Flavin Oxidase-Induced ROS Generation Modulates PKC Biphasic Effect of Resveratrol on Endothelial Cell Survival


 , ,
, ,  ,
, 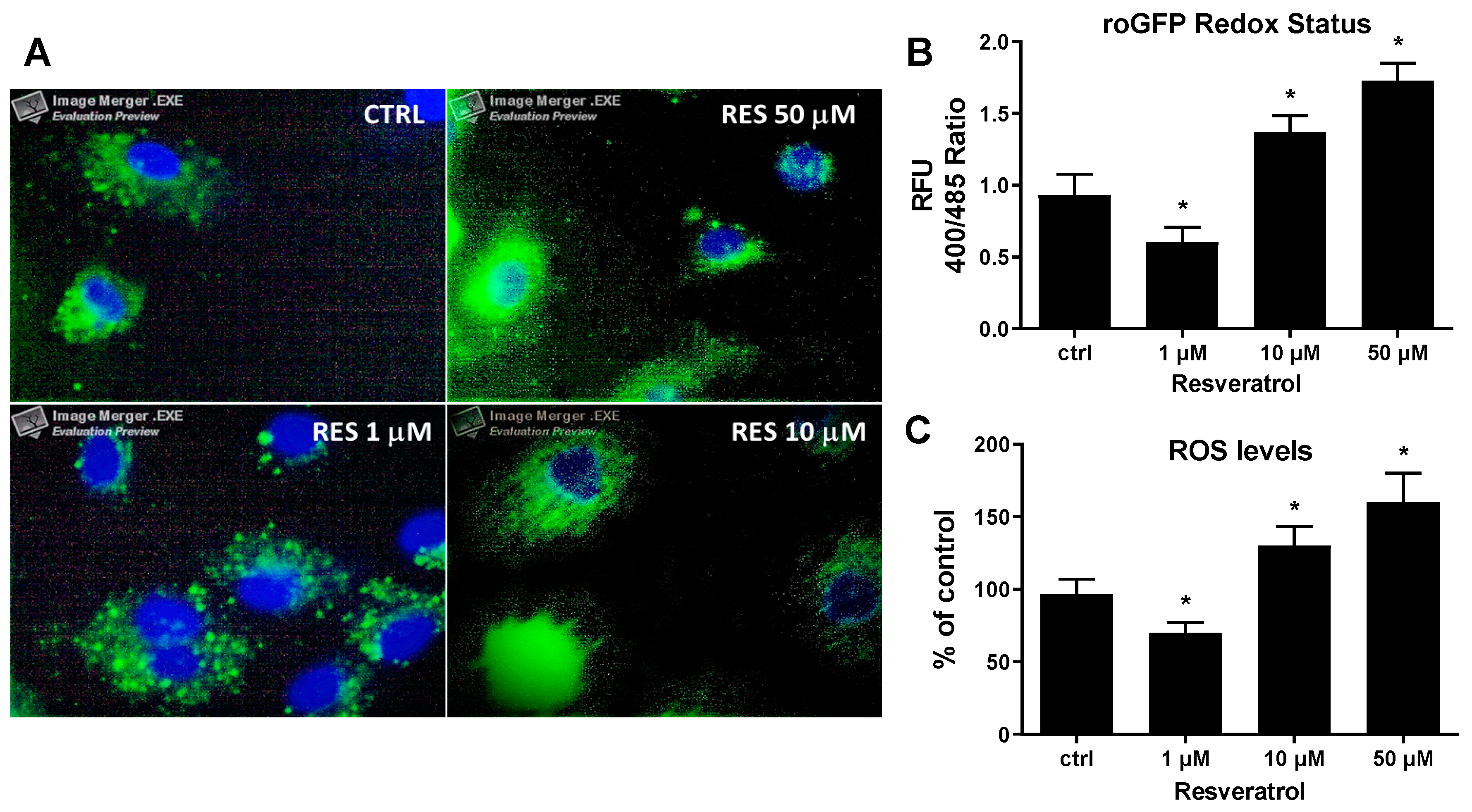{kind=link}
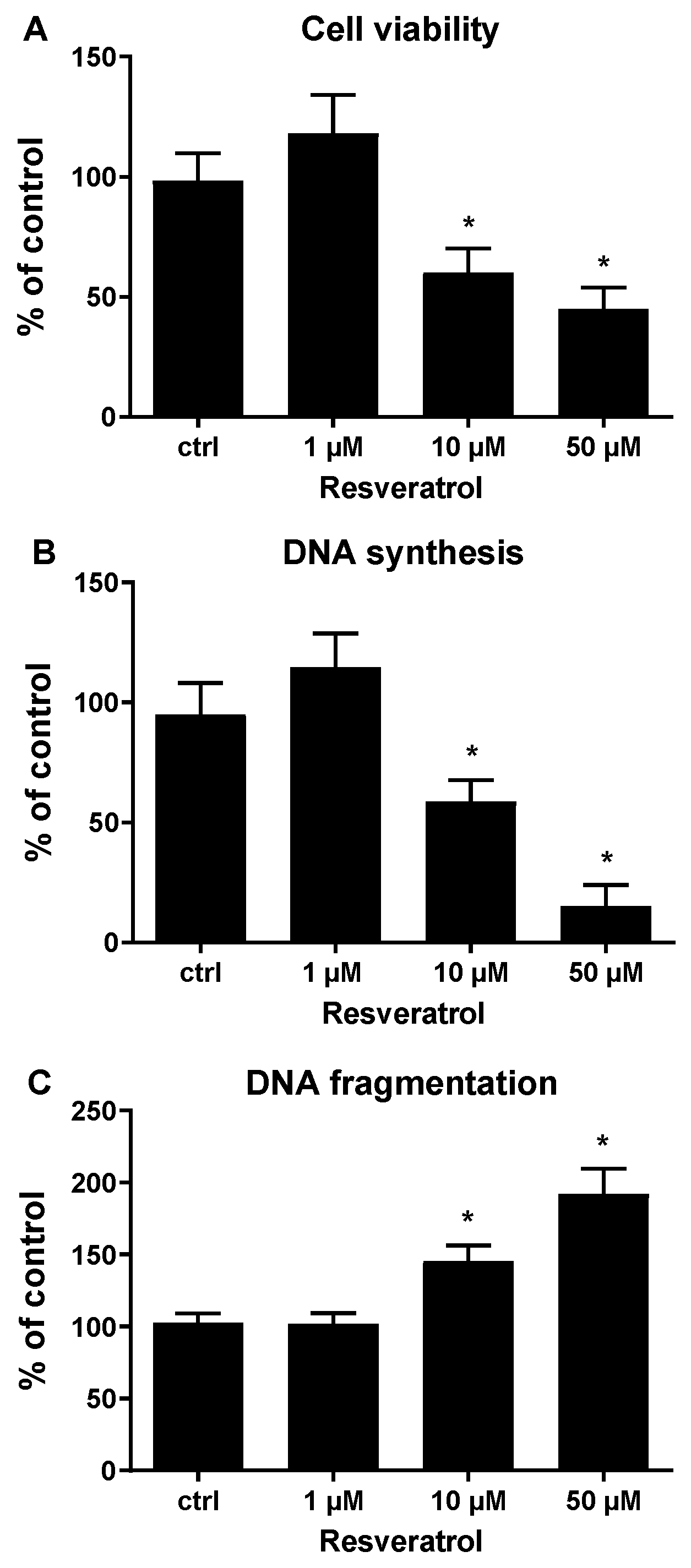{kind=link}
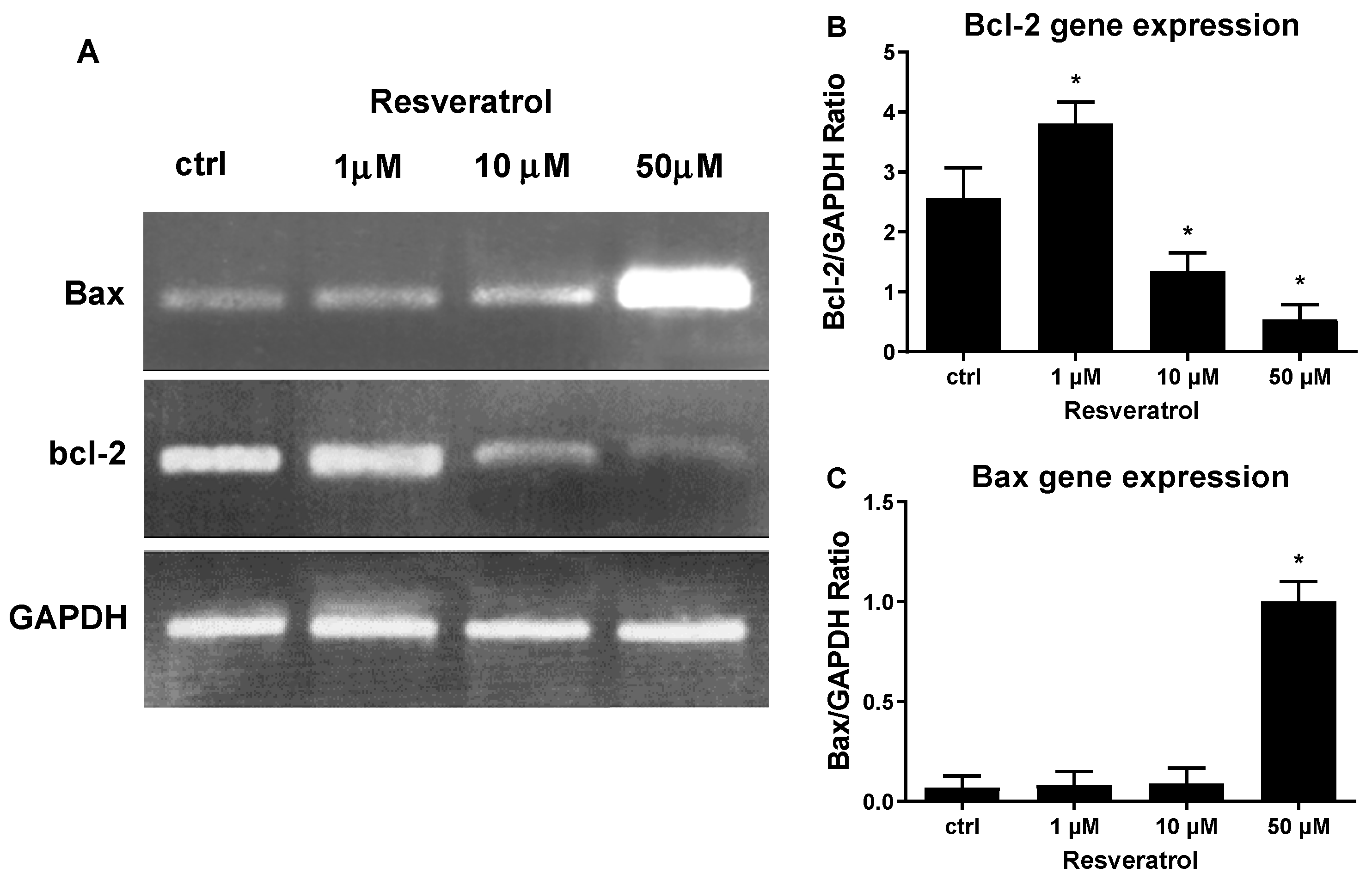{kind=link}
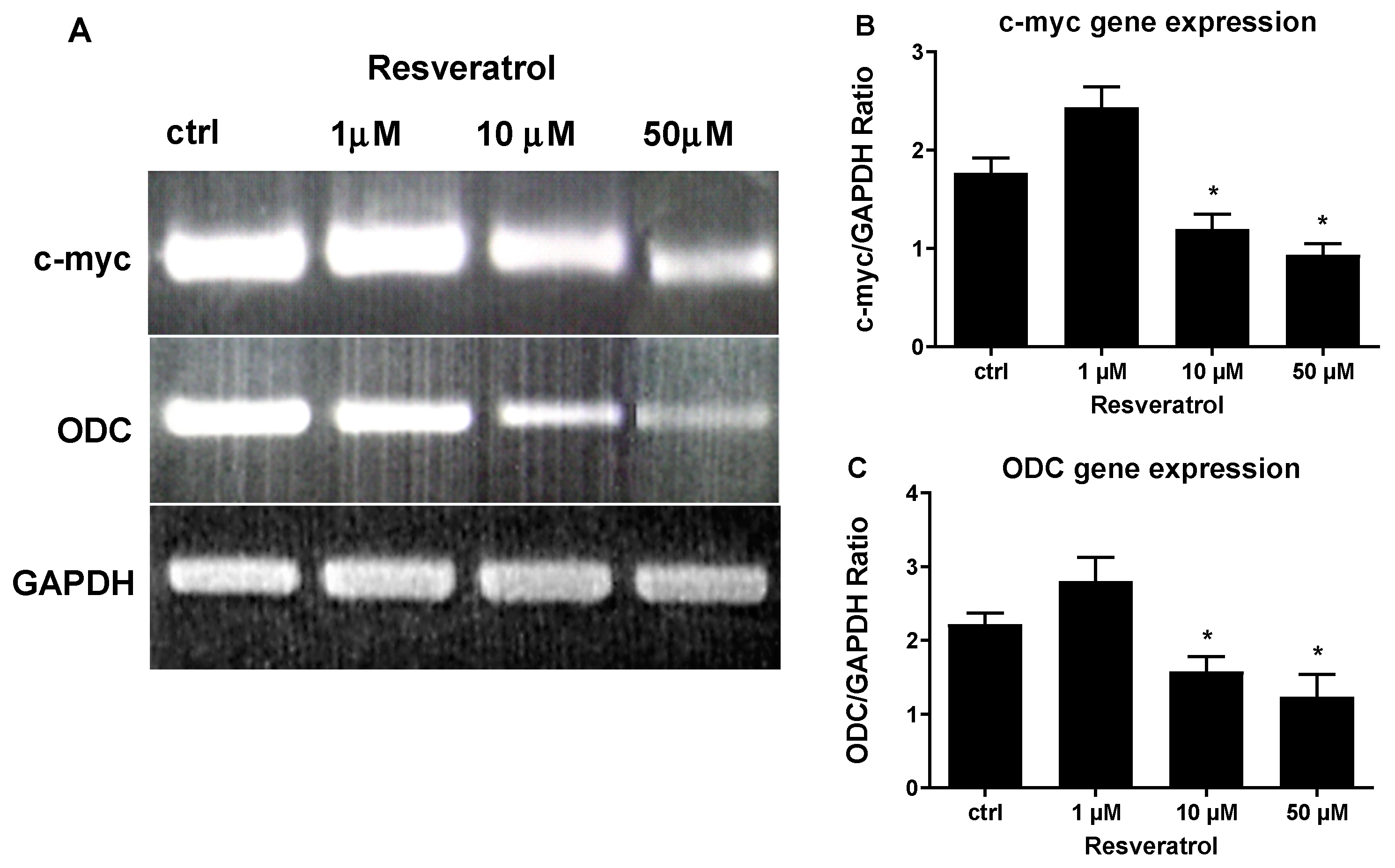{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Measurement of Intracellular ROS
2.3. Determination of the Intracellular Redox State
2.4. Determination of Cell Viability
2.5. Determination of DNA Synthesis
2.6. Immunoblot Analysis
2.7. Determination of DNA Fragmentation
2.8. Cell Cycle Analysis
2.9. Semi-Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.10. Determination of PKC Activity
2.11. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Foyer, C.H.; Noctor, G. Redox homeostasis and antioxidant signaling: A metabolic interface between stress perception and physiological responses. Plant Cell 2005, 17, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Reactive oxygen species. In Encyclopedia of Biophysics; Springer: Cham, Switzerland, 2013; pp. 2198–2200. [Google Scholar]
- Schieber, M.; Chandel, N.S. Ros function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Clarke, T.B. Microbial programming of systemic innate immunity and resistance to infection. PLoS PATHOG. 2014, 10, e1004506. [Google Scholar] [CrossRef]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell Signal 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ros) homeostasis and redox regulation in cellular signaling. Cell Signal 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Iwamoto, N.; Miura, T.; Ishii, T.; Cho, A.K.; Kumagai, Y. Redox cycling of 1,2-naphthoquinone by thioredoxin1 through cys32 and cys35 causes inhibition of its catalytic activity and activation of ask1/p38 signaling. Chem. Res. Toxicol. 2012, 25, 1222–1230. [Google Scholar] [CrossRef]
- Krylatov, A.V.; Maslov, L.N.; Voronkov, N.S.; Boshchenko, A.A.; Popov, S.V.; Gomez, L.; Wang, H.; Jaggi, A.S.; Downey, J.M. Reactive oxygen species as intracellular signaling molecules in the cardiovascular system. Curr. Cardiol. Rev. 2018, 14, 290–300. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Archiv. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Lan, L.; Wei, W.; Zheng, Y.; Niu, L.; Chen, X.; Huang, D.; Gao, Y.; Mo, S.; Lu, J.; Guo, M.; et al. Deferoxamine suppresses esophageal squamous cell carcinoma cell growth via erk1/2 mediated mitochondrial dysfunction. Cancer Lett. 2018, 432, 132–143. [Google Scholar] [CrossRef]
- Wu, G.; Liu, T.; Li, H.; Li, Y.; Li, D.; Li, W. C-myc and reactive oxygen species play roles in tetrandrine-induced leukemia differentiation. Cell Death Dis. 2018, 9, 473. [Google Scholar] [CrossRef]
- Sander, C.S.; Chang, H.; Hamm, F.; Elsner, P.; Thiele, J.J. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int. J. Dermatol. 2004, 43, 326–335. [Google Scholar] [CrossRef]
- Huang, C.; Hsu, P.; Hung, Y.; Liao, Y.; Liu, C.; Hour, C.; Kao, M.; Tsay, G.J.; Hung, H.; Liu, G.Y. Ornithine decarboxylase prevents methotrexate-induced apoptosis by reducing intracellular reactive oxygen species production. Apoptosis Int. J. Program. Cell Death 2005, 10, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Klann, E.; Roberson, E.D.; Knapp, L.T.; Sweatt, J.D. A role for superoxide in protein kinase c activation and induction of long-term potentiation. J. Biol. Chem. 1998, 273, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Knapp, L.T.; Klann, E. Potentiation of hippocampal synaptic transmission by superoxide requires the oxidative activation of protein kinase c. J. Neurosci.Off. J. Soc. Neurosc. 2002, 22, 674–683. [Google Scholar] [CrossRef]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell signaling through protein kinase c oxidation and activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef] [PubMed]
- Moshier, J.A.; Malecka-Panas, E.; Geng, H.; Dosescu, J.; Tureaud, J.; Skunca, M.; Majumdar, A.P. Ornithine decarboxylase transformation of nih/3t3 cells is mediated by altered epidermal growth factor receptor activity. Cancer Res. 1995, 55, 5358–5365. [Google Scholar]
- Thomas, T.; Thomas, T.J. Polyamines in cell growth and cell death: Molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci. CMLS 2001, 58, 244–258. [Google Scholar] [CrossRef]
- Desbarats, L.; Schneider, A.; Muller, D.; Burgin, A.; Eilers, M. Myc: A single gene controls both proliferation and apoptosis in mammalian cells. Experientia 1996, 52, 1123–1129. [Google Scholar] [CrossRef]
- Dang, C.V. Myc, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with myc. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. C-myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- He, W.; Roh, E.; Yao, K.; Liu, K.; Meng, X.; Liu, F.; Wang, P.; Bode, A.M.; Dong, Z. Targeting ornithine decarboxylase (odc) inhibits esophageal squamous cell carcinoma progression. NPJ Precis. Oncol. 2017, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. C-myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef]
- Hibshoosh, H.; Johnson, M.; Weinstein, I.B. Effects of overexpression of ornithine decarboxylase (odc) on growth control and oncogene-induced cell transformation. Oncogene 1991, 6, 739–743. [Google Scholar] [PubMed]
- Steinberg, S.F. Mechanisms for redox-regulation of protein kinase c. Front. Pharmacol. 2015, 6, 128. [Google Scholar] [CrossRef]
- Thallas-Bonke, V.; Jha, J.C.; Gray, S.P.; Barit, D.; Haller, H.; Schmidt, H.H.; Coughlan, M.T.; Cooper, M.E.; Forbes, J.M.; Jandeleit-Dahm, K.A. Nox-4 deletion reduces oxidative stress and injury by pkc-alpha-associated mechanisms in diabetic nephropathy. Physiol. Rep. 2014, 2, e12192. [Google Scholar] [CrossRef]
- Gao, X.; Schottker, B. Reduction-oxidation pathways involved in cancer development: A systematic review of literature reviews. Oncotarget 2017, 8, 51888–51906. [Google Scholar]
- Wang, Y.; Zhao, J.; Yang, W.; Bi, Y.; Chi, J.; Tian, J.; Li, W. High-dose alcohol induces reactive oxygen species-mediated apoptosis via pkc-beta/p66shc in mouse primary cardiomyocytes. Biochem. Biophys. Res. Commun. 2015, 456, 656–661. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Okabe, J.; Gray, S.P.; Hettige, T.; Chow, B.S.M.; Thallas-Bonke, V.; De Vos, L.; Holterman, C.E.; Coughlan, M.T.; et al. Nadph oxidase nox5 accelerates renal injury in diabetic nephropathy. Diabetes 2017, 66, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Gammone, M.A.; Riccioni, G.; D′Orazio, N. Marine carotenoids against oxidative stress: Effects on human health. Mar. Drugs 2015, 13, 6226–6246. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Knock, G.A.; Ward, J.P. Redox regulation of protein kinases as a modulator of vascular function. Antioxid. Redox Signal. 2011, 15, 1531–1547. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.K.; Lee, Y.R.; Choi, S.; Park, M.S.; Kang, G.; Kim, C.S.; Jeon, B.H. Protein kinase c beta ii upregulates intercellular adhesion molecule-1 via mitochondrial activation in cultured endothelial cells. Korean J. Physiol. Pharmacol. 2017, 21, 377–384. [Google Scholar] [CrossRef]
- Gao, L.; Mann, G.E. Vascular nad(p)h oxidase activation in diabetes: A double-edged sword in redox signalling. Cardiovasc. Res. 2009, 82, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Sisein, E.A. Biochemistry of free radicals and antioxidants. Sch. Acad. J. Biosci. 2014, 2, 110–118. [Google Scholar]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev.Drug Dis. 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Li, H.; Xia, N.; Förstermann, U. Cardiovascular effects and molecular targets of resveratrol. Nitric Oxide 2012, 26, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Pervaiz, S.; Holme, A.L. Resveratrol: Its biologic targets and functional activity. Antioxid. Redox Signal. 2009, 11, 2851–2897. [Google Scholar] [CrossRef]
- Ladurner, A.; Schachner, D.; Schueller, K.; Pignitter, M.; Heiss, E.H.; Somoza, V.; Dirsch, V.M. Impact of trans-resveratrol-sulfates and -glucuronides on endothelial nitric oxide synthase activity, nitric oxide release and intracellular reactive oxygen species. Molecules 2014, 19, 16724–16736. [Google Scholar] [CrossRef]
- Willcox, B.J.; Curb, J.D.; Rodriguez, B.L. Antioxidants in cardiovascular health and disease: Key lessons from epidemiologic studies. Am. J. Cardiol. 2008, 101, 75D–86D. [Google Scholar] [CrossRef] [PubMed]
- Pasciu, V.; Posadino, A.M.; Cossu, A.; Sanna, B.; Tadolini, B.; Gaspa, L.; Marchisio, A.; Dessole, S.; Capobianco, G.; Pintus, G. Akt downregulation by flavin oxidase–induced ros generation mediates dose-dependent endothelial cell damage elicited by natural antioxidants. Toxicol. Sci. 2010, 114, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.H.; Nealon, R.S.; Scholey, A.; Howe, P.R. Low dose resveratrol improves cerebrovascular function in type 2 diabetes mellitus. Nutr. Metabol. Cardiovasc. Dis. NMCD 2016, 26, 393–399. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, H.; Zou, Y.; Liu, Q.; Guo, C.; Gao, G.; Shao, C.; Gong, Y. Resveratrol modulates angiogenesis through the gsk3beta/beta-catenin/tcf-dependent pathway in human endothelial cells. Biochem. Pharmacol. 2010, 80, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Posadino, A.M.; Cossu, A.; Giordo, R.; Zinellu, A.; Sotgia, S.; Vardeu, A.; Hoa, P.T.; Nguyen le, H.V.; Carru, C.; Pintus, G. Resveratrol alters human endothelial cells redox state and causes mitochondrial-dependent cell death. Food Chem. Toxicol. 2015, 78, 10–16. [Google Scholar] [CrossRef]
- Heo, J.R.; Kim, S.M.; Hwang, K.A.; Kang, J.H.; Choi, K.C. Resveratrol induced reactive oxygen species and endoplasmic reticulum stressmediated apoptosis, and cell cycle arrest in the a375sm malignant melanoma cell line. Int. J. Mol. Med. 2018, 42, 1427–1435. [Google Scholar]
- Ji, S.; Zheng, Z.; Liu, S.; Ren, G.; Gao, J.; Zhang, Y.; Li, G. Resveratrol promotes oxidative stress to drive dlc1 mediated cellular senescence in cancer cells. Exp. Cell Res. 2018, 370, 292–302. [Google Scholar] [CrossRef]
- Giordo, R.; Cossu, A.; Pasciu, V.; Hoa, P.T.; Posadino, A.M.; Pintus, G. Different redox response elicited by naturally occurring antioxidants in human endothelial cells. Open Biochem. J. 2013, 7, 44–53. [Google Scholar] [CrossRef]
- Perez-Cremades, D.; Bueno-Beti, C.; Garcia-Gimenez, J.L.; Ibanez-Cabellos, J.S.; Hermenegildo, C.; Pallardo, F.V.; Novella, S. Extracellular histones disarrange vasoactive mediators release through a cox-nos interaction in human endothelial cells. J. Cell. Mol. Med. 2017, 21, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Posadino, A.M.; Phu, H.T.; Cossu, A.; Giordo, R.; Fois, M.; Thuan, D.T.B.; Piga, A.; Sotgia, S.; Zinellu, A.; Carru, C.; et al. Oxidative stress-induced akt downregulation mediates green tea toxicity towards prostate cancer cells. Toxicol. In Vitro 2017, 42, 255–262. [Google Scholar] [CrossRef]
- Fois, A.G.; Posadino, A.M.; Giordo, R.; Cossu, A.; Agouni, A.; Rizk, N.M.; Pirina, P.; Carru, C.; Zinellu, A.; Pintus, G. Antioxidant activity mediates pirfenidone antifibrotic effects in human pulmonary vascular smooth muscle cells exposed to sera of idiopathic pulmonary fibrosis patients. Oxid. Med. Cell. Longev. 2018, 2018, 2639081. [Google Scholar] [CrossRef]
- Pintus, G.; Tadolini, B.; Maioli, M.; Posadino, A.M.; Gaspa, L.; Ventura, C. Heparin down-regulates the phorbol ester-induced protein kinase c gene expression in human endothelial cells: Enzyme-mediated autoregulation of protein kinase c-alpha and -delta genes. FEBS Lett. 1999, 449, 135–140. [Google Scholar] [CrossRef]
- Pintus, G.; Tadolini, B.; Maioli, M.; Posadino, A.M.; Bennardini, F.; Bettuzzi, S.; Ventura, C. Heparin inhibits phorbol ester-induced ornithine decarboxylase gene expression in endothelial cells. FEBS Lett. 1998, 423, 98–104. [Google Scholar] [CrossRef]
- Boin, F.; Erre, G.L.; Posadino, A.M.; Cossu, A.; Giordo, R.; Spinetti, G.; Passiu, G.; Emanueli, C.; Pintus, G. Oxidative stress-dependent activation of collagen synthesis is induced in human pulmonary smooth muscle cells by sera from patients with scleroderma-associated pulmonary hypertension. Orphanet J. Rare Dis. 2014, 9. [Google Scholar] [CrossRef]
- Posadino, A.M.; Porcu, M.C.; Marongiu, B.; Cossu, A.; Piras, A.; Porcedda, S.; Falconieri, D.; Cappuccinelli, R.; Biosa, G.; Pintus, G.; et al. Antioxidant activity of supercritical carbon dioxide extracts of salvia desoleana on two human endothelial cell models. Food Res. Int. 2012, 46, 354–359. [Google Scholar] [CrossRef]
- Meyer, A.J.; Brach, T.; Marty, L.; Kreye, S.; Rouhier, N.; Jacquot, J.P.; Hell, R. Redox-sensitive gfp in arabidopsis thaliana is a quantitative biosensor for the redox potential of the cellular glutathione redox buffer. Plant J. 2007, 52, 973–986. [Google Scholar] [CrossRef]
- Dooley, C.T.; Dore, T.M.; Hanson, G.T.; Jackson, W.C.; Remington, S.J.; Tsien, R.Y. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 2004, 279, 22284–22293. [Google Scholar] [CrossRef] [PubMed]
- Cossu, A.; Posadino, A.M.; Giordo, R.; Emanueli, C.; Sanguinetti, A.M.; Piscopo, A.; Poiana, M.; Capobianco, G.; Piga, A.; Pintus, G. Apricot melanoidins prevent oxidative endothelial cell death by counteracting mitochondrial oxidation and membrane depolarization. PLoS ONE 2012, 7, e48817. [Google Scholar] [CrossRef]
- Posadino, A.M.; Cossu, A.; Piga, A.; Madrau, M.A.; Del Caro, A.; Colombino, M.; Paglietti, B.; Rubino, S.; Iaccarino, C.; Crosio, C.; et al. Prune melanoidins protect against oxidative stress and endothelial cell death. Front. Biosci. (Elite Ed) 2011, 3, 1034–1041. [Google Scholar] [CrossRef]
- Rosenwasser, S.; Rot, I.; Meyer, A.J.; Feldman, L.; Jiang, K.; Friedman, H. A fluorometer-based method for monitoring oxidation of redox-sensitive gfp (rogfp) during development and extended dark stress. Physiol. Plant. 2010, 138, 493–502. [Google Scholar] [CrossRef]
- Posadino, A.M.; Biosa, G.; Zayed, H.; Abou-Saleh, H.; Cossu, A.; Nasrallah, G.K.; Giordo, R.; Pagnozzi, D.; Porcu, M.C.; Pretti, L.; et al. Protective effect of cyclically pressurized solid(-)liquid extraction polyphenols from cagnulari grape pomace on oxidative endothelial cell death. Molecules 2018, 23, 2105. [Google Scholar] [CrossRef]
- Posadino, A.M.; Cossu, A.; Giordo, R.; Zinellu, A.; Sotgia, S.; Vardeu, A.; Hoa, P.T.; Deiana, L.; Carru, C.; Pintus, G. Coumaric acid induces mitochondrial damage and oxidative-mediated cell death of human endothelial cells. Cardiovasc. Toxicol. 2013, 13, 301–306. [Google Scholar] [CrossRef]
- Debidda, M.; Sanna, B.; Cossu, A.; Posadino, A.M.; Tadolini, B.; Ventura, C.; Pintus, G. Nami-a inhibits the pma-induced odc gene expression in ecv304 cells: Involvement of pkc/raf/mek/erk signalling pathway. Int. J. Oncol. 2003, 23, 477–482. [Google Scholar] [CrossRef]
- Pintus, G.; Tadolini, B.; Posadino, A.M.; Sanna, B.; Debidda, M.; Carru, C.; Deiana, L.; Ventura, C. Pkc/raf/mek/erk signaling pathway modulates native-ldl-induced e2f-1 gene expression and endothelial cell proliferation. Cardiovasc. Res. 2003, 59, 934–944. [Google Scholar] [CrossRef]
- Sanna, B.; Debidda, M.; Pintus, G.; Tadolini, B.; Posadino, A.M.; Bennardini, F.; Sava, G.; Ventura, C. The anti-metastatic agent imidazolium trans-imidazoledimethylsulfoxide-tetrachlororuthenate induces endothelial cell apoptosis by inhibiting the mitogen-activated protein kinase/extracellular signal-regulated kinase signaling pathway. Arch. Biochem. Biophys. 2002, 403, 209–218. [Google Scholar] [CrossRef]
- Kasibhatla, S.; Amarante-Mendes, G.P.; Finucane, D.; Brunner, T.; Bossy-Wetzel, E.; Green, D.R. Analysis of DNA fragmentation using agarose gel electrophoresis. Cold Spring Harb. Protoc. 2006, 2006, pdb. prot4429. [Google Scholar] [CrossRef]
- Rahbar Saadat, Y.; Saeidi, N.; Zununi Vahed, S.; Barzegari, A.; Barar, J. An update to DNA ladder assay for apoptosis detection. Bioimpacts 2015, 5, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Pozarowski, P.; Darzynkiewicz, Z. Analysis of cell cycle by flow cytometry. In Checkpoint Controls and Cancer; Springer: Cham, Switzerland, 2004; pp. 301–311. [Google Scholar]
- Klimaszewska-Wisniewska, A.; Halas-Wisniewska, M.; Tadrowski, T.; Gagat, M.; Grzanka, D.; Grzanka, A. Paclitaxel and the dietary flavonoid fisetin: A synergistic combination that induces mitotic catastrophe and autophagic cell death in a549 non-small cell lung cancer cells. Cancer Cell Int. 2016, 16, 10. [Google Scholar] [CrossRef]
- Marone, M.; Mozzetti, S.; De Ritis, D.; Pierelli, L.; Scambia, G. Semiquantitative rt-pcr analysis to assess the expression levels of multiple transcripts from the same sample. Biol. Proced. Online 2001, 3, 19–25. [Google Scholar] [CrossRef]
- Cheng, H.; Pan, Y.; Yao, Y.; Zhu, Z.; Chen, J.; Sun, X.; Qiu, Y.; Ding, Y. Expression and significance of caveolin-1 in hepatitis b virus-associated hepatocellular carcinoma. Exp. Ther. Med. 2017, 14, 4356–4362. [Google Scholar] [CrossRef]
- Li, H.; Spagnol, G.; Zheng, L.; Stauch, K.L.; Sorgen, P.L. Regulation of connexin43 function and expression by tyrosine kinase 2. J. Biol. Chem. 2016, 291, 15867–15880. [Google Scholar] [CrossRef]
- Shao, B.; Bayraktutan, U. Hyperglycaemia promotes cerebral barrier dysfunction through activation of protein kinase c-β. Diab. Obes. Metabol. 2013, 15, 993–999. [Google Scholar] [CrossRef]
- Lin, H.M.; Lee, Y.J.; Li, G.; Pestell, R.G.; Kim, H.R. Bcl-2 induces cyclin d1 promoter activity in human breast epithelial cells independent of cell anchorage. Cell Death Differ. 2001, 8, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Tucker, C.A.; Kapanen, A.I.; Chikh, G.; Hoffman, B.G.; Kyle, A.H.; Wilson, I.M.; Masin, D.; Gascoyne, R.D.; Bally, M.; Klasa, R.J. Silencing bcl-2 in models of mantle cell lymphoma is associated with decreases in cyclin d1, nuclear factor-κb, p53, bax, and p27 levels. Mol. Cancer Ther. 2008, 7, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Ciemerych, M.A.; Sicinski, P. Ras and myc can drive oncogenic cell proliferation through individual d-cyclins. Oncogene 2005, 24, 7114. [Google Scholar] [CrossRef]
- Daksis, J.I.; Lu, R.Y.; Facchini, L.M.; Marhin, W.W.; Penn, L.J. Myc induces cyclin d1 expression in the absence of de novo protein synthesis and links mitogen-stimulated signal transduction to the cell cycle. Oncogene 1994, 9, 3635–3645. [Google Scholar] [PubMed]
- Gliki, G.; Wheeler-Jones, C.; Zachary, I. Vascular endothelial growth factor induces protein kinase c (pkc)-dependent akt/pkb activation and phosphatidylinositol 3′-kinase-mediates pkc delta phosphorylation: Role of pkc in angiogenesis. Cell Biol. Int. 2002, 26, 751–759. [Google Scholar] [CrossRef]
- Aronis, K.N.; Chamberland, J.P.; Mantzoros, C.S. Glp-1 promotes angiogenesis in human endothelial cells in a dose-dependent manner, through the akt, src and pkc pathways. Metabolism 2013, 62, 1279–1286. [Google Scholar] [CrossRef]
- Marciniak, A.; Walczyna, B.; Rajtar, G.; Marciniak, S.; Wojtak, A.; Lasiecka, K. Tempol, a membrane-permeable radical scavenger, exhibits anti-inflammatory and cardioprotective effects in the cerulein-induced pancreatitis rat model. Oxid. Med. Cell. Longev. 2016, 2016, 4139851. [Google Scholar] [CrossRef]
- Bernardy, C.C.F.; Zarpelon, A.C.; Pinho-Ribeiro, F.A.; Calixto-Campos, C.; Carvalho, T.T.; Fattori, V.; Borghi, S.M.; Casagrande, R.; Verri, W.A., Jr. Tempol, a superoxide dismutase mimetic agent, inhibits superoxide anion-induced inflammatory pain in mice. Biomed. Res. Int. 2017, 2017, 9584819. [Google Scholar] [CrossRef]
- Batinic-Haberle, I.; Reboucas, J.S.; Spasojevic, I. Superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Signal. 2010, 13, 877–918. [Google Scholar] [CrossRef]
- Carrizzo, A.; Forte, M.; Damato, A.; Trimarco, V.; Salzano, F.; Bartolo, M.; Maciag, A.; Puca, A.A.; Vecchione, C. Antioxidant effects of resveratrol in cardiovascular, cerebral and metabolic diseases. Food Chem. Toxicol. Int. J. Pub. Br. Indust. Biol. Res. Associat. 2013, 61, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Xie, Y.; Meng, Y.; Ma, W.; Tong, Z.; Yang, X.; Lai, S.; Zhou, Y.; He, M.; Liao, Z. Resveratrol protects cardiomyocytes against anoxia/reoxygenation via dephosphorylation of vdac1 by akt-gsk3 beta pathway. Eur. J. Pharmacol. 2019, 843, 80–87. [Google Scholar] [CrossRef]
- Lefevre, J.; Michaud, S.-E.; Haddad, P.; Dussault, S.; Ménard, C.; Groleau, J.; Turgeon, J.; Rivard, A. Moderate consumption of red wine (cabernet sauvignon) improves ischemia-induced neovascularization in apoe-deficient mice: Effect on endothelial progenitor cells and nitric oxide. FASEB J. 2007, 21, 3845–3852. [Google Scholar] [CrossRef]
- Gadacha, W.; Ben-Attia, M.; Bonnefont-Rousselot, D.; Aouani, E.; Ghanem-Boughanmi, N.; Touitou, Y. Resveratrol opposite effects on rat tissue lipoperoxidation: Pro-oxidant during day-time and antioxidant at night. Redox Rep. 2009, 14, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Finckh, B.; Karten, B.; Kohlschütter, A.; Beisiegel, U. Antioxidant and prooxidant activity of alpha-tocopherol in human plasma and low density lipoprotein. J. Lipid Res. 1996, 37, 1436–1448. [Google Scholar] [PubMed]
- Mukherjee, S.; Dudley, J.I.; Das, D.K. Dose-dependency of resveratrol in providing health benefits. Dose Response 2010, 8, 478–500. [Google Scholar] [CrossRef]
- Schilder, Y.; Heiss, E.; Schachner, D.; Ziegler, J.; Reznicek, G.; Sorescu, D.; Dirsch, V. Nadph oxidases 1 and 4 mediate cellular senescence induced by resveratrol in human endothelial cells. Free Radic. Biol. Med. 2009, 46, 1598–1606. [Google Scholar] [CrossRef]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef]
- Biroccio, A.; Benassi, B.; Amodei, S.; Gabellini, C.; Del Bufalo, D.; Zupi, G. C-myc down-regulation increases susceptibility to cisplatin through reactive oxygen species-mediated apoptosis in m14 human melanoma cells. Mol. Pharmacol. 2001, 60, 174–182. [Google Scholar] [CrossRef]
- Atten, M.J.; Attar, B.M.; Milson, T.; Holian, O. Resveratrol-induced inactivation of human gastric adenocarcinoma cells through a protein kinase c-mediated mechanism. Biochem. Pharmacol. 2001, 62, 1423–1432. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, Y.; Dusting, G.J. Nadph oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Baldini, C.; Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Redox control of protein kinase c: Cell- and disease-specific aspects. Antioxid. Redox Signal. 2010, 13, 1051–1085. [Google Scholar] [CrossRef]
- Bouwman, R.A.; Musters, R.J.; van Beek-Harmsen, B.J.; de Lange, J.J.; Boer, C. Reactive oxygen species precede protein kinase c-delta activation independent of adenosine triphosphate-sensitive mitochondrial channel opening in sevoflurane-induced cardioprotection. Anesthesiology 2004, 100, 506–514. [Google Scholar] [CrossRef]
- Inoguchi, T.; Sonta, T.; Tsubouchi, H.; Etoh, T.; Kakimoto, M.; Sonoda, N.; Sato, N.; Sekiguchi, N.; Kobayashi, K.; Sumimoto, H.; et al. Protein kinase c-dependent increase in reactive oxygen species (ros) production in vascular tissues of diabetes: Role of vascular nad(p)h oxidase. J. Am. Soc. Nephrol. 2003, 14, S227–S232. [Google Scholar] [CrossRef]
- Stein, J.; Steven, S.; Bros, M.; Sudowe, S.; Hausding, M.; Oelze, M.; Munzel, T.; Grabbe, S.; Reske-Kunz, A.; Daiber, A. Role of protein kinase c and nox2-derived reactive oxygen species formation in the activation and maturation of dendritic cells by phorbol ester and lipopolysaccharide. Oxidat. Med. Cell. Longev. 2017, 2017, 4157213. [Google Scholar] [CrossRef]
- Gray, R.D.; Lucas, C.D.; MacKellar, A.; Li, F.; Hiersemenzel, K.; Haslett, C.; Davidson, D.J.; Rossi, A.G. Activation of conventional protein kinase c (pkc) is critical in the generation of human neutrophil extracellular traps. J. Inflamm. (London, England) 2013, 10, 12. [Google Scholar] [CrossRef]
- Liou, J.S.; Chen, C.Y.; Chen, J.S.; Faller, D.V. Oncogenic ras mediates apoptosis in response to protein kinase c inhibition through the generation of reactive oxygen species. J. Biol. Chem. 2000, 275, 39001–39011. [Google Scholar] [CrossRef]
- Wu, W.S.; Tsai, R.K.; Chang, C.H.; Wang, S.; Wu, J.R.; Chang, Y.X. Reactive oxygen species mediated sustained activation of protein kinase c alpha and extracellular signal-regulated kinase for migration of human hepatoma cell hepg2. Mol. Cancer Res. MCR 2006, 4, 747–758. [Google Scholar] [CrossRef]
- Lee, H.B.; Yu, M.R.; Song, J.S.; Ha, H. Reactive oxygen species amplify protein kinase c signaling in high glucose-induced fibronectin expression by human peritoneal mesothelial cells. Kidney Int. 2004, 65, 1170–1179. [Google Scholar] [CrossRef]
- Gresele, P.; Pignatelli, P.; Guglielmini, G.; Carnevale, R.; Mezzasoma, A.M.; Ghiselli, A.; Momi, S.; Violi, F. Resveratrol, at concentrations attainable with moderate wine consumption, stimulates human platelet nitric oxide production. J. Nutr. 2008, 138, 1602–1608. [Google Scholar] [CrossRef]
- Sanchez, M.; Galisteo, M.; Vera, R.; Villar, I.C.; Zarzuelo, A.; Tamargo, J.; Perez-Vizcaino, F.; Duarte, J. Quercetin downregulates nadph oxidase, increases enos activity and prevents endothelial dysfunction in spontaneously hypertensive rats. J. Hypertens. 2006, 24, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sepulveda, R.; Jimenez, R.; Romero, M.; Zarzuelo, M.J.; Sanchez, M.; Gomez-Guzman, M.; Vargas, F.; O′Valle, F.; Zarzuelo, A.; Perez-Vizcaino, F.; et al. Wine polyphenols improve endothelial function in large vessels of female spontaneously hypertensive rats. Hypertension 2008, 51, 1088–1095. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A double-edged sword in health benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, N.; Zhang, Z.; Li, W.; Zhu, W. Resveratrol induces cell apoptosis in adipocytes via ampk activation. Biochem. Biophys. Res. Commun. 2015, 457, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xiao, X.; Feng, X.; Li, W.; Zhou, N.; Zheng, L.; Sun, Y.; Zhang, Z.; Zhu, W. Resveratrol induces sirt1-dependent apoptosis in 3t3-l1 preadipocytes by activating ampk and suppressing akt activity and survivin expression. J. Nutr. Biochem. 2012, 23, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, Z.; Li, W.; Shan, Z.; Zhu, W. Resveratrol inhibits cell differentiation in 3t3-l1 adipocytes via activation of ampk. Can. J. Physiol. Pharmacol. 2011, 89, 793–799. [Google Scholar]
- Chen, S.; Zhao, Z.; Ke, L.; Li, Z.; Li, W.; Zhang, Z.; Zhou, Y.; Feng, X.; Zhu, W. Resveratrol improves glucose uptake in insulin-resistant adipocytes via sirt1. J. Nutr. Biochem. 2018, 55, 209–218. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, M.; Zhou, Y.; Wang, C.; Yuan, Y.; Li, L.; Bresette, W.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Resveratrol exerts dose-dependent anti-fibrotic or pro-fibrotic effects in kidneys: A potential risk to individuals with impaired kidney function. Phytomedicine 2019, 57, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.H.X.; Howe, P.R.C. Resveratrol counteracts insulin resistance-potential role of the circulation. Nutrients 2018, 10, 1160. [Google Scholar] [CrossRef] [PubMed]
- Wicinski, M.; Socha, M.; Walczak, M.; Wodkiewicz, E.; Malinowski, B.; Rewerski, S.; Gorski, K.; Pawlak-Osinska, K. Beneficial effects of resveratrol administration-focus on potential biochemical mechanisms in cardiovascular conditions. Nutrients 2018, 10, 1813. [Google Scholar] [CrossRef]
- Bagul, P.K.; Deepthi, N.; Sultana, R.; Banerjee, S.K. Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of nfkb-p65 and histone 3. J. Nutr. Biochem. 2015, 26, 1298–1307. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Wang, Z.; Xu, Z.; Zhang, Q.; Yin, M. Effects of dietary resveratrol on excess-iron-induced bone loss via antioxidative character. J. Nutr. Biochem. 2015, 26, 1174–1182. [Google Scholar] [CrossRef]
- Curro, M.; Trovato-Salinaro, A.; Gugliandolo, A.; Koverech, G.; Lodato, F.; Caccamo, D.; Calabrese, V.; Ientile, R. Resveratrol protects against homocysteine-induced cell damage via cell stress response in neuroblastoma cells. J. Neurosci. Res. 2015, 93, 149–156. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posadino, A.M.; Giordo, R.; Cossu, A.; Nasrallah, G.K.; Shaito, A.; Abou-Saleh, H.; Eid, A.H.; Pintus, G. Flavin Oxidase-Induced ROS Generation Modulates PKC Biphasic Effect of Resveratrol on Endothelial Cell Survival. Biomolecules 2019, 9, 209. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9060209
Posadino AM, Giordo R, Cossu A, Nasrallah GK, Shaito A, Abou-Saleh H, Eid AH, Pintus G. Flavin Oxidase-Induced ROS Generation Modulates PKC Biphasic Effect of Resveratrol on Endothelial Cell Survival. Biomolecules. 2019; 9(6):209. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9060209
Chicago/Turabian StylePosadino, Anna Maria, Roberta Giordo, Annalisa Cossu, Gheyath K. Nasrallah, Abdullah Shaito, Haissam Abou-Saleh, Ali H. Eid, and Gianfranco Pintus. 2019. "Flavin Oxidase-Induced ROS Generation Modulates PKC Biphasic Effect of Resveratrol on Endothelial Cell Survival" Biomolecules 9, no. 6: 209. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9060209