LCAT, ApoD, and ApoA1 Expression and Review of Cholesterol Deposition in the Cornea

Abstract
:1. Introduction
2. Materials and Methods
2.1. Western Blot Assay
2.2. RNA Sequencing Analysis
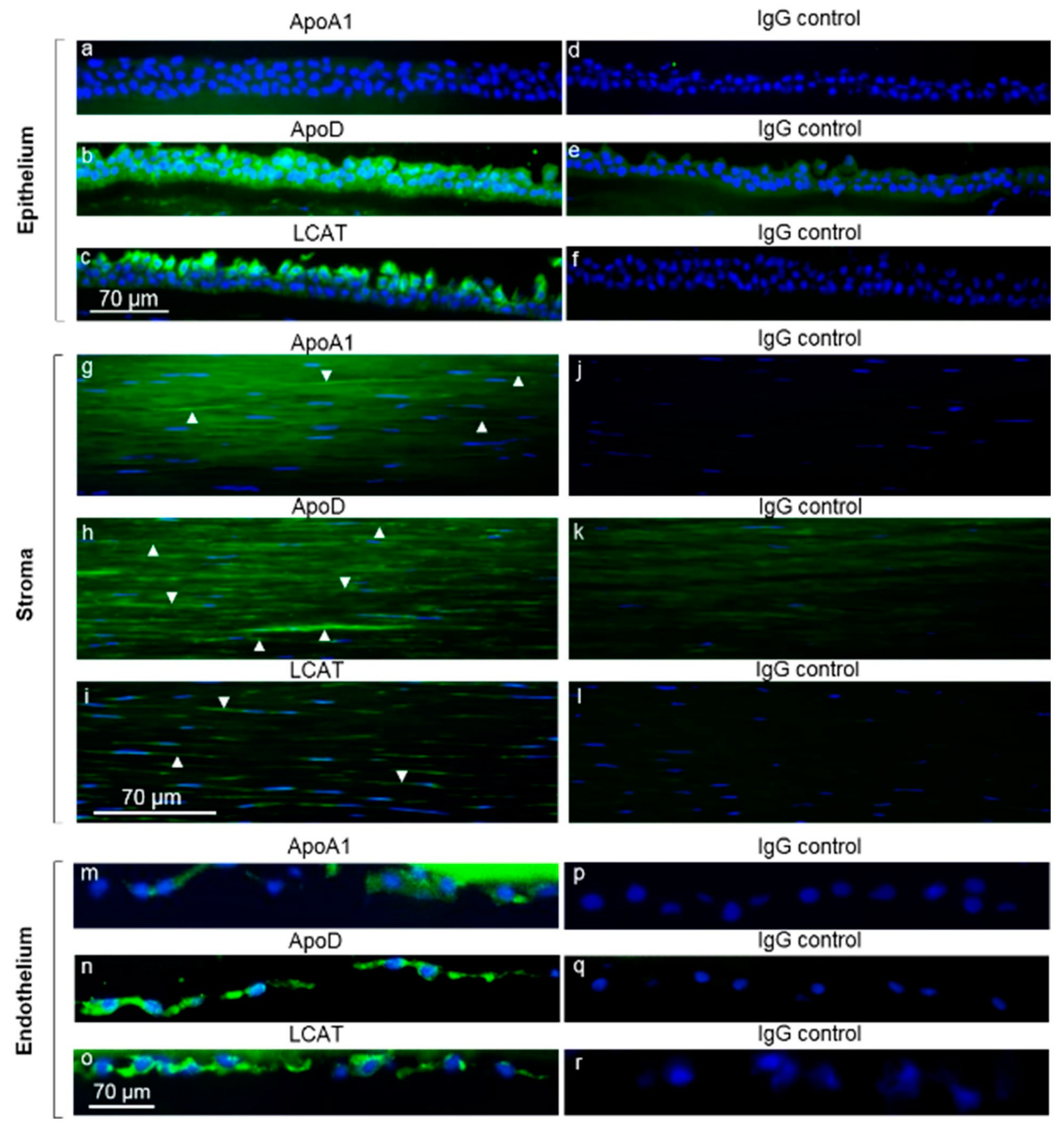
2.3. Immunofluorescence Analysis
2.4. Lipid Analysis of Corneas
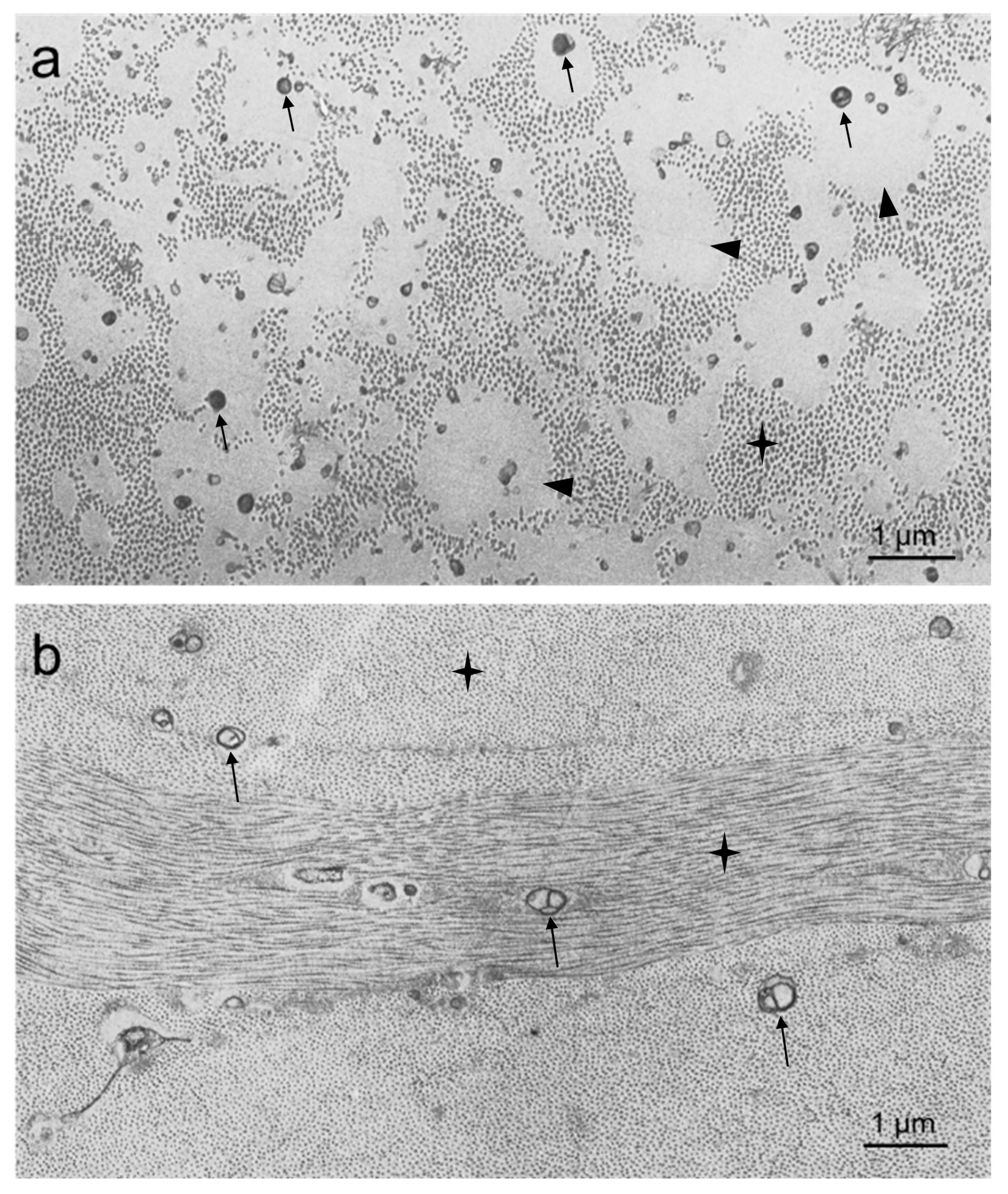
2.5. Electron Microscopy
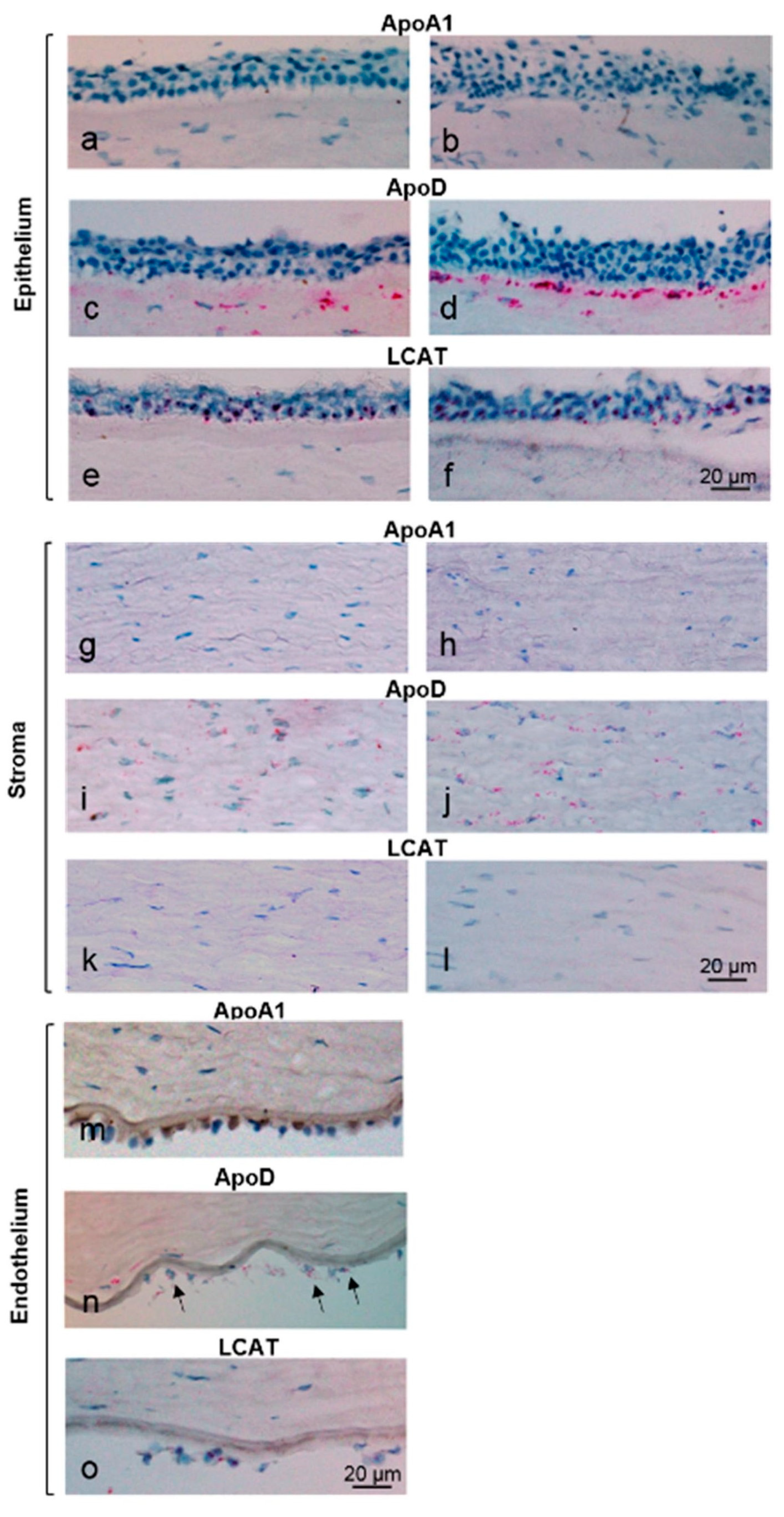
2.6. In Situ Hybridization
3. Results
4. Discussion
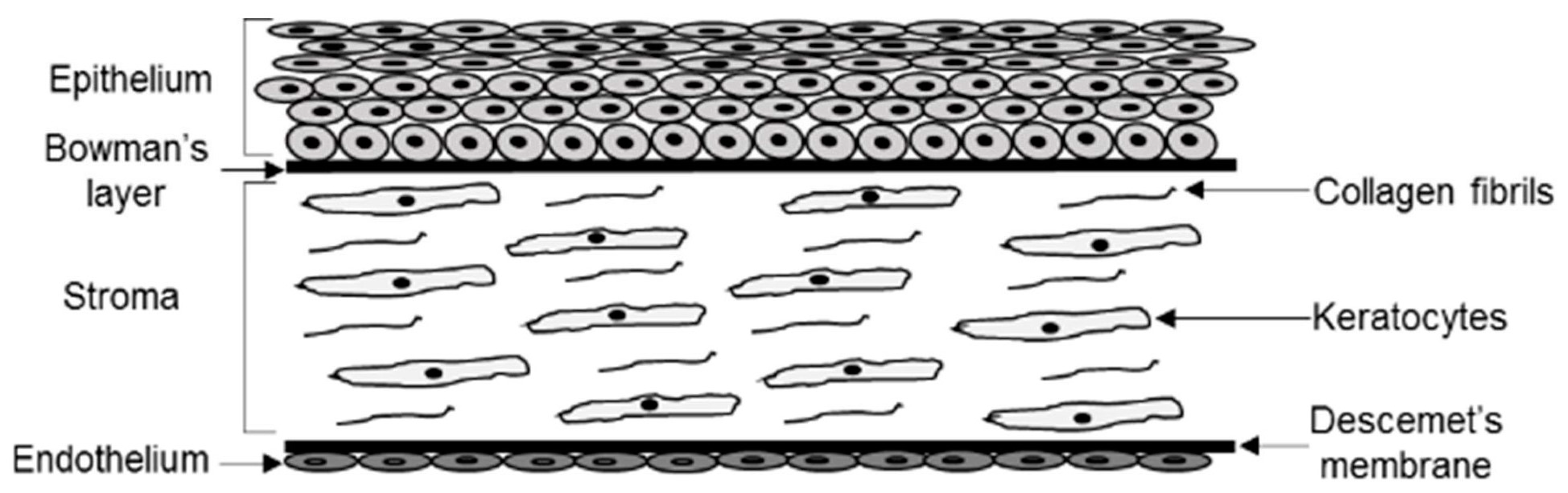
4.1. Corneal Expression of LCAT, ApoD, and ApoA1
4.2. Morphology of Corneal Lipid Deposits
4.3. Source of Cholesterol that Accumulates in Cornea
4.4. Cholesterol Removal from the Cornea
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Apo | apolipoprotein |
| BSA | bovine serum albumin |
| DPBS | Dulbecco’s phosphate-buffered saline |
| FBS | fetal bovine serum |
| HDL | high-density lipoprotein |
| LCAT | lecithin:cholesterol acyltransferase |
| LDL | low-density lipoprotein |
| TBS | tris-buffered saline |
References
- Winder, A.F.; Borysiewicz, L.K. Corneal opacification and familial disorders affecting plasma high-density lipoprotein. Birth Defects Orig. Artic. Ser. 1982, 18, 433–440. [Google Scholar]
- Barchiesi, B.J.; Eckel, R.H.; Ellis, P.P. The cornea and disorders of lipid metabolism. Surv. Ophthalmol. 1991, 36, 1–22. [Google Scholar] [CrossRef]
- Bron, A.J. Corneal changes in the dislipoproteinaemias. Cornea 1989, 8, 135–140. [Google Scholar] [CrossRef]
- Schaefer, E.J.; Anthanont, P.; Diffenderfer, M.R.; Polisecki, E.; Asztalos, B.F. Diagnosis and treatment of high density lipoprotein deficiency. Prog. Cardiovasc. Dis. 2016, 59, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S. Assembly of high-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 20–27. [Google Scholar] [CrossRef]
- Hamilton, R.L.; Williams, M.C.; Fielding, C.J.; Havel, R.J. Discoidal bilayer structure of nascent high density lipoproteins from perfused rat liver. J. Clin. Investig. 1976, 58, 667–680. [Google Scholar] [CrossRef]
- Green, P.H.; Tall, A.R.; Glickman, R.M. Rat intestine secretes discoid high density lipoprotein. J. Clin. Investig. 1978, 61, 528–534. [Google Scholar] [CrossRef]
- Wang, S.; Smith, J.D. ABCA1 and nascent HDL biogenesis. Biofactors 2014, 40, 547–554. [Google Scholar] [CrossRef]
- Jin, X.; Dimitriadis, E.K.; Liu, Y.; Combs, C.A.; Chang, J.; Varsano, N.; Stempinski, E.; Flores, R.; Jackson, S.N.; Muller, L.; et al. Macrophages Shed Excess Cholesterol in Unique Extracellular Structures Containing Cholesterol Microdomains. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1504–1518. [Google Scholar] [CrossRef]
- Czarnecka, H.; Yokoyama, S. Regulation of cellular cholesterol efflux by lecithin:cholesterol acyltransferase reaction through nonspecific lipid exchange. J. Biol. Chem. 1996, 271, 2023–2028. [Google Scholar] [CrossRef]
- Smith, K.M.; Lawn, R.M.; Wilcox, J.N. Cellular localization of apolipoprotein D and lecithin:cholesterol acyltransferase mRNA in rhesus monkey tissues by in situ hybridization. J. Lipid Res. 1990, 31, 995–1004. [Google Scholar] [PubMed]
- Warden, C.H.; Langner, C.A.; Gordon, J.I.; Taylor, B.A.; McLean, J.W.; Lusis, A.J. Tissue-specific expression, developmental regulation, and chromosomal mapping of the lecithin: Cholesterol acyltransferase gene. Evidence for expression in brain and testes as well as liver. J. Biol. Chem. 1989, 264, 21573–21581. [Google Scholar] [PubMed]
- McLean, J.; Wion, K.; Drayna, D.; Fielding, C.; Lawn, R. Human lecithin-cholesterol acyltransferase gene: Complete gene sequence and sites of expression. Nucleic Acids Res. 1986, 14, 9397–9406. [Google Scholar] [CrossRef] [PubMed]
- Rye, K.A.; Bursill, C.A.; Lambert, G.; Tabet, F.; Barter, P.J. The metabolism and anti-atherogenic properties of HDL. J. Lipid Res. 2009, 50, S195–S200. [Google Scholar] [CrossRef]
- Zorich, N.; Jonas, A.; Pownall, H.J. Activation of lecithin cholesterol acyltransferase by human apolipoprotein E in discoidal complexes with lipids. J. Biol. Chem. 1985, 260, 8831–8837. [Google Scholar]
- Soutar, A.K.; Garner, C.W.; Baker, H.N.; Sparrow, J.T.; Jackson, R.L.; Gotto, A.M.; Smith, L.C. Effect of the human plasma apolipoproteins and phosphatidylcholine acyl donor on the activity of lecithin: Cholesterol acyltransferase. Biochemistry 1975, 14, 3057–3064. [Google Scholar] [CrossRef]
- Gordon, V.; Innerarity, T.L.; Mahley, R.W. Formation of cholesterol- and apoprotein E-enriched high density lipoproteins in vitro. J. Biol. Chem. 1983, 258, 6202–6212. [Google Scholar]
- Schaefer, E.J.; Gregg, R.E.; Ghiselli, G.; Forte, T.M.; Ordovas, J.M.; Zech, L.A.; Brewer, H.B., Jr. Familial apolipoprotein E deficiency. J. Clin. Investig. 1986, 78, 1206–1219. [Google Scholar] [CrossRef]
- Rifkind, B.M. The Incidence of Arcus Senilis in Ischaemic Heart-Disease Its Relation to Serum-Lipid Levels. Lancet 1965, 1, 312–314. [Google Scholar] [CrossRef]
- Zech, L.A., Jr.; Hoeg, J.M. Correlating corneal arcus with atherosclerosis in familial hypercholesterolemia. Lipids Health Dis. 2008, 7, 7. [Google Scholar] [CrossRef]
- Walton, K.W. Studies on the pathogenesis of corneal arcus formation. I. The human corneal arcus and its relation to atherosclerosis as studied by immunofluorescence. J. Pathol. 1973, 111, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, P.M.; Zhang, W.Y.; Salehizadeh, B.; Pettiford, B.; Kruth, H.S. Cholesterol accumulation in human cornea: Evidence that extracellular cholesteryl ester-rich lipid particles deposit independently of foam cells. J. Lipid Res. 1996, 37, 1849–1861. [Google Scholar] [PubMed]
- Ashraf, F.; Cogan, D.G.; Kruth, H.S. Apolipoprotein A-I and B distribution in the human cornea. Investig. Ophthalmol. Vis. Sci. 1993, 34, 3574–3578. [Google Scholar]
- McCulley, J.P. The circulation of fluid at the limbus (flow and diffusion at the limbus). Eye 1989, 3(Pt. 2), 114–120. [Google Scholar] [CrossRef]
- Maurice, D.M. The Use of Permeability Studies in the Investigation of Submicroscopic Structure. In Proceedings of the Seventh International Congress of Anatomists, New York, NY, USA, 1960; pp. 381–391. [Google Scholar]
- Steyrer, E.; Kostner, G.M. Activation of lecithin-cholesterol acyltransferase by apolipoprotein D: Comparison of proteoliposomes containing apolipoprotein D, A-I or C-I. Biochim. Biophys. Acta 1988, 958, 484–491. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequence from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Cogan, D.G.; Kruth, H.S.; Datilis, M.B.; Martin, N. Corneal opacity in LCAT disease. Cornea 1992, 11, 595–599. [Google Scholar] [CrossRef]
- Mautner, S.L.; Sanchez, J.A.; Rader, D.J.; Mautner, G.C.; Ferrans, V.J.; Fredrickson, D.S.; Brewer, H.B., Jr.; Roberts, W.C. The heart in Tangier disease. Severe coronary atherosclerosis with near absence of high-density lipoprotein cholesterol. Am. J. Clin. Pathol. 1992, 98, 191–198. [Google Scholar] [CrossRef]
- Chu, F.C.; Kuwabara, T.; Cogan, D.G.; Schaefer, E.J.; Brewer, H.B., Jr. Ocular manifestations of familial high-density lipoprotein deficiency (Tangier disease). Arch. Ophthalmol. 1979, 97, 1926–1928. [Google Scholar] [CrossRef]
- Gaynor, P.M.; Zhang, W.Y.; Weiss, J.S.; Skarlatos, S.I.; Rodrigues, M.M.; Kruth, H.S. Accumulation of HDL apolipoproteins accompanies abnormal cholesterol accumulation in Schnyder’s corneal dystrophy. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Gamble, W.; Vaughan, M.; Kruth, H.S.; Avigan, J. Procedure for determination of free and total cholesterol in micro- or nanogram amounts suitable for studies with cultured cells. J. Lipid Res. 1978, 19, 1068–1070. [Google Scholar] [PubMed]
- Bartlett, G.R. Phosphorus assay in column chromatography. J. Biol. Chem. 1959, 234, 466–468. [Google Scholar]
- Ordovas, J.M.; Cassidy, D.K.; Civeira, F.; Bisgaier, C.L.; Schaefer, E.J. Familial apolipoprotein A-I, C-III, and A-IV deficiency and premature atherosclerosis due to deletion of a gene complex on chromosome 11. J. Biol. Chem. 1989, 264, 16339–16342. [Google Scholar]
- Guyton, J.R.; Klemp, K.F. Ultrastructural discrimination of lipid droplets and vesicles in atherosclerosis: Value of osmium-thiocarbohydrazide-osmium and tannic acid-paraphenylenediamine techniques. J. Histochem. Cytochem. 1988, 36, 1319–1328. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.T.; Ma, X.J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Philipson, B.T. Fish eye disease. Birth Defects Orig. Artic. Ser. 1982, 18, 441–448. [Google Scholar]
- Bethell, W.; McCulloch, C.; Ghosh, M. Lecithin cholesterol acyl transferase deficiency. Light and electron microscopic finding from two corneas. Can. J. Ophthalmol. 1975, 10, 494–501. [Google Scholar]
- Viestenz, A.; Schlotzer-Schrehardt, U.; Hofmann-Rummelt, C.; Seitz, B.; Kuchle, M. Histopathology of corneal changes in lecithin-cholesterol acyltransferase deficiency. Cornea 2002, 21, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Koster, H.; Savoldelli, M.; Dumon, M.F.; Dubourg, L.; Clerc, M.; Pouliquen, Y. A fish-eye disease-like familial condition with massive corneal clouding and dyslipoproteinemia. Report of clinical, histologic, electron microscopic, and biochemical features. Cornea 1992, 11, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Winder, A.F.; Alexander, R.; Garner, A.; Johnston, D.; Vallance, D.; McCreanor, G.; Frohlich, J. The pathology of cornea in Tangier disease (familial high density lipoprotein deficiency). J. Clin. Pathol. 1996, 49, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Funderburgh, J.L.; Mann, M.M.; Funderburgh, M.L. Keratocyte phenotype mediates proteoglycan structure: A role for fibroblasts in corneal fibrosis. J. Biol. Chem. 2003, 278, 45629–45637. [Google Scholar] [CrossRef] [Green Version]
- Winder, A.F.; Garner, A.; Sheraidah, G.A.; Barry, P. Familial lecithin:cholesterol acyltransferase deficiency. Biochemistry of the cornea. J. Lipid Res. 1985, 26, 283–287. [Google Scholar]
- Jonas, A. Lecithin cholesterol acyltransferase. Biochim. Biophys. Acta 2000, 1529, 245–256. [Google Scholar] [CrossRef]
- Chen, C.H.; Albers, J.J. Distribution of lecithin-cholesterol acyltransferase (LCAT) in human plasma lipoprotein fractions. Evidence for the association of active LCAT with low density lipoproteins. Biochem. Biophys. Res. Commun. 1982, 107, 1091–1096. [Google Scholar] [CrossRef]
- Ruiz, M.; Sanchez, D.; Correnti, C.; Strong, R.K.; Ganfornina, M.D. Lipid-binding properties of human ApoD and Lazarillo-related lipocalins: Functional implications for cell differentiation. FEBS J. 2013, 280, 3928–3943. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.C.; Lange, D.; McConathy, W.J.; Patel, Y.C.; Patel, S.C. Probing the structure of the ligand binding cavity of lipocalins by fluorescence spectroscopy. Protein Eng. 1997, 10, 621–625. [Google Scholar] [CrossRef] [Green Version]
- Holzfeind, P.; Merschak, P.; Dieplinger, H.; Redl, B. The human lacrimal gland synthesizes apolipoprotein D mRNA in addition to tear prealbumin mRNA, both species encoding members of the lipocalin superfamily. Exp. Eye Res. 1995, 61, 495–500. [Google Scholar] [CrossRef]
- McCulley, J.P.; Shine, W. A compositional based model for the tear film lipid layer. Trans. Am. Ophthalmol. Soc. 1997, 95, 79–88, discussion 88–93. [Google Scholar]
- Rassart, E.; Bedirian, A.; Do Carmo, S.; Guinard, O.; Sirois, J.; Terrisse, L.; Milne, R. Apolipoprotein D. Biochim. Biophys. Acta 2000, 1482, 185–198. [Google Scholar] [CrossRef]
- Glasgow, B.J.; Marshall, G.; Gasymov, O.K.; Abduragimov, A.R.; Yusifov, T.N.; Knobler, C.M. Tear lipocalins: Potential lipid scavengers for the corneal surface. Investig. Ophthalmol. Vis. Sci. 1999, 40, 3100–3107. [Google Scholar]
- Breustedt, D.A.; Schonfeld, D.L.; Skerra, A. Comparative ligand-binding analysis of ten human lipocalins. Biochim. Biophys. Acta 2006, 1764, 161–173. [Google Scholar] [CrossRef]
- Rohrl, C.; Stangl, H. HDL endocytosis and resecretion. Biochim. Biophys. Acta 2013, 1831, 1626–1633. [Google Scholar] [CrossRef] [Green Version]
- Uchida, Y.; Uchida, Y.; Shimoyama, E.; Hiruta, N.; Tabata, T.; Kobayashi, T. Deposition patterns and localization of apolipoprotein A1 and their relation to plaque morphology in human coronary artery. JSM Atheroscler 2017, 2, 1025. [Google Scholar]
- Vollmer, E.; Brust, J.; Roessner, A.; Bosse, A.; Burwikel, F.; Kaesberg, B.; Harrach, B.; Robenek, H.; Bocker, W. Distribution patterns of apolipoproteins A1, A2, and B in the wall of atherosclerotic vessels. Virchows Arch. A Pathol. Anat. Histopathol. 1991, 419, 79–88. [Google Scholar] [CrossRef]
- Kaesberg, B.; Harrach, B.; Dieplinger, H.; Robenek, H. In situ immunolocalization of lipoproteins in human arteriosclerotic tissue. Arterioscler. Thromb. 1993, 13, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Velagapudi, S.; Schraml, P.; Yalcinkaya, M.; Bolck, H.A.; Rohrer, L.; Moch, H.; von Eckardstein, A. Scavenger receptor BI promotes cytoplasmic accumulation of lipoproteins in clear-cell renal cell carcinoma. J. Lipid Res. 2018, 59, 2188–2201. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.S. Schnyder crystalline dystrophy sine crystals. Recommendation for a revision of nomenclature. Ophthalmology 1996, 103, 465–473. [Google Scholar] [CrossRef]
- Rodrigues, M.M.; Kruth, H.S.; Krachmer, J.H.; Willis, R. Unesterified cholesterol in Schnyder’s corneal crystalline dystrophy. Am. J. Ophthalmol. 1987, 104, 157–163. [Google Scholar] [CrossRef]
- Weiss, J.S.; Rodrigues, M.M.; Kruth, H.S.; Rajagopalan, S.; Rader, D.J.; Kachadoorian, H. Panstromal Schnyder’s corneal dystrophy. Ultrastructural and histochemical studies. Ophthalmology 1992, 99, 1072–1081. [Google Scholar] [CrossRef]
- Blanco-Vaca, F.; Qu, S.J.; Fiol, C.; Fan, H.Z.; Pao, Q.; Marzal-Casacuberta, A.; Albers, J.J.; Hurtado, I.; Gracia, V.; Pinto, X.; et al. Molecular basis of fish-eye disease in a patient from Spain. Characterization of a novel mutation in the LCAT gene and lipid analysis of the cornea. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Freeman, S.R.; Vaisman, B.; Liu, Y.; Chang, J.; Varsano, N.; Addadi, L.; Remaley, A.; Kruth, H.S. ABCA1 contributes to macrophage deposition of extracellular cholesterol. J. Lipid Res. 2015, 56, 1720–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, S.R.; Jin, X.; Anzinger, J.J.; Xu, Q.; Purushothaman, S.; Fessler, M.B.; Addadi, L.; Kruth, H.S. ABCG1-mediated generation of extracellular cholesterol microdomains. J. Lipid Res. 2014, 55, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Forte, T.M.; Nichols, A.V.; Gong, E.L.; Levy, R.I.; Lux, S. Electron microscopic study on reassembly of plasma high density apoprotein with various lipids. Biochim. Biophys. Acta 1971, 248, 381–386. [Google Scholar] [CrossRef]
- Forte, T.; Norum, K.R.; Glomset, J.A.; Nichols, A.V. Plasma lipoproteins in familial lecithin: Cholesterol acyltransferase deficiency: Structure of low and high density lipoproteins as revealed by elctron microscopy. J. Clin. Investig. 1971, 50, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Seidel, D.; Gjone, E.; Blomhoff, J.P.; Geisen, H.P. Plasma lipoproteins in patients with familial plasma lecithin: Cholesterol acyltransferase (LCAT) deficiency--studies on the apolipoprotein composition of isolated fractions with identification of LP-X. Horm. Metab. Res. 1974, Suppl 4, 6–11. [Google Scholar]
- Glomset, J.A.; Nichols, A.V.; Norum, K.R.; King, W.; Forte, T. Plasma lipoproteins in familial lecithin: Cholesterol acyltransferase deficiency. Further studies of very low and low density lipoprotein abnormalities. J. Clin. Investig. 1973, 52, 1078–1092. [Google Scholar] [CrossRef]
- Torsvik, H.; Berg, K.; Magnani, H.N.; McConathy, W.J.; Alaupovic, P.; Gjone, E. Identification of the abnormal cholestatic lipoprotein (LP-X) in familial lecithin:Cholesterol acyltransferase deficiency. FEBS Lett. 1972, 24, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Ossoli, A.; Neufeld, E.B.; Thacker, S.G.; Vaisman, B.; Pryor, M.; Freeman, L.A.; Brantner, C.A.; Baranova, I.; Francone, N.O.; Demosky, S.J., Jr.; et al. Lipoprotein X Causes Renal Disease in LCAT Deficiency. PLoS ONE 2016, 11, e0150083. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Herzenberg, A.M.; Eskandarian, M.; Maguire, G.F.; Scholey, J.W.; Connelly, P.W.; Ng, D.S. A novel in vivo lecithin-cholesterol acyltransferase (LCAT)-deficient mouse expressing predominantly LpX is associated with spontaneous glomerulopathy. Am. J. Pathol. 2004, 165, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Forte, T.M.; Carlson, L.A. Electron microscopic structure of serum lipoproteins from patients with fish eye disease. Arteriosclerosis 1984, 4, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Rye, K.A.; Clay, M.A.; Barter, P.J. Remodelling of high density lipoproteins by plasma factors. Atherosclerosis 1999, 145, 227–238. [Google Scholar] [CrossRef]
- Assmann, G.; Herbert, P.N.; Fredrickson, D.S.; Forte, T. Isolation and characterization of an abnormal high density lipoprotein in Tangier Diesase. J. Clin. Investig. 1977, 60, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Cenedella, R.J.; Fleschner, C.R. Cholesterol biosynthesis by the cornea. Comparison of rates of sterol synthesis with accumulation during early development. J. Lipid Res. 1989, 30, 1079–1084. [Google Scholar]
- Weiss, J.S.; Kruth, H.S.; Kuivaniemi, H.; Tromp, G.; Karkera, J.; Mahurkar, S.; Lisch, W.; Dupps, W.J., Jr.; White, P.S.; Winters, R.S.; et al. Genetic analysis of 14 families with Schnyder crystalline corneal dystrophy reveals clues to UBIAD1 protein function. Am. J. Med. Genet. A 2008, 146, 271–283. [Google Scholar] [CrossRef]
- Schumacher, M.M.; Jun, D.J.; Johnson, B.M.; DeBose-Boyd, R.A. UbiA prenyltransferase domain-containing protein-1 modulates HMG-CoA reductase degradation to coordinate synthesis of sterol and nonsterol isoprenoids. J. Biol. Chem. 2018, 293, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Thrift, R.N.; Forte, T.M.; Cahoon, B.E.; Shore, V.G. Characterization of lipoproteins produced by the human liver cell line, Hep G2, under defined conditions. J. Lipid Res. 1986, 27, 236–250. [Google Scholar]
- Kruth, H.S.; Skarlatos, S.I.; Gaynor, P.M.; Gamble, W. Production of cholesterol-enriched nascent high density lipoproteins by human monocyte-derived macrophages is a mechanism that contributes to macrophage cholesterol efflux. J. Biol. Chem. 1994, 269, 24511–24518. [Google Scholar]
- Basu, S.K.; Ho, Y.K.; Brown, M.S.; Bilheimer, D.W.; Anderson, R.G.; Goldstein, J.L. Biochemical and genetic studies of the apoprotein E secreted by mouse macrophages and human monocytes. J. Biol. Chem. 1982, 257, 9788–9795. [Google Scholar]
- Hughes, T.E.; Sasak, W.V.; Ordovas, J.M.; Forte, T.M.; Lamon-Fava, S.; Schaefer, E.J. A novel cell line (Caco-2) for the study of intestinal lipoprotein synthesis. J. Biol. Chem. 1987, 262, 3762–3767. [Google Scholar]
- LaDu, M.J.; Gilligan, S.M.; Lukens, J.R.; Cabana, V.G.; Reardon, C.A.; Van Eldik, L.J.; Holtzman, D.M. Nascent astrocyte particles differ from lipoproteins in CSF. J. Neurochem. 1998, 70, 2070–2081. [Google Scholar] [CrossRef] [Green Version]
- Kruth, H.S. Cholesterol deposition in atherosclerotic lesions. Subcell. Biochem. 1997, 28, 319–362. [Google Scholar]
- Jonas, A.; Sweeny, S.A.; Herbert, P.N. Discoidal complexes of A and C apolipoproteins with lipids and their reactions with lecithin: Cholesterol acyltransferase. J. Biol. Chem. 1984, 259, 6369–6375. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | FPKM ± SD |
|---|---|
| APOA1 | 0 |
| APOA2 | 0 |
| APOA4 | 0 |
| APOC1 | 2 ± 1 |
| APOC2 | 0 |
| APOC3 | 0 |
| APOD | 72 ± 6 |
| APOE | 12 ± 1 |
| ABCA1 | 20 ± 0 |
| ABCG1 | 0 |
| PLTP | 24 ± 4 |
| CETP | 0 |
| LCAT | 3 ± 1 |
| LIPC | 0 |
| LIPG | 0 |
| LPL | 0 |
| SCARB1 | 7 ± 1 |
| Gene | Degree of Central Corneal Cloudiness | Peripheral Arcus | Effect on Coronary Artery Disease |
|---|---|---|---|
| ABCA1 | mild | absent | mild increase |
| APOA1 | moderate | present | severe increase |
| LCAT | severe | present | no consistent effect |
| Disease | Affected Gene | Lipid Content, µmol/g Tissue (Wet Weight) | |||||
|---|---|---|---|---|---|---|---|
| TC | UC | EC | PL | UC/PL | % UC | ||
| Normal | - | 2 ± 0.6 | 1 ± 0.3 | 1 ± 0.3 | 2 ± 0.3 | 0.5 ± 0.03 | 50 ± 5 |
| SCD | UBIAD1 | 27 ± 10 | 16 ± 3 | 11 ± 5 | 11 ± 1 | 1.5 ± 0.2 | 63 ± 7 |
| Tangier | ABCA1 | 3 | 3 | 0 | 2 | 1.3 | 100 * |
| Fish-eye | LCAT | 20 | 19 | 1 | 15 | 1.3 | 97 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores, R.; Jin, X.; Chang, J.; Zhang, C.; Cogan, D.G.; Schaefer, E.J.; Kruth, H.S. LCAT, ApoD, and ApoA1 Expression and Review of Cholesterol Deposition in the Cornea. Biomolecules 2019, 9, 785. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9120785
Flores R, Jin X, Chang J, Zhang C, Cogan DG, Schaefer EJ, Kruth HS. LCAT, ApoD, and ApoA1 Expression and Review of Cholesterol Deposition in the Cornea. Biomolecules. 2019; 9(12):785. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9120785
Chicago/Turabian StyleFlores, Rhonda, Xueting Jin, Janet Chang, Connie Zhang, David G. Cogan, Ernst J. Schaefer, and Howard S. Kruth. 2019. "LCAT, ApoD, and ApoA1 Expression and Review of Cholesterol Deposition in the Cornea" Biomolecules 9, no. 12: 785. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9120785