Interplay between BMPs and Reactive Oxygen Species in Cell Signaling and Pathology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
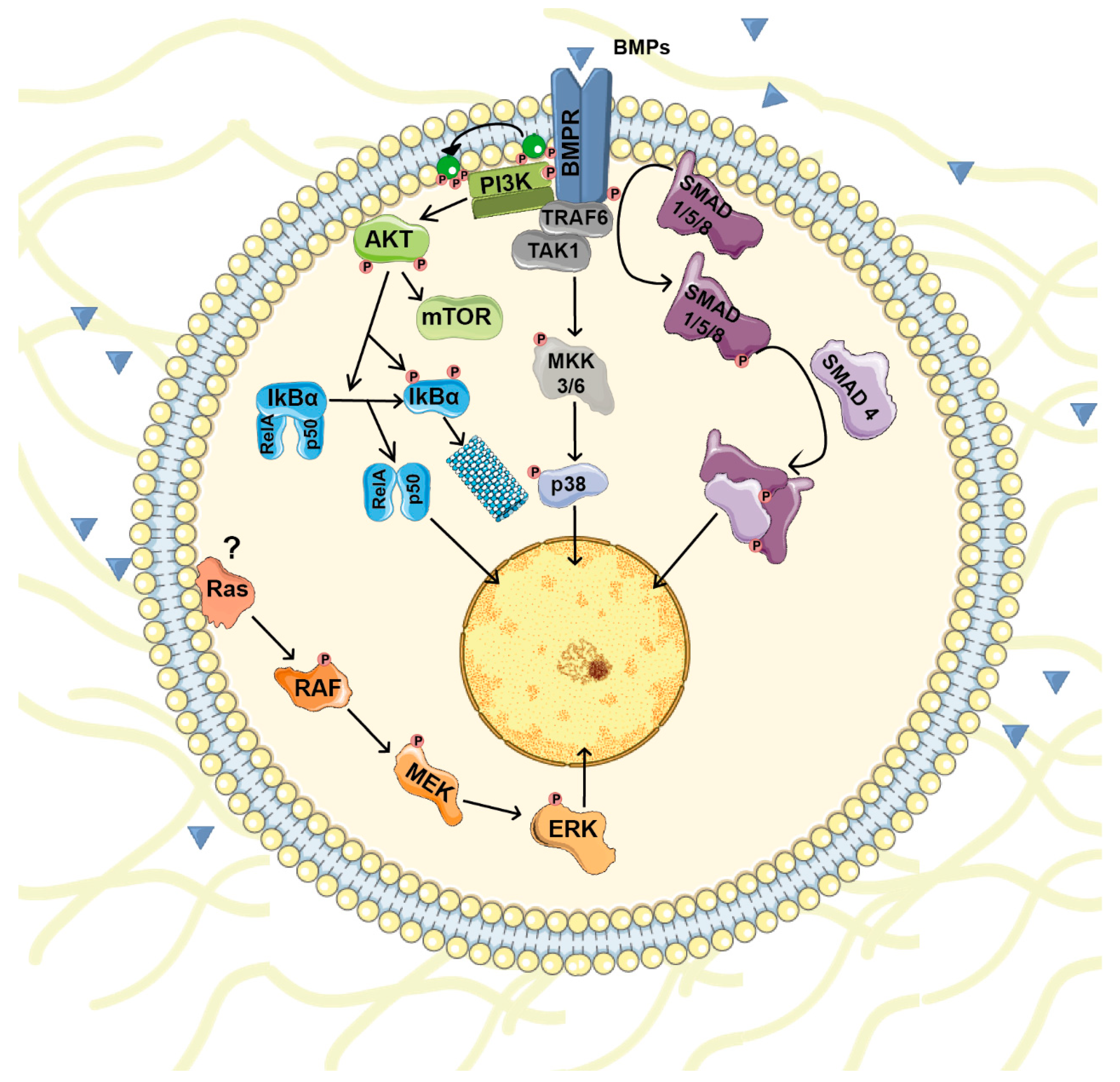
2. BMP Signals
3. ROS
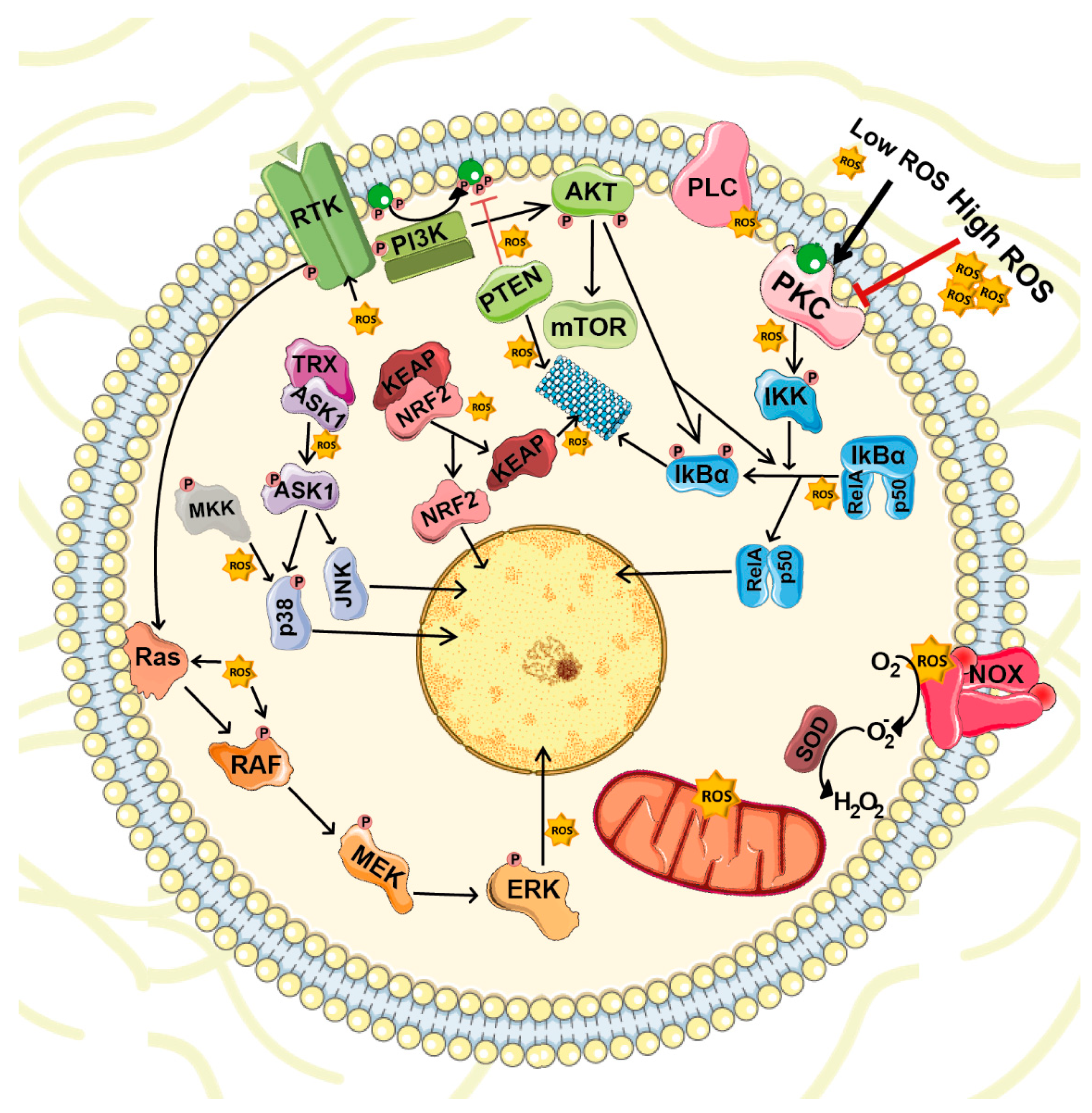
4. ROS-Mediated Cellular Signaling
5. NOXs
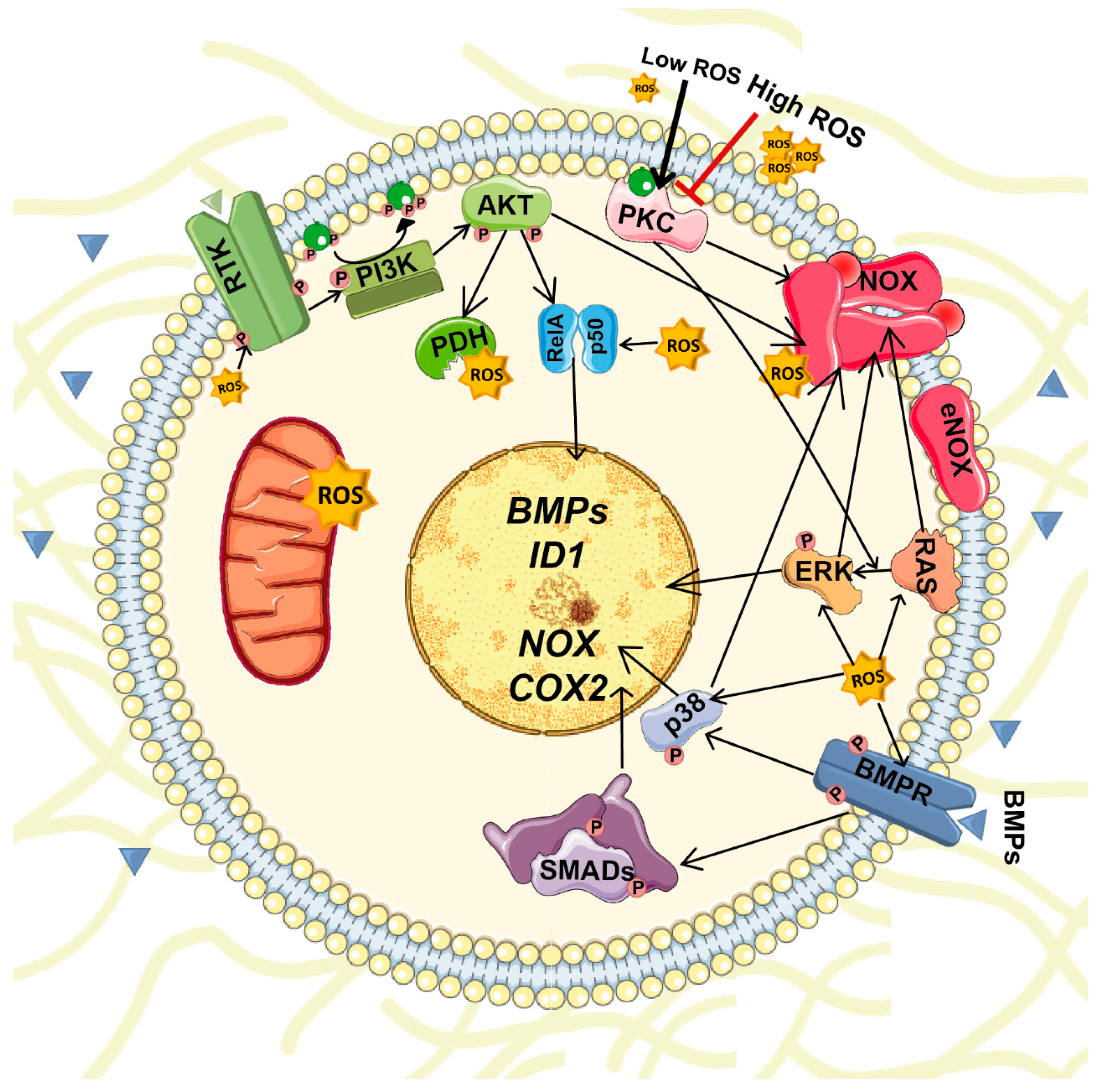
6. Interplay between BMPs and ROS Signaling
7. BMPs and ROS in Cell Specification
8. BMPs and ROS in Pathology
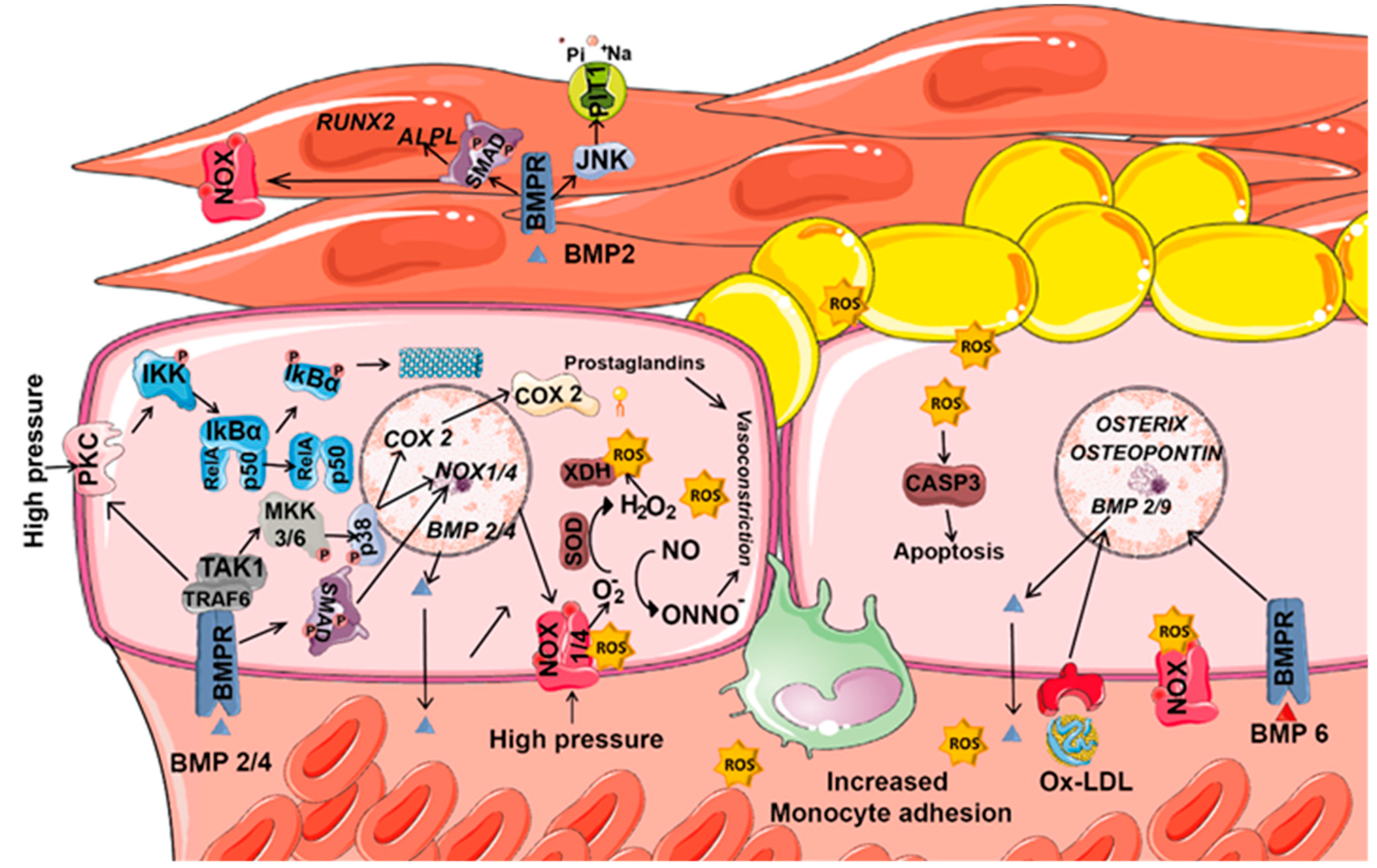
8.1. ROS and BMPs in Vascular Diseases
8.2. ROS and BMPs in Obesity and Diabetes
8.3. ROS and BMPs in Kidney Injury
8.4. ROS and BMPs in Musculoskeletal Diseases
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Urist, M.R.; Rosen, V.; Celeste, A.; Mitsock, L.; Whitters, M.; Kriz, R.; Hewick, R.; Wang, E. Bone: Formation by Autoinduction; American Association for the Advancement of Science: Washington, DC, USA, 1965; Volume 150, pp. 893–899. [Google Scholar]
- Winnier, G.; Blessing, M.; Labosky, P.A.; Hogan, B.L. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995, 9, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Dehart, D.B.; Sulik, K.K.; Hogan, B.L.M. Distinct requirements for extra-embryonic and embryonic bone morphogenetic protein 4 in the formation of the node and primitive streak and coordination of left-right asymmetry in the mouse. Development 2002, 129, 4685–4696. [Google Scholar] [PubMed]
- Hébert, J.M.; Mishina, Y.; McConnell, S.K. BMP Signaling Is Required Locally to Pattern the Dorsal Telencephalic Midline. Neuron 2002, 35, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Mehler, M.F.; Mabie, P.C.; Zhang, D.; Kessler, J.A. Bone morphogenetic proteins in the nervous system. Trends Neurosci. 1997, 20, 309–317. [Google Scholar] [CrossRef]
- Storm, E.E.; Huynh, T.V.; Copeland, N.G.; Jenkins, N.A.; Kingsley, D.M.; Lee, S.J. Limb alterations in brachypodism mice due to mutations in a new member of the TGFβ-superfamily. Nature 1994, 368, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Harris, S.E.; Horn, D.; Geng, Z.; Nishimura, R.; Mundy, G.R.; Chen, D. Bone morphogenetic protein receptor signaling is necessary for normal murine postnatal bone formation. J. Cell Biol. 2002, 157, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Pizette, S.; Niswander, L. BMPs Are Required at Two Steps of Limb Chondrogenesis: Formation of Prechondrogenic Condensations and Their Differentiation into Chondrocytes. Dev. Biol. 2000, 219, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozney, J.M. The bone morphogenetic protein family and osteogenesis. Mol. Reprod. Dev. 1992, 32, 160–167. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Oshima, K.; Fogo, A.; Hogan, B.L.; Ichikawa, I. Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J. Clin. Investig. 2000, 105, 863–873. [Google Scholar] [CrossRef] [Green Version]
- Lowery, J.W.; De Caestecker, M.P. BMP signaling in vascular development and disease. Cytokine Growth Factor Rev. 2010, 21, 287–298. [Google Scholar] [CrossRef] [Green Version]
- De Vinuesa, A.G.; Abdelilah-Seyfried, S.; Knaus, P.; Zwijsen, A.; Bailly, S.; Information, P.E.K.F.C. BMP signaling in vascular biology and dysfunction. Cytokine Growth Factor Rev. 2016, 27, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Bloch, D.B.; Ten Dijke, P.; Goumans, M.J.T.H.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signaling in cardiovascular disease and anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.Q.; Liaw, L.; Hogan, B.L. Bone morphogenetic protein 8A plays a role in the maintenance of spermatogenesis and the integrity of the epididymis. Development 1998, 125, 1103–1112. [Google Scholar] [PubMed]
- Otsuka, F.; Yao, Z.; Lee, T.; Yamamoto, S.; Erickson, G.F.; Shimasaki, S. Bone morphogenetic protein-15. Identification of target cells and biological functions. J. Biol. Chem. 2000, 275, 39523–39528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zou, M.H. Roles of Reactive Oxygen Species in Physiology and Pathology. In Atherosclerosis: Risks, Mechanisms, and Therapies; Wiley: Hoboken, NJ, USA, 2015; pp. 379–392. [Google Scholar]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Galano, J.M.; Durand, T.; Le Guennec, J.Y.; Lee, J.C.Y. Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef] [Green Version]
- Reid, M.B.; Khawli, F.A.; Moody, M.R. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol. 1993, 75, 1081–1087. [Google Scholar]
- Kraaij, M.D.; Savage, N.D.L.; Van Der Kooij, S.W.; Koekkoek, K.; Wang, J.; Berg, J.M.V.D.; Ottenhoff, T.H.M.; Kuijpers, T.W.; Holmdahl, R.; Van Kooten, C.; et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 17686–17691. [Google Scholar] [CrossRef] [Green Version]
- Sellak, H.; Franzini, E.; Hakim, J.; Pasquier, C. Reactive oxygen species rapidly increase endothelial ICAM-1 ability to bind neutrophils without detectable upregulation. Blood 1994, 83, 2669–2677. [Google Scholar] [Green Version]
- Kawabata, M. Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev. 1998, 9, 49–61. [Google Scholar] [CrossRef]
- Nohe, A. Signal transduction of bone morphogenetic protein receptors. Cell. Signal. 2004, 16, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P. Structural studies of the TGF-βs and their receptors—Insights into evolution of the TGF-β superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGF-β Signal Transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Gámez, B.; Rodríguez-Carballo, E.; Ventura, F. BMP signaling in telencephalic neural cell specification and maturation. Front. Cell. Neurosci. 2013, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef]
- Brazil, D.P.; Church, R.H.; Surae, S.; Godson, C.; Martin, F. BMP signalling: Agony and antagony in the family. Trends Cell Biol. 2015, 25, 249–264. [Google Scholar] [CrossRef]
- Rider, C.C.; Mulloy, B. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem. J. 2010, 429, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone Morphogenetic Proteins: A critical review. Cell. Signal. 2011, 23, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoonbroodt, S.; Ferreira, V.; Best-Belpomme, M.; Boelaert, J.R.; Legrand-Poels, S.; Korner, M.; Piette, J. Crucial Role of the Amino-Terminal Tyrosine Residue 42 and the Carboxyl-Terminal PEST Domain of I B in NF- B Activation by an Oxidative Stress. J. Immunol. 2000, 164, 4292–4300. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [PubMed]
- León-Buitimea, A.; Rodríguez-Fragoso, L.; Lauer, F.T.; Bowles, H.; Thompson, T.A.; Burchiel, S.W. Ethanol-induced oxidative stress is associated with EGF receptor phosphorylation in MCF-10A cells overexpressing CYP2E1. Toxicol. Lett. 2012, 209, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Wentworth, C.C.; Alam, A.; Jones, R.M.; Nusrat, A.; Neish, A.S. Enteric Commensal Bacteria Induce Extracellular Signal-regulated Kinase Pathway Signaling via Formyl Peptide Receptor-dependent Redox Modulation of Dual Specific Phosphatase 3. J. Biol. Chem. 2011, 286, 38448–38455. [Google Scholar] [CrossRef] [Green Version]
- Banan, A.; Fields, J.Z.; Zhang, Y.; Keshavarzian, A. Phospholipase C-γ inhibition prevents EGF protection of intestinal cytoskeleton and barrier against oxidants. Am. J. Physiol. Liver Physiol. 2001, 281, G412–G423. [Google Scholar] [CrossRef]
- Matsukawa, J.; Matsuzawa, A.; Takeda, K.; Ichijo, H. The ASK1-MAP Kinase Cascades in Mammalian Stress Response. J. Biochem. 2004, 136, 261–265. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Yue, P.; Tao, H.; Ramalingam, S.S.; Owonikoko, T.K.; Deng, X.; Wang, Y.; Fu, H.; Khuri, F.R.; et al. Protein Phosphatase 2A and DNA-dependent Protein Kinase Are Involved in Mediating Rapamycin-induced Akt Phosphorylation. J. Biol. Chem. 2013, 288, 13215–13224. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Jeong, W.; Rhee, S.G.; Lee, C. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef]
- Shimura, T.; Sasatani, M.; Kamiya, K.; Kawai, H.; Inaba, Y.; Kunugita, N. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in lowdose irradiated human fibroblasts. Oncotarget 2016, 7, 3559–3570. [Google Scholar] [CrossRef]
- Srinivasan, S.; Spear, J.; Chandran, K.; Joseph, J.; Kalyanaraman, B.; Avadhani, N.G. Oxidative Stress Induced Mitochondrial Protein Kinase A Mediates Cytochrome C Oxidase Dysfunction. PLoS ONE 2013, 8, e77129. [Google Scholar] [CrossRef]
- Thompson, J.W.; Narayanan, S.V.; Perez-Pinzon, M.A. Redox Signaling Pathways Involved in Neuronal Ischemic Preconditioning. Curr. Neuropharmacol. 2012, 10, 354–369. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Kimchi, A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 2012, 19, 788–797. [Google Scholar] [CrossRef]
- Kruk, J.S.; Vasefi, M.S.; Heikkila, J.J.; Beazely, M.A. Reactive Oxygen Species Are Required for 5-HT-Induced Transactivation of Neuronal Platelet-Derived Growth Factor and TrkB Receptors, but Not for ERK1/2 Activation. PLoS ONE 2013, 8, e77027. [Google Scholar] [CrossRef]
- Luczak, E.D.; Anderson, M.E. CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell. Cardiol. 2014, 73, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schröder, E.; Wait, R.; Begum, S.; Kentish, J.C.; Eaton, P. Oxidant-induced Activation of Type I Protein Kinase A Is Mediated by RI Subunit Interprotein Disulfide Bond Formation. J. Biol. Chem. 2006, 281, 21827–21836. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.C. Protein Kinase C: Structure, Function, and Regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free. Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell Signaling through Protein Kinase C Oxidation and Activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef] [Green Version]
- Cowell, C.F.; Döppler, H.; Yan, I.K.; Hausser, A.; Umezawa, Y.; Storz, P. Mitochondrial diacylglycerol initiates protein-kinase D1-mediated ROS signaling. J. Cell Sci. 2009, 122, 919–928. [Google Scholar] [CrossRef]
- Wang, Q.J. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol. Sci. 2006, 27, 317–323. [Google Scholar] [CrossRef]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by Protein Kinase C Regulates Antioxidant Response Element-mediated Transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [Green Version]
- Nakaso, K.; Yano, H.; Fukuhara, Y.; Takeshima, T.; Wada-Isoe, K.; Nakashima, K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003, 546, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef]
- Jain, A.; Jaiswal, A.K. GSK-3beta Acts Upstream of Fyn Kinase in Regulation of Nuclear Export and Degradation of NF-E2 Related Factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants 2017, 6, 90. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free. Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Hoyal, C.R.; Gutierrez, A.; Young, B.M.; Catz, S.D.; Lin, J.H.; Tsichlis, P.N.; Babior, B.M. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 5130–5135. [Google Scholar] [CrossRef]
- Kanai, F.; Liu, H.; Field, S.J.; Akbary, H.; Matsuo, T.; Brown, G.E.; Cantley, L.C.; Yaffe, M.B. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nature 2001, 3, 675–678. [Google Scholar] [CrossRef]
- Zhan, Y.; Virbasius, J.V.; Song, X.; Pomerleau, D.P.; Zhou, G.W. The p40phox and p47phox PX domains of NADPH oxidase target cell membranes via direct and indirect recruitment by phosphoinositides. J. Biol. Chem. 2002, 277, 4512–4518. [Google Scholar] [CrossRef]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Wingler, K.; Wünsch, S.; Kreutz, R.; Rothermund, L.; Paul, M.; Schmidt, H.H. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic. Biol. Med. 2001, 31, 1456–1464. [Google Scholar] [CrossRef]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H Oxidase Homolog Nox4 Modulates Insulin-Stimulated Generation of H2O2 and Plays an Integral Role in Insulin Signal Transduction. Mol. Cell. Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef]
- Simone, S.; Cosola, C.; Loverre, A.; Cariello, M.; Sallustio, F.; Rascio, F.; Gesualdo, L.; Schena, F.P.; Grandaliano, G.; Pertosa, G. BMP-2 induces a profibrotic phenotype in adult renal progenitor cells through Nox4 activation. Am. J. Physiol. Physiol. 2012, 303, F23–F34. [Google Scholar] [CrossRef] [Green Version]
- Mandal, C.C.; Ganapathy, S.; Gorin, Y.; Mahadev, K.; Block, K.; Abboud, H.E.; Harris, S.E.; Ghosh-Choudhury, G.; Ghosh-Choudhury, N. Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem. J. 2011, 433, 393–402. [Google Scholar] [CrossRef]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2016, 10, 301. [Google Scholar] [CrossRef]
- Chandrasekaran, V.; Lea, C.; Sosa, J.C.; Higgins, D.; Lein, P.J. Reactive oxygen species are involved in BMP-induced dendritic growth in cultured rat sympathetic neurons. Mol. Cell. Neurosci. 2015, 67, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Sorescu, G.P.; Song, H.; Tressel, S.L.; Hwang, J.; Dikalov, S.; Smith, D.A.; Boyd, N.L.; Platt, M.O.; Lassègue, B.; Griendling, K.K.; et al. Bone Morphogenic Protein 4 Produced in Endothelial Cells by Oscillatory Shear Stress Induces Monocyte Adhesion by Stimulating Reactive Oxygen Species Production From a Nox1-Based NADPH Oxidase. Circ. Res. 2004, 95, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Pache, G.; Schäfer, C.; Wiesemann, S.; Springer, E.; Liebau, M.; Reinhardt, H.C.; August, C.; Pavenstädt, H.; Bek, M.J. Upregulation of Id-1 via BMP-2 receptors induces reactive oxygen species in podocytes. Am. J. Physiol. Physiol. 2006, 291, F654–F662. [Google Scholar] [CrossRef] [Green Version]
- Susperregui, A.R.G.; Gamell, C.; Rodríguez-Carballo, E.; Ortuño, M.J.; Bartrons, R.; Rosa, J.L.; Ventura, F. Noncanonical BMP Signaling Regulates Cyclooxygenase-2 Transcription. Mol. Endocrinol. 2011, 25, 1006–1017. [Google Scholar] [CrossRef]
- Hernanz, R.; Briones, A.M.; Salaices, M.; Alonso, M.J. New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension. Clin. Sci. 2014, 126, 111–121. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef]
- Sampson, N.; Berger, P.; Zenzmaier, C. Redox Signaling as a Therapeutic Target to Inhibit Myofibroblast Activation in Degenerative Fibrotic Disease. BioMed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Csiszar, A.; Smith, K.E.; Koller, A.; Kaley, G.; Edwards, J.G.; Ungvari, Z. Regulation of Bone Morphogenetic Protein-2 Expression in Endothelial Cells. Circulation 2005, 111, 2364–2372. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, T.; Kuroyanagi, N.; Yamaguchi, K.; Gotoh, Y.; Irie, K.; Kano, T.; Shirakabe, K.; Muro, Y.; Shibuya, H.; Matsumoto, K. A Novel Kinase Cascade Mediated by Mitogen-activated Protein Kinase Kinase 6and MKK3. J. Biol. Chem. 1996, 271, 13675–13679. [Google Scholar] [CrossRef]
- Chen, X.; Liao, J.; Lu, Y.; Duan, X.; Sun, W. Activation of the PI3K/Akt Pathway Mediates Bone Morphogenetic Protein 2-Induced Invasion of Pancreatic Cancer Cells Panc-1. Pathol. Oncol. Res. 2011, 17, 257–261. [Google Scholar] [CrossRef]
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin Exerts an Antiapoptotic Effect by Regulating the Redox State of Akt. J. Biol. Chem. 2003, 278, 50226–50233. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef]
- Hu, C.; Zhao, L.; Peng, C.; Li, L. Regulation of the mitochondrial reactive oxygen species: Strategies to control mesenchymal stem cell fates ex vivo and in vivo. J. Cell. Mol. Med. 2018, 22, 5196–5207. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, S. The Signaling Pathways Involved in Chondrocyte Differentiation and Hypertrophic Differentiation. Stem Cells Int. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kang, I.S.; Kim, C. NADPH oxidase gp91phox contributes to RANKL-induced osteoclast differentiation by upregulating NFATc1. Sci. Rep. 2016, 6, 38014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, H.; Kwak, H.B.; Lee, S.W.; Jin, H.M.; Kim, H.M.; Kim, H.H.; Lee, Z.H. Reactive oxygen species mediate RANK signaling in osteoclasts. Exp. Cell Res. 2004, 301, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stouffs, M.; Serrander, L.; Banfi, B.; Bettiol, E.; Charnay, Y.; Steger, K.; Krause, K.H.; Jaconi, M.E. The NADPH Oxidase NOX4 Drives Cardiac Differentiation: Role in Regulating Cardiac Transcription Factors and MAP Kinase Activation. Mol. Biol. Cell 2006, 17, 3978–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Gong, F. BMP-2 and icariin synergistically promote p38MAPK-mediated cardiomyocyte differentiation of mesenchymal stem cells via enhanced NOX4-driven ROS generation. Med. Chem. Res. 2017, 26, 2547–2556. [Google Scholar] [CrossRef]
- Lein, P.; Johnson, M.; Guo, X.; Rueger, D.; Higgins, D. Osteogenic protein-1 induces dendritic growth in rat sympathetic neurons. Neuron 1995, 15, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Charette, M.; Banker, G.; Withers, G.S.; Higgins, D. Bone morphogenetic protein-7 enhances dendritic growth and receptivity to innervation in cultured hippocampal neurons. Eur. J. Neurosci. 2000, 12, 106–116. [Google Scholar]
- Le Roux, P.; Behar, S.; Higgins, D.; Charette, M. OP-1 Enhances Dendritic Growth from Cerebral Cortical Neurons in Vitro. Exp. Neurol. 1999, 160, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Hocking, J.C.; Hehr, C.L.; Chang, R.Y.; Johnston, J.; McFarlane, S. TGFβ ligands promote the initiation of retinal ganglion cell dendrites in vitro and in vivo. Mol. Cell. Neurosci. 2008, 37, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Thangnipon, W.; Puangmalai, N.; Suwanna, N.; Soi-Ampornkul, R.; Phonchai, R.; Kotchabhakdi, N.; Mukda, S.; Phermthai, T.; Julavijitphong, S.; Tuchinda, P.; et al. Potential role of N-benzylcinnamide in inducing neuronal differentiation from human amniotic fluid mesenchymal stem cells. Neurosci. Lett. 2016, 610, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Salmon, R.M.; Upton, P.D.; Morrell, N.W.; Li, W. Regulation of bone morphogenetic protein 9 (BMP9) by redox-dependent proteolysis. J. Biol. Chem. 2014, 289, 31150–31159. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Weissman, I.L.; Reya, T.; Clarke, M.F. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [Green Version]
- Wang, L.; Park, P.; Zhang, H.; La Marca, F.; Claeson, A.; Valdivia, J.; Lin, C.Y. BMP-2 inhibits the tumorigenicity of cancer stem cells in human osteosarcoma OS99-1 cell line. Cancer Biol. Ther. 2011, 11, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, Y.; Scopelliti, A.; Cammareri, P.; Todaro, M.; Iovino, F.; Ricci–Vitiani, L.; Gulotta, G.; Dieli, F.; De Maria, R.; Stassi, G. Bone Morphogenetic Protein 4 Induces Differentiation of Colorectal Cancer Stem Cells and Increases Their Response to Chemotherapy in Mice. Gastroenterology 2011, 140, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Wu, Q.; Yan, D.H.; Lee, C.H.; Rahim, N.; Tritschler, I.; DeVecchio, J.; Kalady, M.F.; Hjelmeland, A.B.; Rich, J.N. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014, 28, 1085–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxidative Med. Cell. Longev. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, F.H.; Gibbons, G.H.; Dzau, V.J. The Emerging Concept of Vascular Remodeling. N. Engl. J. Med. 1994, 330, 1431–1438. [Google Scholar] [CrossRef]
- Mueller, C.F.H.; Laude, K.; McNally, J.S.; Harrison, D.G. Redox Mechanisms in Blood Vessels. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 274–278. [Google Scholar] [CrossRef]
- Kojda, G.; Harrison, D. Interactions between NO and reactive oxygen species: Pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc. Res. 1999, 43, 562–571. [Google Scholar] [PubMed]
- Csiszar, A.; Lehoux, S.; Ungvari, Z. Hemodynamic Forces, Vascular Oxidative Stress, and Regulation of BMP-2/4 Expression. Antioxid. Redox Signal. 2009, 11, 1683–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruska, K.A.; Mathew, S.; Saab, G. Bone Morphogenetic Proteins in Vascular Calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Sara, J.D.; Wang, F.L.; Liu, L.P.; Su, L.X.; Zhe, J.; Wu, X.; Liu, J.H. Increased plasma BMP-2 levels are associated with atherosclerosis burden and coronary calcification in type 2 diabetic patients. Cardiovasc. Diabetol. 2015, 14, 64. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Liberman, M.; Johnson, R.C.; Handy, D.E.; Loscalzo, J.; Leopold, J.A. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem. Biophys. Res. Commun. 2011, 413, 436–441. [Google Scholar] [PubMed] [Green Version]
- Csiszar, A.; Ahmad, M.; Smith, K.E.; Labinskyy, N.; Gao, Q.; Kaley, G.; Edwards, J.G.; Wolin, M.S.; Ungvari, Z. Bone Morphogenetic Protein-2 Induces Proinflammatory Endothelial Phenotype. Am. J. Pathol. 2006, 168, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.Y.; Yung, L.H.; Wong, W.T.; Liu, J.; Leung, F.P.; Liu, L.; Chen, Y.; Kong, S.K.; Kwan, K.M.; Ng, S.M.; et al. Bone morphogenic protein-4 induces endothelial cell apoptosis through oxidative stress-dependent p38MAPK and JNK pathway. J. Mol. Cell. Cardiol. 2012, 52, 237–244. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Jo, H.; Ballabh, P.; Ungvari, Z. Differential proinflammatory and prooxidant effects of bone morphogenetic protein-4 in coronary and pulmonary arterial endothelial cells. Am. J. Physiol. Circ. Physiol. 2008, 295, H569–H577. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.T.; Tian, X.Y.; Chen, Y.; Leung, F.P.; Liu, L.; Lee, H.K.; Ng, C.F.; Xu, A.; Yao, X.; Vanhoutte, P.M.; et al. Bone Morphogenic Protein-4 Impairs Endothelial Function Through Oxidative Stress–Dependent Cyclooxygenase-2 Upregulation. Circ. Res. 2010, 107, 984–991. [Google Scholar] [CrossRef]
- Luo, J.Y.; Zhang, Y.; Wang, L.; Huang, Y. Regulators and effectors of bone morphogenetic protein signalling in the cardiovascular system. J. Physiol. 2015, 593, 2995–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.M.; Zhang, Y.; Huang, Y. Bone morphogenic protein-4-induced oxidant signaling via protein carbonylation for endothelial dysfunction. Free Radic. Biol. Med. 2014, 75, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Grgurevic, L.; Christensen, G.L.; Schulz, T.J.; Vukicevic, S.; Information, P.E. Bone morphogenetic proteins in inflammation, glucose homeostasis and adipose tissue energy metabolism. Cytokine Growth Factor Rev. 2016, 27, 105–118. [Google Scholar] [CrossRef]
- Yung, L.M.; Sánchez-Duffhues, G.; Dijke, P.T.; Yu, P.B. Bone morphogenetic protein 6 and oxidized low-density lipoprotein synergistically recruit osteogenic differentiation in endothelial cells. Cardiovasc. Res. 2015, 108, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derwall, M.; Malhotra, R.; Lai, C.S.; Beppu, Y.; Aikawa, E.; Seehra, J.S.; Zapol, W.M.; Bloch, K.D.; Yu, P.B. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arter. Thromb. Vasc. Biol. 2012, 32, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Duffhues, G.; De Vinuesa, A.G.; Van De Pol, V.; Geerts, M.E.; De Vries, M.R.; Janson, S.G.T.; Van Dam, H.; Lindeman, J.H.; Goumans, M.J.; Dijke, P.T. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J. Pathol. 2019, 247, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Huo, R.; Sheng, Y.; Li, Y.; Xie, X.; Chen, C.; Liu, H.B.; Li, N.; Li, C.B.; Guo, W.T.; et al. Bone Morphogenetic Protein-4 Mediates Cardiac Hypertrophy, Apoptosis, and Fibrosis in Experimentally Pathological Cardiac Hypertrophy. Hypertension 2013, 61, 352–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Tao, T.; Wang, X.; Liu, M.; Song, D.; Liu, X.; Shi, D. Zedoarondiol Attenuates Endothelial Cells Injury Induced by Oxidized Low-Density Lipoprotein via Nrf2 Activation. Cell. Physiol. Biochem. 2018, 48, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.P.; Cui, H.J.; Yang, A.L.; Luo, J.K.; Tang, T. Astragaloside IV Improves Vasodilatation Function by Regulating the PI3K/Akt/eNOS Signaling Pathway in Rat Aorta Endothelial Cells. J. Vasc. Res. 2018, 55, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Rösen, P.; Nawroth, P.P.; King, G.; Möller, W.; Tritschler, H.J.; Packer, L. The role of oxidative stress in the onset and progression of diabetes and its complications: A summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes. Metab. Res. Rev. 2001, 17, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Asaba, K.; Tojo, A.; Onozato, M.L.; Goto, A.; Quinn, M.T.; Fujita, T.; Wilcox, C.S. Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int. 2005, 67, 1890–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, A.S.; Du, P.; Dikalova, A.; Lassegue, B.; Aleman, M.; Góngora, M.C.; Brown, K.; Joseph, G.; Harrison, D.G.; Taylor, W.R.; et al. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes. Am. J. Physiol. Circ. Physiol. 2007, 292, H2073–H2082. [Google Scholar] [CrossRef] [PubMed]
- Zhen, D.; Chen, Y.; Tang, X. Metformin reverses the deleterious effects of high glucose on osteoblast function. J. Diabetes Complicat. 2010, 24, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Diabetic Retinopathy: Mitochondrial Dysfunction and Retinal Capillary Cell Death. Antioxid. Redox Signal. 2005, 7, 1581. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef]
- Wang, H.T.; Liu, C.F.; Tsai, T.H.; Chen, Y.L.; Chang, H.W.; Tsai, C.Y.; Leu, S.; Zhen, Y.Y.; Chai, H.T.; Chung, S.Y.; et al. Effect of obesity reduction on preservation of heart function and attenuation of left ventricular remodeling, oxidative stress and inflammation in obese mice. J. Transl. Med. 2012, 10, 145. [Google Scholar] [CrossRef]
- Saeed, O.; Otsuka, F.; Polavarapu, R.; Karmali, V.; Weiss, D.; Davis, T.; Rostad, B.; Pachura, K.; Adams, L.; Elliott, J.; et al. Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Hoffmann, J.M.; Svensson, P.A.; Grimsby, J.; Rondinone, C.; Smith, U. BMP4 and BMP Antagonists Regulate Human White and Beige Adipogenesis. Diabetes 2015, 64, 1670–1681. [Google Scholar] [CrossRef] [Green Version]
- De Villiers, D.; Potgieter, M.; Ambele, M.A.; Adam, L.; Durandt, C.; Pepper, M.S. The Role of Reactive Oxygen Species in Adipogenic Differentiation. In Genome Editing; Springer: Cham, Switzerland, 2017; Volume 1083, pp. 125–144. [Google Scholar]
- Youn, J.Y.; Gao, L.; Cai, H. The p47phox-and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetology 2012, 55, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Zhou, J.; Cai, H. Bone Morphogenic Protein 4 Mediates NOX1-Dependent eNOS Uncoupling, Endothelial Dysfunction, and COX2 Induction in Type 2 Diabetes Mellitus. Mol. Endocrinol. 2015, 29, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhong, H.; Liang, J.Y.; Fu, P.; Luo, Z.J.; Zhou, L.; Gou, R.; Huang, J. Effect of high glucose levels on the calcification of vascular smooth muscle cells by inducing osteoblastic differentiation and intracellular calcium deposition via BMP-2/Cbfα-1 pathway. J. Zhejiang Univ. Sci. B 2010, 11, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Song, H.; Kumar, S.; Nam, D.; Kwon, H.S.; Chang, K.H.; Son, D.J.; Kang, D.W.; Brodie, S.A.; Weiss, D.; et al. Anti-inflammatory and antiatherogenic role of BMP receptor II in endothelial cells. Arter. Thromb. Vasc. Biol. 2013, 33, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.S.; Aly, Z.A.; Lai, C.F.; Cheng, S.L.; Cai, J.; Huang, E.; Behrmann, A.; Towler, D.A. Vascular Bmp Msx2 Wnt Signaling and Oxidative Stress in Arterial Calcification. Ann. N. Y. Acad. Sci. 2007, 1117, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Boström, K.I.; Jumabay, M.; Matveyenko, A.; Nicholas, S.B.; Yao, Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ. Res. 2011, 108, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Kolset, S.O.; Reinholt, F.P.; Jenssen, T. Diabetic Nephropathy and Extracellular Matrix. J. Histochem. Cytochem. 2012, 60, 976–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.H.; Chang, C.K.; Cheng, M.F.; Lin, H.J.; Cheng, J.T. The antioxidative effect of bone morphogenetic protein-7 against high glucose-induced oxidative stress in mesangial cells. Biochem. Biophys. Res. Commun. 2009, 382, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Díez, R.; Lavoz, C.; Carvajal, G.; Rayego-Mateos, S.; Diez, R.R.; Arduan, A.O.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Gremlin Is a Downstream Profibrotic Mediator of Transforming Growth Factor-Beta in Cultured Renal Cells. Nephron Exp. Nephrol. 2012, 122, 62–74. [Google Scholar] [CrossRef]
- Kane, R.; Stevenson, L.; Godson, C.; Stitt, A.W.; O’Brien, C. Gremlin gene expression in bovine retinal pericytes exposed to elevated glucose. Br. J. Ophthalmol 2005, 89, 1638–1642. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Chan, P.S. Oxidative Stress and Diabetic Retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Q.; Simon, T.C.; Strebeck, F.; Chaudhary, L.; Morrissey, J.; Liapis, H.; Klahr, S.; Hruska, K.A. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy11Professor Robert Chevalier served as a guest editor for this paper. Kidney Int. 2003, 63, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.I.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-β1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Hsing, C.H.; Chou, W.; Wang, J.J.; Chen, H.W.; Yeh, C.H. Propofol increases bone morphogenetic protein-7 and decreases oxidative stress in sepsis-induced acute kidney injury. Nephrol. Dial. Transplant. 2011, 26, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Ha, H. Mechanisms of Epithelial-Mesenchymal Transition of Peritoneal Mesothelial Cells During Peritoneal Dialysis. J. Korean Med. Sci. 2007, 22, 943–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolan, V.; Murphy, M.; Sadlier, D.; Lappin, D.; Doran, P.; Godson, C.; Martin, F.; O’Meara, Y.; Schmid, H.; Henger, A.; et al. Expression of gremlin, a bone morphogenetic protein antagonist, in human diabetic nephropathy. Am. J. Kidney Dis. 2005, 45, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Q. Bone morphogenetic protein-7 and Gremlin: New emerging therapeutic targets for diabetic nephropathy. Biochem. Biophys. Res. Commun. 2009, 383, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Nezu, M.; Suzuki, N.; Yamamoto, M. Targeting the KEAP1-NRF2 System to Prevent Kidney Disease Progression. Am. J. Nephrol. 2017, 45, 473–483. [Google Scholar] [CrossRef]
- Choi, B.H.; Kang, K.S.; Kwak, M.K. Effect of Redox Modulating NRF2 Activators on Chronic Kidney Disease. Molecules 2014, 19, 12727–12759. [Google Scholar] [CrossRef] [Green Version]
- Verdijk, L.B.; Snijders, T.; Drost, M.; Delhaas, T.; Kadi, F.; van Loon, L.J.C. Satellite cells in human skeletal muscle; from birth to old age. Age (Omaha) 2014, 36, 545–557. [Google Scholar] [CrossRef]
- Frühbeck, G.; Sesma, P.; Burrell, M.A. PRDM16: The interconvertible adipo-myocyte switch. Trends Cell Biol. 2009, 19, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Morozzi, G.; Beccafico, S.; Bianchi, R.; Riuzzi, F.; Bellezza, I.; Giambanco, I.; Arcuri, C.; Minelli, A.; Donato, R. Oxidative stress-induced S100B accumulation converts myoblasts into brown adipocytes via an NF-κB/YY1/miR-133 axis and NF-κB/YY1/BMP-7 axis. Cell Death Differ. 2017, 24, 2077–2088. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Bian, H.; Aya-Ay, J.; Garces, A.; Morgan, E.F.; Gilbert, S.R. Hypoxia and HIF-1α expression in the epiphyseal cartilage following ischemic injury to the immature femoral head. Bone 2009, 45, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Nakase, T.; Miyaji, T.; Tomita, T.; Kaneko, M.; Kuriyama, K.; Myoui, A.; Sugamoto, K.; Ochi, T.; Yoshikawa, H. Localization of bone morphogenetic protein-2 in human osteoarthritic cartilage and osteophyte. Osteoarthr. Cartil. 2003, 11, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, N.; Shafer, S.; Oxendine, I.; Mortlock, D.P.; Chandler, R.L.; Oxburgh, L.; Kim, H.K. Acute BMP2 upregulation following induction of ischemic osteonecrosis in immature femoral head. Bone 2013, 53, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Ku, W.Y.; Zhou, Z.; Dellon, E.S.; Falk, G.W.; Nakagawa, H.; Wang, M.L.; Liu, K.; Wang, J.; Katzka, D.A.; et al. BMP-driven NRF2 activation in esophageal basal cell differentiation and eosinophilic esophagitis. J. Clin. Investig. 2015, 125, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Tardif, G.; Hum, D.; Pelletier, J.P.; Boileau, C.; Ranger, P.; Martel-Pelletier, J. Differential gene expression and regulation of the bone morphogenetic protein antagonists follistatin and gremlin in normal and osteoarthritic human chondrocytes and synovial fibroblasts. Arthritis Rheum. 2004, 50, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Tardif, G.; Pelletier, J.P.; Boileau, C.; Martel-Pelletier, J. The BMP antagonists follistatin and gremlin in normal and early osteoarthritic cartilage: An immunohistochemical study. Osteoarthr. Cartil. 2009, 17, 263–270. [Google Scholar] [CrossRef]
- Han, E.J.; Yoo, S.A.; Kim, G.M.; Hwang, D.; Cho, C.S.; You, S.; Kim, W.U. GREM1 Is a Key Regulator of Synoviocyte Hyperplasia and Invasiveness. J. Rheumatol. 2016, 43, 474–485. [Google Scholar] [CrossRef]
- Yi, J.; Jin, Q.; Zhang, B.; Wu, X.; Ge, D. Gremlin-1 Concentrations Are Correlated with the Severity of Knee Osteoarthritis. Med. Sci. Monit. 2016, 22, 4062–4065. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-de-Diego, C.; Valer, J.A.; Pimenta-Lopes, C.; Rosa, J.L.; Ventura, F. Interplay between BMPs and Reactive Oxygen Species in Cell Signaling and Pathology. Biomolecules 2019, 9, 534. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100534
Sánchez-de-Diego C, Valer JA, Pimenta-Lopes C, Rosa JL, Ventura F. Interplay between BMPs and Reactive Oxygen Species in Cell Signaling and Pathology. Biomolecules. 2019; 9(10):534. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100534
Chicago/Turabian StyleSánchez-de-Diego, Cristina, José Antonio Valer, Carolina Pimenta-Lopes, José Luis Rosa, and Francesc Ventura. 2019. "Interplay between BMPs and Reactive Oxygen Species in Cell Signaling and Pathology" Biomolecules 9, no. 10: 534. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9100534