
Managing Single-Stranded DNA during Replication Stress in Fission Yeast


Abstract
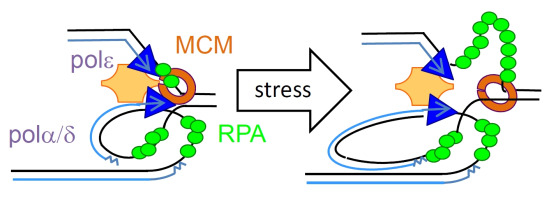
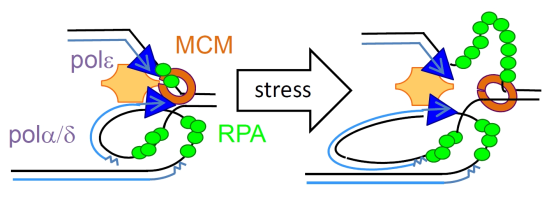
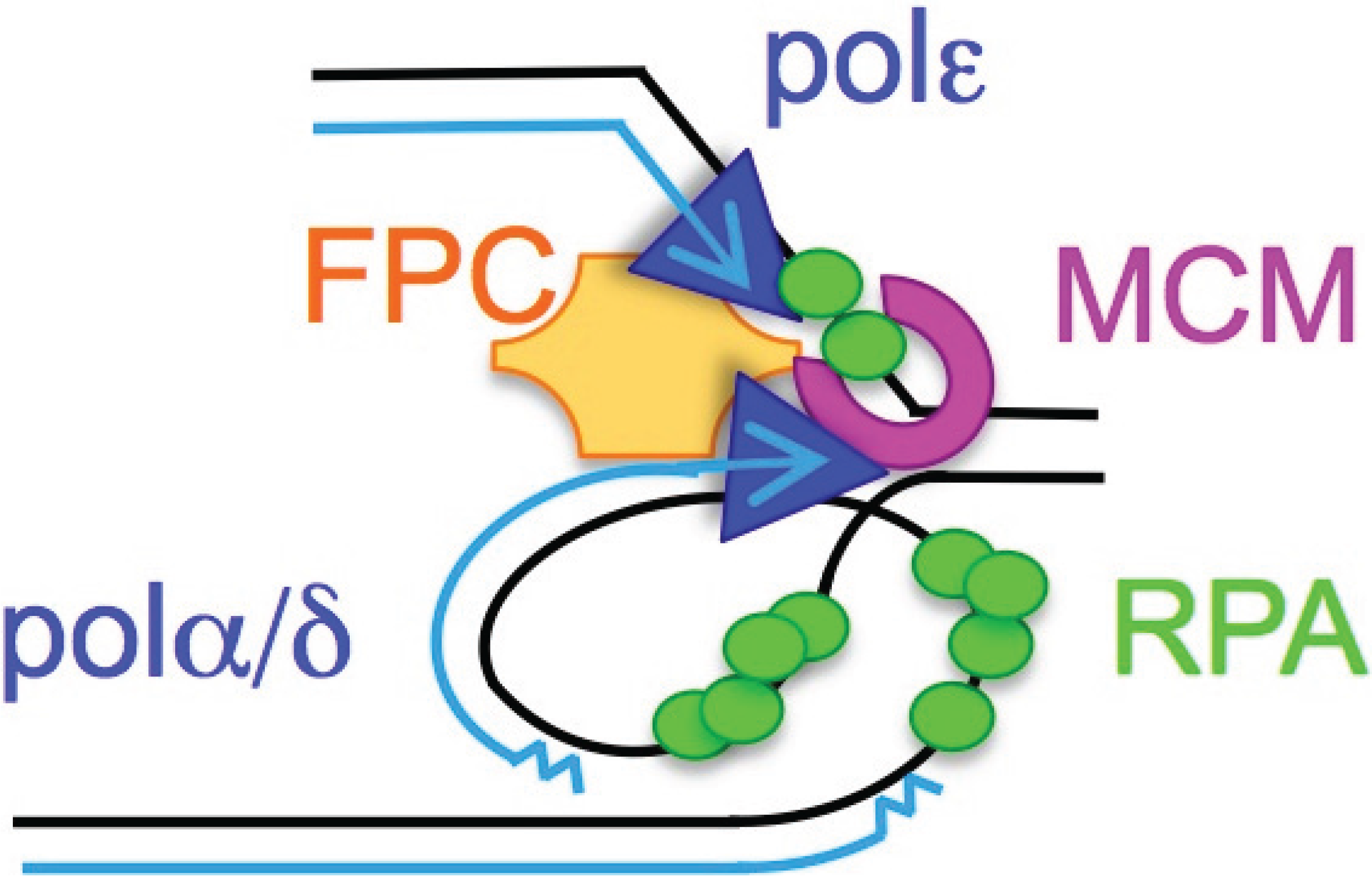
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

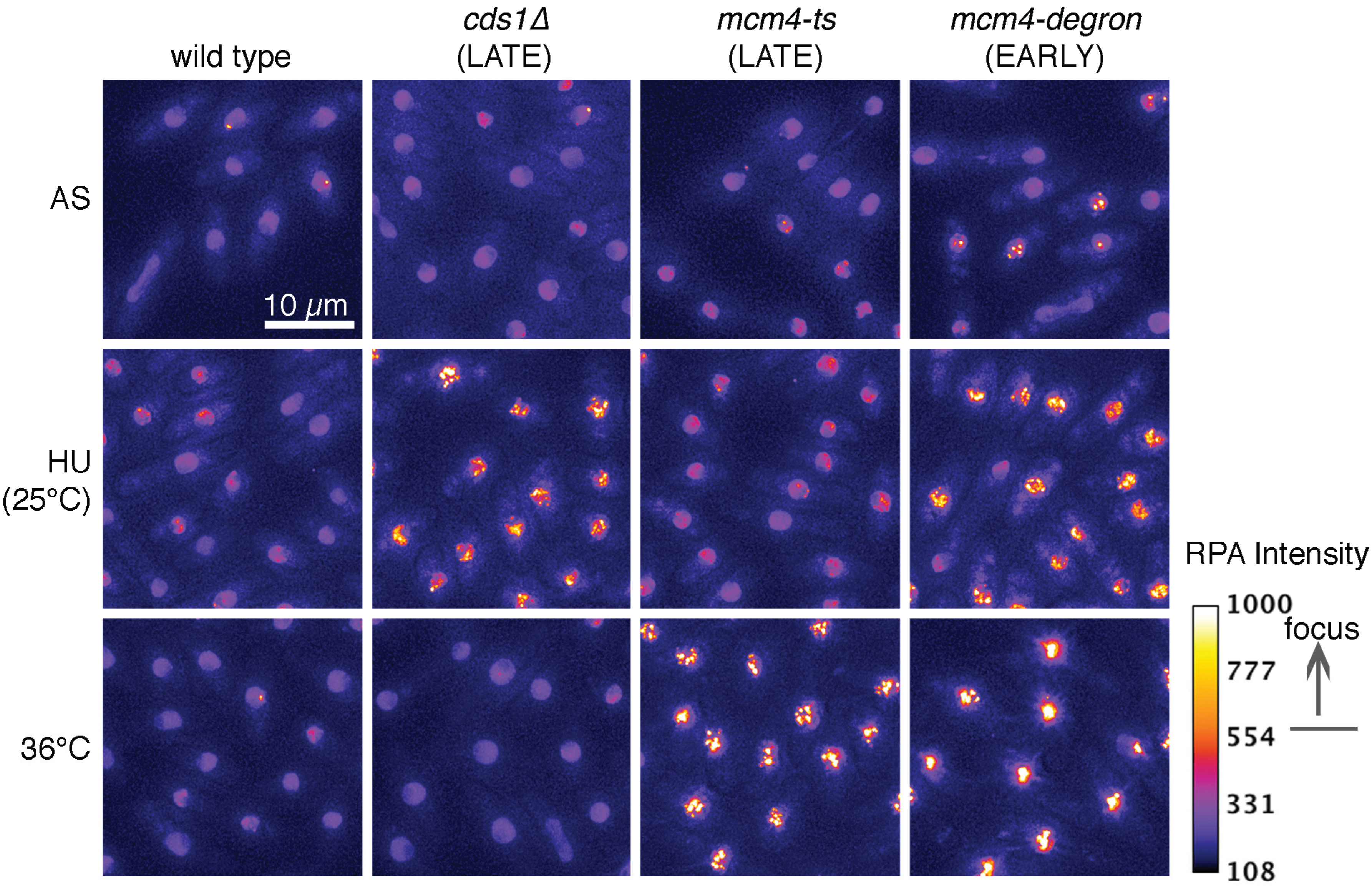
2. ssDNA Is a Hallmark of Stress
3. RPA Is the ssDNA Sensor

4. Association between ssDNA and DNA Damage

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Khajavi, M.; Connolly, A.M.; Towne, C.F.; Batish, S.D.; Lupski, J.R. The DNA replication fostes/mmbir mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009, 41, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Klassen, R.; Prakash, L.; Prakash, S. A major role of DNA polymerase delta in replication of both the leading and lagging DNA strands. Mol. Cell 2015, 59, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Sabatinos, S.A.; Forsburg, S.L. Preserving the replication fork in response to nucleotide starvation: Evading the replication fork collapse point. In The Mechanisms of DNA Replication; Stuart, D., Ed.; Intech: Rijeka, Croatia, 2013. [Google Scholar]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common fragile sites: Mechanisms of instability revisited. Trends Genet. 2012, 28, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Beeharry, N.; Rattner, J.B.; Caviston, J.P.; Yen, T. Centromere fragmentation is a common mitotic defect of s and g 2 checkpoint override. Cell Cycle 2013, 12, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.M.; Lambert, S. Replication stress-induced genome instability: The dark side of replication maintenance by homologous recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.; Kaochar, S.; Jones, H.; Paek, A.; Clark, A.J. The replication fork’s five degrees of freedom, their failure and genome rearrangements. Curr. Opin. Cell Biol. 2009, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Sperka, T.; Wang, J.; Rudolph, K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012, 13, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Dubrana, K.; van Attikum, H.; Hediger, F.; Gasser, S.M. The processing of double-strand breaks and binding of single-strand-binding proteins RPA and Rad51 modulate the formation of atr-kinase foci in yeast. J. Cell Sci. 2007, 120, 4209–4220. [Google Scholar] [CrossRef] [PubMed]
- Morin, I.; Ngo, H.P.; Greenall, A.; Zubko, M.K.; Morrice, N.; Lydall, D. Checkpoint-dependent phosphorylation of exo1 modulates the DNA damage response. EMBO J. 2008, 27, 2400–2410. [Google Scholar] [CrossRef] [PubMed]
- Schaetzlein, S.; Kodandaramireddy, N.R.; Ju, Z.; Lechel, A.; Stepczynska, A.; Lilli, D.R.; Clark, A.B.; Rudolph, C.; Kuhnel, F.; Wei, K.; et al. Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomere-dysfunctional mice. Cell 2007, 130, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Van, C.; Yan, S.; Michael, W.M.; Waga, S.; Cimprich, K.A. Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J. Cell Biol. 2010, 189, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through atrip recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Newport, J. Initiation of eukaryotic DNA replication: Origin unwinding and sequential chromatin association of cdc45, RPA, and DNA polymerase alpha. Mol. Cell 2000, 5, 617–627. [Google Scholar] [CrossRef]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of mcm helicase and DNA polymerase activities activates the atr-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Pacek, M.; Walter, J.C. A requirement for mcm7 and cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J. 2004, 23, 3667–3676. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable uv lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Collingwood, D.; Boeck, M.E.; Fox, L.A.; Alvino, G.M.; Fangman, W.L.; Raghuraman, M.K.; Brewer, B.J. Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat. Cell Biol. 2006, 8, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Lucca, C.; Vanoli, F.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Haber, J.; Foiani, M. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 2004, 23, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Namiki, Y.; Zou, L. Atrip associates with replication protein a-coated ssDNA through multiple interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Katou, Y.; Kanoh, Y.; Bando, M.; Noguchi, H.; Tanaka, H.; Ashikari, T.; Sugimoto, K.; Shirahige, K. S-phase checkpoint proteins tof1 and mrc1 form a stable replication-pausing complex. Nature 2003, 424, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Sofueva, S.; Osman, F.; Lorenz, A.; Steinacher, R.; Castagnetti, S.; Ledesma, J.; Whitby, M.C. Ultrafine anaphase bridges, broken DNA and illegitimate recombination induced by a replication fork barrier. Nucleic Acids Res. 2011, 39, 6568–6584. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; di Rienzi, S.C.; Raghuraman, M.K.; Brewer, B.J. Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 2011, 1, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. Apobec3b is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of apobec3b and 3a in breast cancer kataegis. eLife 2013, 2, e00534. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An apobec cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Sabatinos, S.A.; Ranatunga, N.; Yuan, J.-P.; Green, M.D.; Forsburg, S.L. Replication stress in early s phase generates apparent micronuclei and chromosome rearrangement in fission yeast. Mol. Biol. Cell 2015. [Google Scholar] [CrossRef] [PubMed]
- Palter, K.B.; Foe, V.E.; Alberts, B.M. Evidence for the formation of nucleosome-like histone complexes on single-stranded DNA. Cell 1979, 18, 451–467. [Google Scholar] [CrossRef]
- Tsunaka, Y.; Kajimura, N.; Tate, S.; Morikawa, K. Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle. Nucleic Acids Res. 2005, 33, 3424–3434. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S.; Kelly, T. Purification and characterization of replication protein a, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 2523–2527. [Google Scholar] [CrossRef] [PubMed]
- Umezu, K.; Sugawara, N.; Chen, C.; Haber, J.E.; Kolodner, R.D. Genetic analysis of yeast RPA1 its multiple functions in DNA metabolism. Genetics 1998, 148, 989–1005. [Google Scholar] [PubMed]
- Longhese, M.P.; Plevani, P.; Lucchini, G. Replication factor a is required in vivo for DNA replication, repair, and recombination. Mol. Cell. Biol. 1994, 14, 7884–7890. [Google Scholar] [PubMed]
- Deng, S.K.; Chen, H.; Symington, L.S. Replication protein a prevents promiscuous annealing between short sequence homologies: Implications for genome integrity. Bioessays 2015, 37, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.K.; Gibb, B.; de Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Firmenich, A.A.; Elias-Arnanz, M.; Berg, P. A novel allele of saccharomyces cerevisiae rfa1 that is deficient in recombination and repair and suppressible by Rad52. Mol. Cell. Biol. 1995, 15, 1620–1631. [Google Scholar] [PubMed]
- Wang, X.; Haber, J.E. Role of saccharomyces single-stranded DNA-binding protein RPA in the strand invasion step of double-strand break repair. PLoS Biol. 2004, 2, e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolner, B.; van Komen, S.; Sung, P.; Peterson, C.L. Recruitment of the recombinational repair machinery to a DNA double-strand break in yeast. Mol. Cell 2003, 12, 221–232. [Google Scholar] [CrossRef]
- Parker, A.E.; Clyne, R.K.; Carr, A.M.; Kelly, T.J. The schizosaccharomyces pombe Rad11+ gene encodes the large subunit of replication protein A. Mol. Cell. Biol. 1997, 17, 2381–2390. [Google Scholar] [PubMed]
- Audry, J.; Maestroni, L.; Delagoutte, E.; Gauthier, T.; Nakamura, T.M.; Gachet, Y.; Saintome, C.; Geli, V.; Coulon, S. RPA prevents g-rich structure formation at lagging-strand telomeres to allow maintenance of chromosome ends. EMBO J. 2015, 34, 1942–1958. [Google Scholar] [CrossRef] [PubMed]
- Kibe, T.; Ono, Y.; Sato, K.; Ueno, M. Fission yeast taz1 and RPA are synergistically required to prevent rapid telomere loss. Mol. Biol. Cell 2007, 18, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Luciano, P.; Coulon, S.; Faure, V.; Corda, Y.; Bos, J.; Brill, S.J.; Gilson, E.; Simon, M.N.; Geli, V. RPA facilitates telomerase activity at chromosome ends in budding and fission yeasts. EMBO J. 2012, 31, 2034–2046. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.R.; Sabouri, N.; Webb, C.J.; Zakian, V.A. The pif1 family helicase pfh1 facilitates telomere replication and has an RPA-dependent role during telomere lengthening. DNA Repair (Amst) 2014, 24, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Baumann, P. Chromosome fusions following telomere loss are mediated by single-strand annealing. Mol. Cell 2008, 31, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Tomita, K.; Matsuura, A.; Nakagawa, T.; Masukata, H.; Uritani, M.; Ushimaru, T.; Ueno, M. A novel allele of fission yeast Rad11 that causes defects in DNA repair and telomere length regulation. Nucleic Acids Res. 2003, 31, 7141–7149. [Google Scholar] [CrossRef] [PubMed]
- New, J.H.; Sugiyama, T.; Zaitseva, E.; Kowalczykowski, S.C. RAD52 protein stimulates DNA strand exchange by Rad51 and replication protein a. Nature 1998, 391, 407–410. [Google Scholar] [PubMed]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell 2004, 118, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with brca2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Sung, P. Function of yeast Rad52 protein as a mediator between replication protein a and the Rad51 recombinase. J. Biol. Chem. 1997, 272, 28194–28197. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, Z.; Liu, Y.; Zou, Y. Preferential localization of hyperphosphorylated replication protein a to double-strand break repair and checkpoint complexes upon DNA damage. Biochem. J. 2005, 391, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Vaithiyalingam, S.; Glick, G.G.; Mordes, D.A.; Chazin, W.J.; Cortez, D. The basic cleft of RPA70n binds multiple checkpoint proteins, including Rad9, to regulate atr signaling. Mol. Cell. Biol. 2008, 28, 7345–7353. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lindsey-Boltz, L.A.; Kemp, M.; Mason, A.C.; Wold, M.S.; Sancar, A. Reconstitution of RPA-covered single-stranded DNA-activated atr-chk1 signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 13660–13665. [Google Scholar] [CrossRef] [PubMed]
- Anantha, R.W.; Sokolova, E.; Borowiec, J.A. RPA phosphorylation facilitates mitotic exit in response to mitotic DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 12903–12908. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Moore, J.K.; Holmes, A.; Umezu, K.; Kolodner, R.D.; Haber, J.E. Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 1998, 94, 399–409. [Google Scholar] [CrossRef]
- Longhese, M.P.; Neecke, H.; Paciotti, V.; Lucchini, G.; Plevani, P. The 70 Kda subunit of replication protein A is required for the G1/S and intra-S DNA damage checkpoints in budding yeast. Nucleic Acids Res. 1996, 24, 3533–3537. [Google Scholar] [CrossRef] [PubMed]
- Brush, G.S.; Morrow, D.M.; Hieter, P.; Kelly, T.J. The ATM homologue Mec1 is required for phosphorylation of replication protein A in yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 15075–15080. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Stillman, B. Cdc2 family kinases phosphorylate a human cell DNA replication factor, RPA, and activate DNA replication. EMBO J. 1992, 11, 2189–2199. [Google Scholar] [PubMed]
- Fotedar, R.; Roberts, J.M. Cell cycle regulated phosphorylation of RPA-32 occurs within the replication initiation complex. EMBO J. 1992, 11, 2177–2187. [Google Scholar] [PubMed]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-pk, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef]
- Olson, E.; Nievera, C.J.; Klimovich, V.; Fanning, E.; Wu, X. RPA2 is a direct downstream target for atr to regulate the s-phase checkpoint. J. Biol. Chem. 2006, 281, 39517–39533. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Feng, Z.; Zhang, J.; Gonzalez-Suarez, I.; Vanderwaal, R.P.; Wu, X.; Powell, S.N.; Roti Roti, J.L.; Gonzalo, S.; Zhang, J. The role of RPA2 phosphorylation in homologous recombination in response to replication arrest. Carcinogenesis 2010, 31, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Treuner, K.; Findeisen, M.; Strausfeld, U.; Knippers, R. Phosphorylation of replication protein a middle submit (RPA32) leads to a disassembly of the RPA heterotrimer. J. Biol. Chem. 1999, 274, 15556–15561. [Google Scholar] [CrossRef] [PubMed]
- Brush, G.S.; Kelly, T.J. Phosphorylation of the replication protein a large subunit in the saccharomyces cerevisiae checkpoint response. Nucleic Acids Res. 2000, 28, 3725–3732. [Google Scholar] [CrossRef] [PubMed]
- Din, S.; Brill, S.J.; Fairman, M.P.; Stillman, B. Cell-cycle-regulated phosphorylation of DNA replication factor a from human and yeast cells. Genes Dev. 1990, 4, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Brill, S.J. Mec1-dependent phosphorylation of yeast RPA1 in vitro. DNA Repair 2003, 2, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.S.; Kuo, S.R.; Melendy, T. Phosphorylation of replication protein a by S-phase checkpoint kinases. DNA Repair 2006, 5, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kvaratskhelia, M.; Hess, S.; Qu, Y.; Zou, Y. Modulation of replication protein a function by its hyperphosphorylation-induced conformational change involving DNA binding domain b. J. Biol. Chem. 2005, 280, 32775–32783. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guan, J.; Wang, H.; Perrault, A.R.; Wang, Y.; Iliakis, G. Replication protein A2 phosphorylation after DNA damage by the coordinated action of ataxia telangiectasia-mutated and DNA-dependent protein kinase. Cancer Res. 2001, 61, 8554–8563. [Google Scholar] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.C.; Rahman, S.; Lisby, M.; Rothstein, R.; Zhao, X. The Slx5-Slx8 complex affects sumoylation of DNA repair proteins and negatively regulates recombination. Mol. Cell. Biol. 2007, 27, 6153–6162. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Huang, C.; Singh, M.; Carpenter, P.B.; Yeh, E.T. Regulation of DNA repair through desumoylation and sumoylation of replication protein a complex. Mol. Cell 2010, 39, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Pellicioli, A.; Vaze, M.B.; Sugawara, N.; Malkova, A.; Foiani, M.; Haber, J.E. Yeast Rad52 and Rad51 recombination proteins define a second pathway of DNA damage assessment in response to a single double-strand break. Mol. Cell. Biol. 2003, 23, 8913–8923. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ira, G.; Tercero, J.A.; Holmes, A.M.; Diffley, J.F.; Haber, J.E. Role of DNA replication proteins in double-strand break-induced recombination in saccharomyces cerevisiae. Mol. Cell. Biol. 2004, 24, 6891–6899. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Rushton, M.D.; Maringele, L. A novel checkpoint and RPA inhibitory pathway regulated by rif1. PLoS Genet. 2011, 7, e1002417. [Google Scholar] [CrossRef] [PubMed]
- Hass, C.S.; Gakhar, L.; Wold, M.S. Functional characterization of a cancer causing mutation in human replication protein a. Mol. Cancer Res. 2010, 8, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Furuya, K.; Miyabe, I.; Tsutsui, Y.; Paderi, F.; Kakusho, N.; Masai, H.; Niki, H.; Carr, A.M. Ddk phosphorylates checkpoint clamp component Rad9 and promotes its release from damaged chromatin. Mol. Cell 2010, 40, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Fanning, E.; Klimovich, V.; Nager, A.R. A dynamic model for replication protein a (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Huberman, J.A. Regulation of replication timing in fission yeast. EMBO J. 2001, 20, 6115–6126. [Google Scholar] [CrossRef] [PubMed]
- Sabatinos, S.A.; Green, M.D.; Forsburg, S.L. Continued DNA synthesis in replication checkpoint mutants leads to fork collapse. Mol. Cell. Biol. 2012, 32, 4986–4997. [Google Scholar] [CrossRef] [PubMed]
- Enoch, T.; Carr, A.M.; Nurse, P. Fission yeast genes involved in coupling mitosis to completion of DNA replication. Genes Dev. 1992, 6, 2035–2046. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Capasso, H.; Walworth, N.C. The topoisomerase I poison camptothecin generates a Chk1-dependent DNA damage checkpoint signal in fission yeast. Yeast 1999, 15, 821–828. [Google Scholar] [CrossRef]
- Kumar, S.; Huberman, J.A. Checkpoint-dependent regulation of origin firing and replication fork movement in response to DNA damage in fission yeast. Mol. Cell. Biol. 2009, 29, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Willis, N.; Rhind, N. Regulation of DNA replication by the S-phase DNA damage checkpoint. Cell Div. 2009. [Google Scholar] [CrossRef] [PubMed]
- Bagley, B.N.; Keane, T.M.; Maklakova, V.I.; Marshall, J.G.; Lester, R.A.; Cancel, M.M.; Paulsen, A.R.; Bendzick, L.E.; Been, R.A.; Kogan, S.C.; et al. A dominantly acting murine allele of MCM4 causes chromosomal abnormalities and promotes tumorigenesis. PLoS Genet. 2012, 8, e1003034. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.T.; Hodson, J.A.; Forsburg, S.L. Reduced dosage of a single fission yeast MCM protein causes genetic instability and S phase delay. J. Cell Sci. 1999, 112, 559–567. [Google Scholar] [PubMed]
- Lambert, S.; Mizuno, K.; Blaisonneau, J.; Martineau, S.; Chanet, R.; Freon, K.; Murray, J.M.; Carr, A.M.; Baldacci, G. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell 2010, 39, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Kaochar, S.; Shanks, L.; Weinert, T. Checkpoint genes and EXO1 regulate nearby inverted repeat fusions that form dicentric chromosomes in saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2010, 107, 21605–21610. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Lambert, S.; Baldacci, G.; Murray, J.M.; Carr, A.M. Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev. 2009, 23, 2876–2886. [Google Scholar] [CrossRef] [PubMed]
- Bass, K.L.; Murray, J.M.; O’Connell, M.J. Brc1-dependent recovery from replication stress. J. Cell Sci. 2012, 125, 2753–2764. [Google Scholar] [CrossRef] [PubMed]
- Sabatinos, S.A.; Mastro, T.L.; Green, M.D.; Forsburg, S.L. A mammalian-like DNA damage response of fission yeast to nucleoside analogs. Genetics 2013, 193, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Sharma, S.; Rozenzhak, S.; Roguev, A.; Krogan, N.J.; Chabes, A.; Russell, P. Replication fork collapse and genome instability in a deoxycytidylate deaminase mutant. Mol. Cell. Biol. 2012, 32, 4445–4454. [Google Scholar] [CrossRef] [PubMed]
- Ukimori, S.; Kawabata, N.; Shimada, H.; Imano, R.; Takahashi, K.; Yukawa, M.; Tsuchiya, E.; Ueno, M. A double mutant between fission yeast telomerase and RECQ helicase is sensitive to thiabendazole, an anti-microtubule drug. Biosci. Biotechnol. Biochem. 2012, 76, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Cavero, S.; Limbo, O.; Russell, P. Critical functions of RPA3/SSB3 in S-phase DNA damage responses in fission yeast. PLoS Genet. 2010, 6, e1001138. [Google Scholar] [CrossRef] [PubMed]
- Meister, P.; Taddei, A.; Vernis, L.; Poidevin, M.; Gasser, S.M.; Baldacci, G. Temporal separation of replication and recombination requires the intra-S checkpoint. J. Cell Biol. 2005, 168, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Meister, P.; Taddei, A.; Ponti, A.; Baldacci, G.; Gasser, S.M. Replication foci dynamics: Replication patterns are modulated by S-phase checkpoint kinases in fission yeast. EMBO J. 2007, 26, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Lisby, M.; Rothstein, R.; Mortensen, U.H. Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl. Acad. Sci. USA 2001, 98, 8276–8282. [Google Scholar] [CrossRef] [PubMed]
- Irmisch, A.; Ampatzidou, E.; Mizuno, K.; O’Connell, M.J.; Murray, J.M. Smc5/6 maintains stalled replication forks in a recombination-competent conformation. EMBO J. 2009, 28, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.M.; Du, L.L.; Redon, C.; Russell, P. Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol. Cell. Biol. 2004, 24, 6215–6230. [Google Scholar] [CrossRef] [PubMed]
- Bailis, J.M.; Luche, D.D.; Hunter, T.; Forsburg, S.L. Minichromosome maintenance proteins interact with checkpoint and recombination proteins to promote S-phase genome stability. Mol. Cell. Biol. 2008, 28, 1724–1738. [Google Scholar] [CrossRef] [PubMed]
- Steinacher, R.; Osman, F.; Dalgaard, J.Z.; Lorenz, A.; Whitby, M.C. The DNA helicase PFH1 promotes fork merging at replication termination sites to ensure genome stability. Genes Dev. 2012, 26, 594–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different Rad51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Bartkova, J.; Lukas, J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007, 26, 7773–7779. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; De, S. Loss of heterozygosity preferentially occurs in early replicating regions in cancer genomes. Nucleic Acids Res. 2013, 41, 7615–7624. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.E.; Kaback, D.B. The role of centromere alignment in meiosis I segregation of homologous chromosomes in saccharomyces cerevisiae. Genetics 1999, 153, 1547–1560. [Google Scholar] [PubMed]
- Hashimoto, Y.; Puddu, F.; Costanzo, V. Rad51- and Mre11-dependent reassembly of uncoupled Cmg helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 2012, 19, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sun, L.; Shen, F.; Chen, Y.; Hua, Y.; Liu, Y.; Zhang, M.; Hu, Y.; Wang, Q.; Xu, W.; et al. The intra-S phase checkpoint targets DNA2 to prevent stalled replication forks from reversing. Cell 2012, 149, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Froget, B.; Blaisonneau, J.; Lambert, S.; Baldacci, G. Cleavage of stalled forks by fission yeast MUS81/EME1 in absence of DNA replication checkpoint. Mol. Biol. Cell 2008, 19, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Cotta-Ramusino, C.; Fachinetti, D.; Lucca, C.; Doksani, Y.; Lopes, M.; Sogo, J.; Foiani, M. EXO1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell 2005, 17, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Janes, S.; Schmidt, U.; Ashour Garrido, K.; Ney, N.; Concilio, S.; Zekri, M.; Caspari, T. Heat induction of a novel Rad9 variant from a cryptic translation initiation site reduces mitotic commitment. J. Cell Sci. 2012, 125, 4487–4497. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.P.; Resnick, J.; Sussman, R. Interaction of single-strand binding protein and RECA protein at the single-stranded DNA site. J. Mol. Biol. 1983, 167, 901–909. [Google Scholar] [CrossRef]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grofte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53bp1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.L.; Ormerod, G.; Durley, S.C.; Sing, T.L.; Brown, G.W.; Kent, N.A.; Downs, J.A. The Ino80 chromatin remodeling complex prevents polyploidy and maintains normal chromatin structure at centromeres. Genes Dev. 2012, 26, 2590–2603. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.L.; Downs, J.A. The RSC and Ino80 chromatin-remodeling complexes in DNA double-strand break repair. Prog. Mol. Biol. Transl. Sci. 2012, 110, 229–261. [Google Scholar] [PubMed]
- Feng, W.; Bachant, J.; Collingwood, D.; Raghuraman, M.K.; Brewer, B.J. Centromere replication timing determines different forms of genomic instability in saccharomyces cerevisiae checkpoint mutants during replication stress. Genetics 2009, 183, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.T.; Karlseder, J. DNA damage associated with mitosis and cytokinesis failure. Oncogene 2013, 32, 4593–4601. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, H.W.; Huttner, D.; Hickson, I.D. How unfinished business from S-phase affects mitosis and beyond. EMBO J. 2013, 32, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabatinos, S.A.; Forsburg, S.L. Managing Single-Stranded DNA during Replication Stress in Fission Yeast. Biomolecules 2015, 5, 2123-2139. https://0-doi-org.brum.beds.ac.uk/10.3390/biom5032123
Sabatinos SA, Forsburg SL. Managing Single-Stranded DNA during Replication Stress in Fission Yeast. Biomolecules. 2015; 5(3):2123-2139. https://0-doi-org.brum.beds.ac.uk/10.3390/biom5032123
Chicago/Turabian StyleSabatinos, Sarah A., and Susan L. Forsburg. 2015. "Managing Single-Stranded DNA during Replication Stress in Fission Yeast" Biomolecules 5, no. 3: 2123-2139. https://0-doi-org.brum.beds.ac.uk/10.3390/biom5032123