, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Araki, M.; Yamada, A.; Kusumoto, R.; Nogimori, T.; Maekawa, T.; Iwai, S.; Hanaoka, F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999, 18, 3491–3501. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999, 399, 700–704. [Google Scholar] [PubMed]
- Masutani, C.; Kusumoto, R.; Iwai, S.; Hanaoka, F. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. EMBO J. 2000, 19, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, S.D.; Kokoska, R.J.; Masutani, C.; Iwai, S.; Hanaoka, F.; Kunkel, T.A. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature 2004, 428, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Zou, L. Dual functions of DNA replication forks in checkpoint signaling and PCNA ubiquitination. Cell Cycle 2009, 8, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Tissier, A.; Kannouche, P.; Reck, M.-P.; Lehmann, A.R.; Fuchs, R.P.P.; Cordonnier, A. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase pol eta and REVl protein. DNA Repair 2004, 3, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Karras, G.I.; Jentsch, S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S Phase. Cell 2010, 141, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Busuttil, R.A.; Lin, Q.; Stambrook, R.J.; Kucherlapati, R.; Vijg, J. Mutation frequencies and spectra in DNA polymerase η-deficient mice. Cancer Res. 2008, 68, 2081–2084. [Google Scholar] [CrossRef] [PubMed]
- Gondo, Y.; Shioyama, Y.; Nakao, K.; Katsuki, M. A novel positive detection system of in vivo mutations in rpsL (strA) transgenic mice. Mutat. Res. 1996, 360, 1–14. [Google Scholar] [CrossRef]
- Moldovan, G.-L.; D’Andrea, A.D. How the Fanconi anemia pathway guards the genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Taniguchi, T.; Ranganathan, V.; New, H.V.; Moreau, L.A.; Stotsky, M.; Mathew, C.G.; Kastan, M.B.; Weaver, D.T.; D’Andrea, A.D. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat. Cell Biol. 2002, 4, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Gennery, A.R.; Slatter, M.A.; Bhattacharya, A.; Barge, D.; Haigh, S.; O’Driscoll, M.; Coleman, R.; Abinun, M.; Flood, T.J.; Cant, A.J.; et al. The clinical and biological overlap between Nijmegen Breakage Syndrome and Fanconi anemia. Clin. Immunol. 2004, 113, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Price, B.D.; Tetradis, S.; Chakrabarti, S.; Maulik, G.; Makrigiorgos, G.M. Reproducible and inexpensive probe preparation for oligonucleotide arrays. Nucleic Acids Res. 2001, 29. [Google Scholar] [CrossRef]
- Tsuchida, K.; Komatsu, K. Impaired removal of DNA interstrand cross-link in Nijmegen breakage syndrome and Fanconi anemia, but not in BRCA-defective group. Cancer Sci. 2008, 99, 2238–2243. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Gregory, M.T.; Yang, W. Human Polz purified with accessory subunits is active in translesion DNA synthesis and complements Polh in cisplatin bypass. Proc. Natl. Acad. Sci. USA 2014, 111, 2954–2959. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J.; Siraki, A.G.; Shangari, N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol. 2005, 35, 609–662. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; McIntee, E.J.; Cheng, G.; Shi, Y.; Villalta, P.W.; Hecht, S.S. Identification of DNA adducts of acetaldehyde. Chem. Res. Toxicol. 2000, 13, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Becker, P. Alcohol metabolism and cancer risk. Available online: http://pubs.niaaa.nih.gov/publications/arh301/38-47.pdf (accessed on 3 March 2015).
- Nagayoshi, H.; Matsumoto, A.; Nishi, R.; Kawamoto, T.; Ichiba, M.; Matsuda, T. Increased formation of gastric N2-ethylidene-2'-deoxyguanosine DNA adducts in aldehyde dehydrogenase-2 knockout mice treated with ethanol. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2009, 673, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Langevin, F.; Crossan, G.P.; Rosado, I.V.; Arends, M.J.; Patel, K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011, 475, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kobayashi, J.; Komatsu, K.; Radiation Biology Center, Kyoto University, Kyoto, Japan. Unpublished data. 2015.
- Chrzanowska, K.H.; Gregorek, H.; Dembowska-Bagińska, B.; Kalina, M.A.; Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, Y.; Komatsu, K. Functional Role of NBS1 in Radiation Damage Response and Translesion DNA Synthesis. Biomolecules 2015, 5, 1990-2002. https://0-doi-org.brum.beds.ac.uk/10.3390/biom5031990
Saito Y, Komatsu K. Functional Role of NBS1 in Radiation Damage Response and Translesion DNA Synthesis. Biomolecules. 2015; 5(3):1990-2002. https://0-doi-org.brum.beds.ac.uk/10.3390/biom5031990
Chicago/Turabian StyleSaito, Yuichiro, and Kenshi Komatsu. 2015. "Functional Role of NBS1 in Radiation Damage Response and Translesion DNA Synthesis" Biomolecules 5, no. 3: 1990-2002. https://0-doi-org.brum.beds.ac.uk/10.3390/biom5031990