Promising Directions in Atherosclerosis Treatment Based on Epigenetic Regulation Using MicroRNAs and Long Noncoding RNAs
.JPG)


Abstract
:1. Introduction
2. Therapeutic Approaches Based on the Use of miRNAs and siRNAs Aimed at the Disruption of Lipid Metabolism
2.1. Therapeutic Approaches Based on the Use of miRNAs
2.2. Technologies for miRNA Delivery
2.3. Therapeutic Approaches Based on the Use of siRNAs Aimed at the Change in Lipid Metabolism
2.4. Technologies for siRNA Delivery
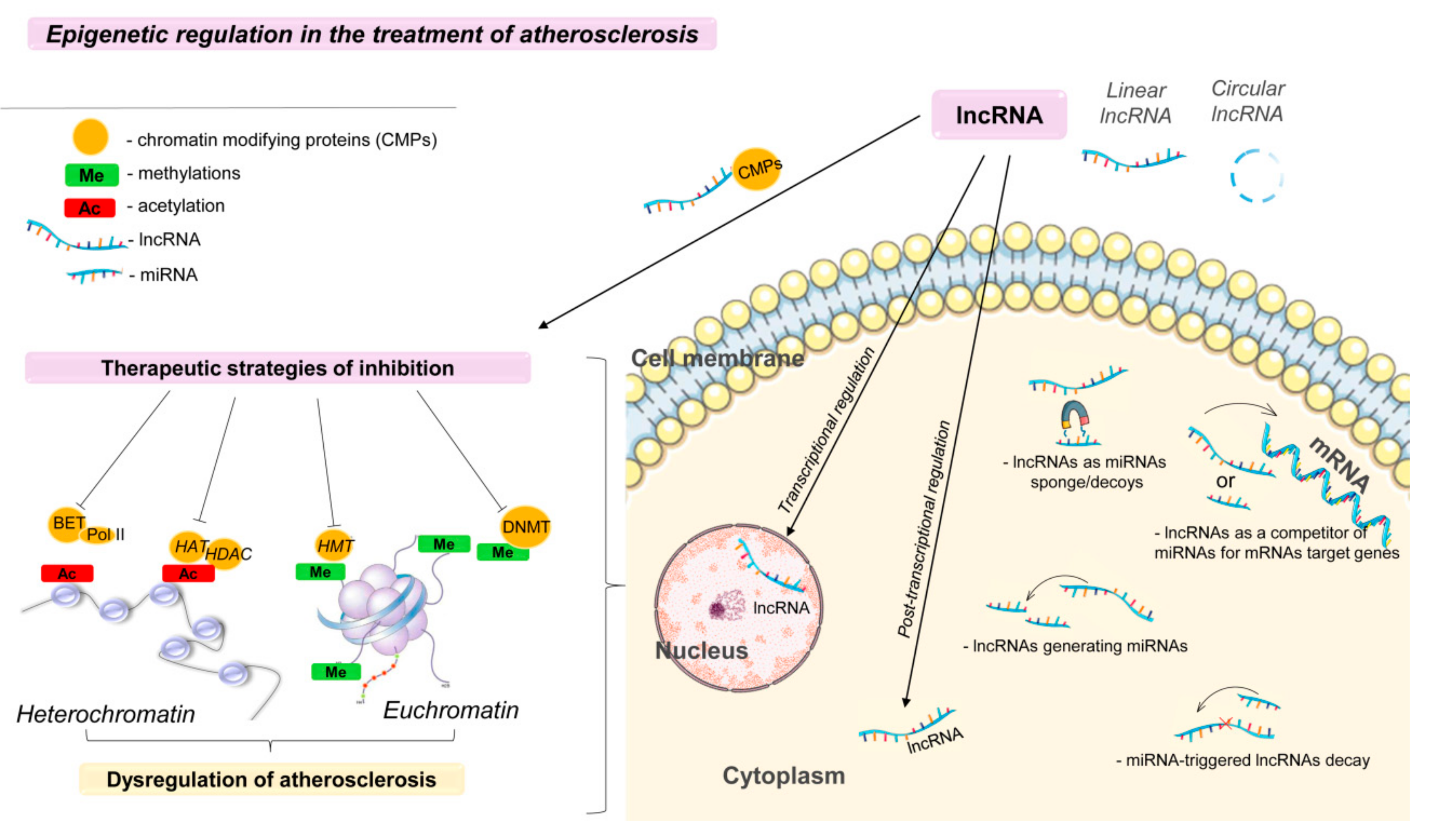
3. Epigenetic Regulation in the Treatment of Atherosclerosis
4. Therapeutic Approaches Based on the Use of Long Noncoding RNA Aimed at Atherosclerosis
- (a)
- miRNA-triggered lncRNA decay;
- (b)
- lncRNAs as miRNA sponges/decoys;
- (c)
- lncRNAs as competitors of miRNAs for mRNAs of target genes;
- (d)
- lncRNAs generating miRNAs [107].
Technologies for lncRNA Delivery
5. Drug Delivery Using Nanoparticles
6. Summary and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vascul. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.; Suárez, Y. Noncoding RNA regulation of endothelial and macrophage functions during atherosclerosis. Vascul. Pharmacol. 2019, 114, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, K.; Boon, R.A. Endothelial cell metabolism in atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Alevizos, I.; Illei, G.G. MicroRNAs as biomarkers in rheumatic diseases. Nat. Rev. Rheumatol. 2010, 6, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.-W.; Wang, Y.; Chen, L.-L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542. [Google Scholar] [CrossRef]
- Rocha, V.Z.; Libby, P. Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 399–409. [Google Scholar] [CrossRef]
- Li, F.; Guo, X.; Chen, S.-Y. Function and therapeutic potential of mesenchymal stem cells in atherosclerosis. Front. Cardiovasc. Med. 2017, 4, 32. [Google Scholar] [CrossRef]
- Marais, A.D. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology (Phila.) 2019, 51, 165–176. [Google Scholar] [CrossRef]
- Wang, Q.; Zheng, D.; Liu, J.; Fang, L.; Li, Q. Atherogenic index of plasma is a novel predictor of non-alcoholic fatty liver disease in obese participants: A cross-sectional study. Lipids Health Dis. 2018, 17. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, S.S.; Hooper, A.J.; Sullivan, D.R.; Burnett, J.R. Non-HDL-cholesterol and apolipoprotein B compared with LDL-cholesterol in atherosclerotic cardiovascular disease risk assessment. Pathology (Phila.) 2019, 51, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Toth, P.P. Triglyceride-rich lipoproteins as a causal factor for cardiovascular disease. Vasc. Health Risk Manag. 2016, 12, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A. Lipoprotein(a)-apheresis in the light of new drug developments. Atheroscler. Suppl. 2017, 30, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Heggermont, W.; Fieuws, S.; Vanhaecke, J.; Van Cleemput, J.; De Geest, B. Endothelium-enriched microRNAs as diagnostic biomarkers for cardiac allograft vasculopathy. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2015, 34, 1376–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryal, B.; Singh, A.K.; Rotllan, N.; Price, N.; Fernández-Hernando, C. MicroRNAs and lipid metabolism. Curr. Opin. Lipidol. 2017, 28, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Zeliadt, N. Big pharma shows signs of renewed interest in RNAi drugs. Nat. Med. 2014, 20, 109. [Google Scholar] [CrossRef]
- Kobayashi, H.; Tomari, Y. RISC assembly: Coordination between small RNAs and Argonaute proteins. Biochim. Biophys. Acta 2016, 1859, 71–81. [Google Scholar] [CrossRef]
- Eulalio, A.; Huntzinger, E.; Nishihara, T.; Rehwinkel, J.; Fauser, M.; Izaurralde, E. Deadenylation is a widespread effect of miRNA regulation. RNA 2009, 15, 21–32. [Google Scholar] [CrossRef]
- Ahmadzada, T.; Reid, G.; McKenzie, D.R. Fundamentals of siRNA and miRNA therapeutics and a review of targeted nanoparticle delivery systems in breast cancer. Biophys. Rev. 2018, 10, 69–86. [Google Scholar] [CrossRef]
- Soh, J.; Iqbal, J.; Queiroz, J.; Fernandez-Hernando, C.; Hussain, M.M. MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat. Med. 2013, 19, 892–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodi, R.; Eastwood, J.; Caslake, M.; Packard, C.J.; Denby, L. Relationship between circulating microRNA-30c with total- and LDL-cholesterol, their circulatory transportation and effect of statins. Clin. Chim. Acta Int. J. Clin. Chem. 2017, 466, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Aryal, B.; Zhang, X.; Fan, Y.; Price, N.L.; Suárez, Y.; Fernández-Hernando, C. Posttranscriptional regulation of lipid metabolism by noncoding RNAs and RNA binding proteins. Semin. Cell Dev. Biol. 2018, 81, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Thrush, A.B.; Nguyen, M.-A.; Richards, L.; Geoffrion, M.; Singaravelu, R.; Ramphos, E.; Shangari, P.; Ouimet, M.; Pezacki, J.P.; et al. Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by anti-miR33 in atherosclerosis. Circ. Res. 2015, 117, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Rotllan, N.; Canfrán-Duque, A.; Zhang, X.; Pati, P.; Arias, N.; Moen, J.; Mayr, M.; Ford, D.A.; Baldán, Á.; et al. Genetic dissection of the impact of mir-33a and mir-33b during the progression of atherosclerosis. Cell Rep. 2017, 21, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, C.M.; Goedeke, L.; Rotllan, N.; Yoon, J.-H.; Cirera-Salinas, D.; Mattison, J.A.; Suárez, Y.; de Cabo, R.; Gorospe, M.; Fernández-Hernando, C. MicroRNA 33 regulates glucose metabolism. Mol. Cell. Biol. 2013, 33, 2891–2902. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Rotllan, N.; Canfrán-Duque, A.; Aranda, J.F.; Ramírez, C.M.; Araldi, E.; Lin, C.-S.; Anderson, N.N.; Wagschal, A.; de Cabo, R.; et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat. Med. 2015, 21, 1280–1289. [Google Scholar] [CrossRef] [Green Version]
- Akinyemiju, T.; Do, A.N.; Patki, A.; Aslibekyan, S.; Zhi, D.; Hidalgo, B.; Tiwari, H.K.; Absher, D.; Geng, X.; Arnett, D.K.; et al. Epigenome-wide association study of metabolic syndrome in African-American adults. Clin. Epigenetics 2018, 10, 49. [Google Scholar] [CrossRef]
- Christopher, A.F.; Kaur, R.P.; Kaur, G.; Kaur, A.; Gupta, V.; Bansal, P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 2016, 7, 68–74. [Google Scholar] [PubMed]
- Chai, F.-N.; Zhang, J.; Xiang, H.-M.; Xu, H.-S.; Li, Y.-F.; Ma, W.-Y.; Li, X.-G.; Ye, X.-L. Protective effect of Coptisine from Rhizoma Coptidis on LPS/D-GalN-induced acute liver failure in mice through up-regulating expression of miR-122. Biomed. Pharmacother. Biomedecine Pharmacother. 2018, 98, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Skroblin, P.; Moschen, A.R.; Yin, X.; Kaudewitz, D.; Zampetaki, A.; Barwari, T.; Whitehead, M.; Ramírez, C.M.; Goedeke, L.; et al. Circulating MicroRNA-122 is associated with the risk of new-onset metabolic syndrome and type 2 diabetes. Diabetes 2017, 66, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Skroblin, P.; Kiechl, S.; Fernández-Hernando, C.; Mayr, M. Liver microRNAs: Potential mediators and biomarkers for metabolic and cardiovascular disease? Eur. Heart J. 2016, 37, 3260–3266. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-L.; Yu, W. Association of circulating microRNA-122 with presence and severity of atherosclerotic lesions. PeerJ 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Wagschal, A.; Najafi-Shoushtari, S.H.; Wang, L.; Goedeke, L.; Sinha, S.; deLemos, A.S.; Black, J.C.; Ramírez, C.M.; Li, Y.; Tewhey, R.; et al. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med. 2015, 21, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-B.; Feng, J.-Y.; Ren, W.-L.; Huang, B.; Zhou, L.; Wen, Y.-J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.-M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Adlakha, Y.K.; Khanna, S.; Singh, R.; Singh, V.P.; Agrawal, A.; Saini, N. Pro-apoptotic miRNA-128-2 modulates ABCA1, ABCG1 and RXRα expression and cholesterol homeostasis. Cell Death Dis. 2013, 4, e780. [Google Scholar] [CrossRef]
- Yang, A.; Sun, Y.; Gao, Y.; Yang, S.; Mao, C.; Ding, N.; Deng, M.; Wang, Y.; Yang, X.; Jia, Y.; et al. Reciprocal regulation between miR-148a/152 and DNA methyltransferase 1 is associated with hyperhomocysteinemia-accelerated atherosclerosis. DNA Cell Biol. 2017, 36, 462–474. [Google Scholar] [CrossRef]
- Naar, A.M. Methods Targeting miR-33 microRNAs for Regulating Lipid Metabolism. U.S. Patent 8,859,519, 14 October 2014. [Google Scholar]
- Fernandez-Hernando, C.; Goedeke, L. Anti-mir-27b and anti-mir-148a Oligonucleotides as Therapeutic Tools for Treating Dyslipidemias and Cardiovascular Diseases. U.S. Patent 2016138018, 19 May 2016. [Google Scholar]
- Naar, A.M.; Najafi-Shoushtari, S.H. Methods Targeting mir-128 for Regulating Cholesterol/Lipid Metabolism. U.S. Patent 13,979,428, 2012. [Google Scholar]
- Laffont, B.; Rayner, K.J. MicroRNAs in the pathobiology and therapy of atherosclerosis. Can. J. Cardiol. 2017, 33, 313–324. [Google Scholar] [CrossRef]
- Baumann, V.; Winkler, J. miRNA-based therapies: Strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med. Chem. 2014, 6, 1967–1984. [Google Scholar] [CrossRef]
- Kota, S.K.; Balasubramanian, S. Cancer therapy via modulation of micro RNA levels: A promising future. Drug Discov. Today 2010, 15, 733–740. [Google Scholar] [CrossRef] [PubMed]
- McKiernan, P.J.; Cunningham, O.; Greene, C.M.; Cryan, S.-A. Targeting miRNA-based medicines to cystic fibrosis airway epithelial cells using nanotechnology. Int. J. Nanomed. 2013, 8, 3907–3915. [Google Scholar] [PubMed] [Green Version]
- Nguyen, D.-D.; Chang, S. Development of novel therapeutic agents by inhibition of oncogenic MicroRNAs. Int. J. Mol. Sci. 2017, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Lindow, M.; Kauppinen, S. Discovering the first microRNA-targeted drug. J. Cell Biol. 2012, 199, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, J.A.; Kramerov, D.A. Noncoding RNAs. Biochem. Biokhimiia 2007, 72, 1161–1178. [Google Scholar] [CrossRef]
- Loyer, X.; Mallat, Z.; Boulanger, C.M.; Tedgui, A. MicroRNAs as therapeutic targets in atherosclerosis. Expert Opin. Ther. Targets 2015, 19, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-Y. PCSK9 as a therapeutic target for cardiovascular disease (Review). Exp. Ther. Med. 2017, 13, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.T.; Turner, T.; Visseren, F.L.J.; et al. Inclisiran in patients at high cardiovascular risk with elevated ldl cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Evaluation of the Circulating Micro-RNA Profile Specificity in Patients with Different Stages of Atherosclerosis According to MSCT Coronary Angiography—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03855891 (accessed on 19 April 2019).
- Zhao, J.; Mi, Y.; Feng, S.-S. siRNA-based nanomedicine. Nanomedicine 2013, 8, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Inclisiran for Participants with Atherosclerotic Cardiovascular Disease and Elevated Low-density Lipoprotein Cholesterol—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03399370 (accessed on 19 April 2019).
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirollo, K.F.; Chang, E.H. Targeted delivery of small interfering RNA: Approaching effective cancer therapies. Cancer Res. 2008, 68, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Nicorescu, I.; Dallinga, G.M.; de Winther, M.P.J.; Stroes, E.S.G.; Bahjat, M. Potential epigenetic therapeutics for atherosclerosis treatment. Atherosclerosis 2019, 281, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series). Pulm. Circ. 2014, 4, 169–174. [Google Scholar] [CrossRef]
- Khyzha, N.; Alizada, A.; Wilson, M.D.; Fish, J.E. Epigenetics of atherosclerosis: Emerging mechanisms and methods. Trends Mol. Med. 2017, 23, 332–347. [Google Scholar] [CrossRef]
- Ganesan, A. Epigenetic drug discovery: A success story for cofactor interference. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Zaina, S.; Heyn, H.; Carmona, F.J.; Varol, N.; Sayols, S.; Condom, E.; Ramírez-Ruz, J.; Gomez, A.; Gonçalves, I.; Moran, S.; et al. DNA methylation map of human atherosclerosis. Circ. Cardiovasc. Genet. 2014, 7, 692–700. [Google Scholar] [CrossRef]
- Fernández-Sanlés, A.; Sayols-Baixeras, S.; Subirana, I.; Degano, I.R.; Elosua, R. Association between DNA methylation and coronary heart disease or other atherosclerotic events: A systematic review. Atherosclerosis 2017, 263, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Muka, T.; Koromani, F.; Portilla, E.; O’Connor, A.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; Franco, O.H. The role of epigenetic modifications in cardiovascular disease: A systematic review. Int. J. Cardiol. 2016, 212, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aavik, E.; Babu, M.; Ylä-Herttuala, S. DNA methylation processes in atheosclerotic plaque. Atherosclerosis 2019, 281, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Jia, L.; Li, Y.; Bian, Y.; Cheng, J.; Li, H.; Xiao, C.; Du, J. Angiotensin II infusion-induced inflammation, monocytic fibroblast precursor infiltration, and cardiac fibrosis are pressure dependent. Cardiovasc. Toxicol. 2011, 11, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Qiu, Y.; Yang, J.; Bian, S.; Chen, G.; Deng, M.; Kang, H.; Huang, L. DNMT1-PPARγ pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci. Rep. 2016, 6, 30053. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tanaka, R.; Hamada, S.; Nakagawa, H.; Miyata, N. Design, synthesis, inhibitory activity, and binding mode study of novel DNA methyltransferase 1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Treatment of cardiovascular pathology with epigenetically active agents: Focus on natural and synthetic inhibitors of DNA methylation and histone deacetylation. Int. J. Cardiol. 2017, 227, 66–82. [Google Scholar] [CrossRef]
- Kim, Y.J.; Chung, S.O.; Kim, J.K.; Park, S.U. Recent studies on resveratrol and its biological and pharmacological activity. EXCLI J. 2017, 16, 602–608. [Google Scholar]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1. [Google Scholar] [CrossRef]
- Raj, P.; Louis, X.L.; Thandapilly, S.J.; Movahed, A.; Zieroth, S.; Netticadan, T. Potential of resveratrol in the treatment of heart failure. Life Sci. 2014, 95, 63–71. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Wang, Z.; Xie, W.; Cai, Y.; Xia, L.; Easwaran, H.; Luo, J.; Yen, R.-W.C.; Li, Y.; Baylin, S.B. Acetylation enhances TET2 function in protecting against abnormal DNA methylation during oxidative stress. Mol. Cell 2017, 65, 323–335. [Google Scholar] [CrossRef]
- Jiang, W.; Agrawal, D.K.; Boosani, C.S. Cell-specific histone modifications in atherosclerosis (Review). Mol. Med. Rep. 2018, 18, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.-S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Issa, J.-P.J.; Kropf, P. DNA Hypomethylating drugs in cancer therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026948. [Google Scholar] [CrossRef] [PubMed]
- Greißel, A.; Culmes, M.; Napieralski, R.; Wagner, E.; Gebhard, H.; Schmitt, M.; Zimmermann, A.; Eckstein, H.-H.; Zernecke, A.; Pelisek, J. Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb. Haemost. 2015, 114, 390–402. [Google Scholar] [PubMed]
- Liu, Y.; Peng, W.; Qu, K.; Lin, X.; Zeng, Z.; Chen, J.; Wei, D.; Wang, Z. TET2: A novel epigenetic regulator and potential intervention target for atherosclerosis. DNA Cell Biol. 2018, 37, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Puri, R.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Brewer, H.B.; Kastelein, J.J.P.; Hu, B.; Uno, K.; Kataoka, Y.; et al. Effect of the BET protein inhibitor, RVX-208, on progression of coronary atherosclerosis: Results of the phase 2b, randomized, double-blind, multicenter, ASSURE Trial. Am. J. Cardiovasc. Drugs Drugs Devices Interv. 2016, 16, 55–65. [Google Scholar] [CrossRef]
- Ghosh, G.C.; Bhadra, R.; Ghosh, R.K.; Banerjee, K.; Gupta, A. RVX 208: A novel BET protein inhibitor, role as an inducer of apo A-I/HDL and beyond. Cardiovasc. Ther. 2017, 35, e12265. [Google Scholar] [CrossRef]
- Schooling, C.M.; Zhao, J.V. How might bromodomain and extra-terminal (BET) inhibitors operate in cardiovascular disease? Am. J. Cardiovasc. Drugs 2019, 19, 107–111. [Google Scholar] [CrossRef]
- Klein, K. Bromodomain protein inhibition: A novel therapeutic strategy in rheumatic diseases. RMD Open 2018, 4, e000744. [Google Scholar] [CrossRef]
- Mujtaba, S.; Zeng, L.; Zhou, M.-M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007, 26, 5521–5527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Yuan, J.; Li, N.; Pei, S.; Xu, J.; Luo, X.; Mao, C.; Liu, J.; Yu, T.; et al. Macrophage/microglial Ezh2 facilitates autoimmune inflammation through inhibition of Socs3. J. Exp. Med. 2018, 215, 1365–1382. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.R.; Quax, P.H.A. Plaque angiogenesis and its relation to inflammation and atherosclerotic plaque destabilization. Curr. Opin. Lipidol. 2016, 27, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Neele, A.E.; Prange, K.H.; Hoeksema, M.A.; van der Velden, S.; Lucas, T.; Dimmeler, S.; Lutgens, E.; Van den Bossche, J.; de Winther, M.P. Macrophage Kdm6b controls the pro-fibrotic transcriptome signature of foam cells. Epigenomics 2017, 9, 383–391. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Brown, M. Epigenetics and Epigenomics. In Hematology, 7th ed.; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Salama, M.E., Abutalib, S.A., Eds.; Elsevier: Toronto, ON, Canada, 2018; Chapter 2; pp. 17–24. ISBN 978-0-323-35762-3. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long noncoding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Evolution and Functions of Long Noncoding RNAs—ScienceDirect. Available online: https://0-www-sciencedirect-com.brum.beds.ac.uk/science/article/pii/S0092867409001421 (accessed on 18 April 2019).
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis is an Epigenetic Disease. Trends Endocrinol. Metab. TEM 2018, 29, 739–742. [Google Scholar] [CrossRef]
- Wilczynska, A.; Bushell, M. The complexity of miRNA-mediated repression. Cell Death Differ. 2015, 22, 22–33. [Google Scholar] [CrossRef]
- Xu, S.; Kamato, D.; Little, P.J.; Nakagawa, S.; Pelisek, J.; Jin, Z.G. Targeting epigenetics and noncoding RNAs in atherosclerosis: From mechanisms to therapeutics. Pharmacol. Ther. 2019, 196, 15–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shi, Z.-M.; Chang, Y.-N.; Hu, Z.-M.; Qi, H.-X.; Hong, W. The ways of action of long noncoding RNAs in cytoplasm and nucleus. Gene 2014, 547, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.R.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long noncoding RNAs in the regulation of gene expression: physiology and disease. Noncoding RNA 2019, 5. [Google Scholar]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long noncoding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Rong, D.; Sun, H.; Li, Z.; Liu, S.; Dong, C.; Fu, K.; Tang, W.; Cao, H. An emerging function of circRNA-miRNAs-mRNA axis in human diseases. Oncotarget 2017, 8, 73271–73281. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; Agarwal, V.; Stefano, J.; Bartel, D.P.; Stoffel, M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol. Cell 2014, 54, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Luo, S.; Lu, J.Y.; Shen, X. Cis- and trans-acting lncRNAs in pluripotency and reprogramming. Curr. Opin. Genet. Dev. 2017, 46, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Beckedorff, F.C.; Ayupe, A.C.; Crocci-Souza, R.; Amaral, M.S.; Nakaya, H.I.; Soltys, D.T.; Menck, C.F.M.; Reis, E.M.; Verjovski-Almeida, S. The intronic long noncoding RNA ANRASSF1 recruits PRC2 to the RASSF1A promoter, reducing the expression of RASSF1A and increasing cell proliferation. PLoS Genet. 2013, 9, e1003705. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Abdelmohsen, K.; Gorospe, M. Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell Dev. Biol. 2014, 34, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, E.J. Cardiovascular disease and long noncoding RNAs: Tools for unraveling the mystery Lnc-ing RNA and phenotype. Circ. Cardiovasc. Genet. 2017, 10, e001556. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, A. The IncRNA CHROME regulates cholesterol homeostasis. Nat. Rev. Cardiol. 2019, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.V.; Silljé, H.H.W.; Sijbesma, J.W.A.; Khan, M.A.F.; Steffensen, K.R.; van Gilst, W.H.; de Boer, R.A. LXRα improves myocardial glucose tolerance and reduces cardiac hypertrophy in a mouse model of obesity-induced type 2 diabetes. Diabetologia 2016, 59, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Theofilatos, D.; Anestis, A.; Hashimoto, K.; Kardassis, D. Transcriptional regulation of the human Liver X receptor α gene by hepatocyte nuclear factor 4α. Biochem. Biophys. Res. Commun. 2016, 469, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Sallam, T.; Jones, M.; Thomas, B.J.; Wu, X.; Gilliland, T.; Qian, K.; Eskin, A.; Casero, D.; Zhang, Z.; Sandhu, J.; et al. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat. Med. 2018, 24, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Invest. 2006, 116, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Rust, S.; Rosier, M.; Funke, H.; Real, J.; Amoura, Z.; Piette, J.C.; Deleuze, J.F.; Brewer, H.B.; Duverger, N.; Denèfle, P.; et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 1999, 22, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ruan, X.; Yang, L.; Kiesewetter, K.; Zhao, Y.; Luo, H.; Chen, Y.; Gucek, M.; Zhu, J.; Cao, H. A liver-enriched long noncoding RNA, lncLSTR, regulates systemic lipid metabolism in mice. Cell Metab. 2015, 21, 455–467. [Google Scholar] [CrossRef]
- Holdt, L.M.; Teupser, D. Long noncoding RNA ANRIL: Lnc-ing genetic variation at the chromosome 9p21 locus to molecular mechanisms of atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 145. [Google Scholar] [CrossRef]
- Congrains, A.; Kamide, K.; Oguro, R.; Yasuda, O.; Miyata, K.; Yamamoto, E.; Kawai, T.; Kusunoki, H.; Yamamoto, H.; Takeya, Y.; et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 2012, 220, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Song, C.-L.; Wang, J.-P.; Xue, X.; Liu, N.; Zhang, X.-H.; Zhao, Z.; Liu, J.-G.; Zhang, C.-P.; Piao, Z.-H.; Liu, Y.; et al. Effect of circular ANRIL on the inflammatory response of vascular endothelial cells in a rat model of coronary atherosclerosis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Hoffmann, S.; Sass, K.; Langenberger, D.; Scholz, M.; Krohn, K.; Finstermeier, K.; Stahringer, A.; Wilfert, W.; Beutner, F.; et al. Alu elements in ANRIL noncoding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 2013, 9, e1003588. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.-S.; Li, J.-Z.; Jia, J.-J.; Zhang, T.; Liu, X.-M.; Yi, L. Long non-coding RNA ANRIL in gene regulation and its duality in atherosclerosis. J. Huazhong Univ. Sci. Technol. Med. 2017, 37, 816–822. [Google Scholar] [CrossRef]
- Arslan, S.; Berkan, Ö.; Lalem, T.; Özbilüm, N.; Göksel, S.; Korkmaz, Ö.; Çetin, N.; Devaux, Y. CardiolincTM network Long noncoding RNAs in the atherosclerotic plaque. Atherosclerosis 2017, 266, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Cremer, S.; Michalik, K.M.; Fischer, A.; Pfisterer, L.; Jaé, N.; Winter, C.; Boon, R.A.; Muhly-Reinholz, M.; John, D.; Uchida, S.; et al. Hematopoietic deficiency of the long noncoding RNA MALAT1 promotes atherosclerosis and plaque inflammation. Circulation 2019, 139, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Rauch, B.H.; Nakagawa, S.; Haghikia, A.; Jasina, A.; Haas, J.; Nath, N.; Jensen, L.; Stroux, A.; Böhm, A.; et al. Immune system-mediated atherosclerosis caused by deficiency of long noncoding RNA MALAT1 in ApoE-/-mice. Cardiovasc. Res. 2019, 115, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Song, T.-F.; Huang, L.-W.; Yuan, Y.; Wang, H.-Q.; He, H.-P.; Ma, W.-J.; Huo, L.-H.; Zhou, H.; Wang, N.; Zhang, T.-C. LncRNA MALAT1 regulates smooth muscle cell phenotype switch via activation of autophagy. Oncotarget 2018, 9, 4411–4426. [Google Scholar] [CrossRef] [PubMed]
- Michalik, K.M.; You, X.; Manavski, Y.; Doddaballapur, A.; Zörnig, M.; Braun, T.; John, D.; Ponomareva, Y.; Chen, W.; Uchida, S.; et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res. 2014, 114, 1389–1397. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, X.; Hamblin, M.H.; Yin, K.-J. Long noncoding RNA malat1 regulates angiogenesis in hindlimb ischemia. Int. J. Mol. Sci. 2018, 19, 1723. [Google Scholar]
- Wu, G.; Cai, J.; Han, Y.; Chen, J.; Huang, Z.-P.; Chen, C.; Cai, Y.; Huang, H.; Yang, Y.; Liu, Y.; et al. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation 2014, 130, 1452–1465. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Ajami, N.E.; Gupta, S.; Maurya, M.R.; Nguyen, P.; Li, J.Y.-S.; Shyy, J.Y.-J.; Chen, Z.; Chien, S.; Subramaniam, S. Systems biology analysis of longitudinal functional response of endothelial cells to shear stress. Proc. Natl. Acad. Sci. USA 2017, 114, 10990–10995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; Ajami, N.E.; Huang, T.-S.; Lin, F.-M.; Lou, C.-H.; Wang, Y.-T.; Li, S.; Kang, J.; Munkacsi, H.; Maurya, M.R.; et al. Enhancer-associated long noncoding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Man, H.S.J.; Sukumar, A.N.; Lam, G.C.; Turgeon, P.J.; Yan, M.S.; Ku, K.H.; Dubinsky, M.K.; Ho, J.J.D.; Wang, J.J.; Das, S.; et al. Angiogenic patterning by STEEL, an endothelial-enriched long noncoding RNA. Proc. Natl. Acad. Sci. USA. 2018, 115, 2401–2406. [Google Scholar] [CrossRef] [Green Version]
- Lyu, Q.; Xu, S.; Lyu, Y.; Choi, M.; Christie, C.K.; Slivano, O.J.; Rahman, A.; Jin, Z.-G.; Long, X.; Xu, Y.; et al. SENCR stabilizes vascular endothelial cell adherens junctions through interaction with CKAP4. Proc. Natl. Acad. Sci. USA 2019, 116, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Filice, M.; Ruiz-Cabello, J. Nucleic Acid Nanotheranostics: Biomedical Applications; Elsevier: Madrid, Spain, 2019; p. 491. ISBN 978-0-12-814471-8. [Google Scholar] [CrossRef]
- Hung, J.; Miscianinov, V.; Sluimer, J.C.; Newby, D.E.; Baker, A.H. Targeting noncoding RNA in vascular biology and disease. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef]
- Kim, D.H.; Rossi, J.J. RNAi mechanisms and applications. BioTechniques 2008, 44, 613–616. [Google Scholar] [CrossRef]
- Haemmig, S.; Feinberg, M.W. Targeting LncRNAs in cardiovascular disease: Options and expeditions. Circ. Res. 2017, 120, 620–623. [Google Scholar] [CrossRef]
- Yang, J.; Meng, X.; Pan, J.; Jiang, N.; Zhou, C.; Wu, Z.; Gong, Z. CRISPR/Cas9-mediated noncoding RNA editing in human cancers. RNA Biol. 2017, 15, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.-T.; Zhou, N.; Huang, J.; Koirala, P.; Xu, M.; Fung, R.; Wu, F.; Mo, Y.-Y. Targeting noncoding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic Acids Res. 2015, 43, e17. [Google Scholar] [CrossRef] [PubMed]
- Cervadoro, A.; Palomba, R.; Vergaro, G.; Cecchi, R.; Menichetti, L.; Decuzzi, P.; Emdin, M.; Luin, S. Targeting inflammation with nanosized drug delivery platforms in cardiovascular diseases: Immune cell modulation in atherosclerosis. Front. Bioeng. Biotechnol. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Nakhlband, A.; Eskandani, M.; Omidi, Y.; Saeedi, N.; Ghaffari, S.; Barar, J.; Garjani, A. Combating atherosclerosis with targeted nanomedicines: Recent advances and future prospective. BioImpacts BI 2018, 8, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Zhang, J.; Martinez-Zaguilan, R.; Sennoune, S.; Hossen, M.N.; Lichtenstein, A.H.; Cao, J.; Meyerrose, G.E.; Paone, R.; Soontrapa, S.; et al. Detection of atherosclerotic lesions and intimal macrophages using CD36-targeted nanovesicles. J. Control. Release Off. J. Control. Release Soc. 2015, 220, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissig, V.; Guzman-Villanueva, D. Nanopharmaceuticals (part 2): Products in the pipeline. Int. J. Nanomed. 2015, 10, 1245–1257. [Google Scholar] [CrossRef] [PubMed]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-based medicines: A review of FDA-approved materials and clinical trials to date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Kharlamov, A.N. Translational exploration and clinical testing of silica–gold nanoparticles in development of multifunctional nanoplatform for theranostics of atherosclerosis. In Biomedical Applications of Functionalized Nanomaterials; Micro and Nano Technologies; Sarmento, B., das Neves, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Chapter 23; pp. 681–741. ISBN 978-0-323-50878-0. [Google Scholar] [CrossRef]
- Ulbrich, K.; Holá, K.; Šubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted drug delivery with polymers and magnetic nanoparticles: Covalent and noncovalent approaches, release control, and clinical studies. Chem. Rev. 2016, 116, 5338–5431. [Google Scholar] [CrossRef]
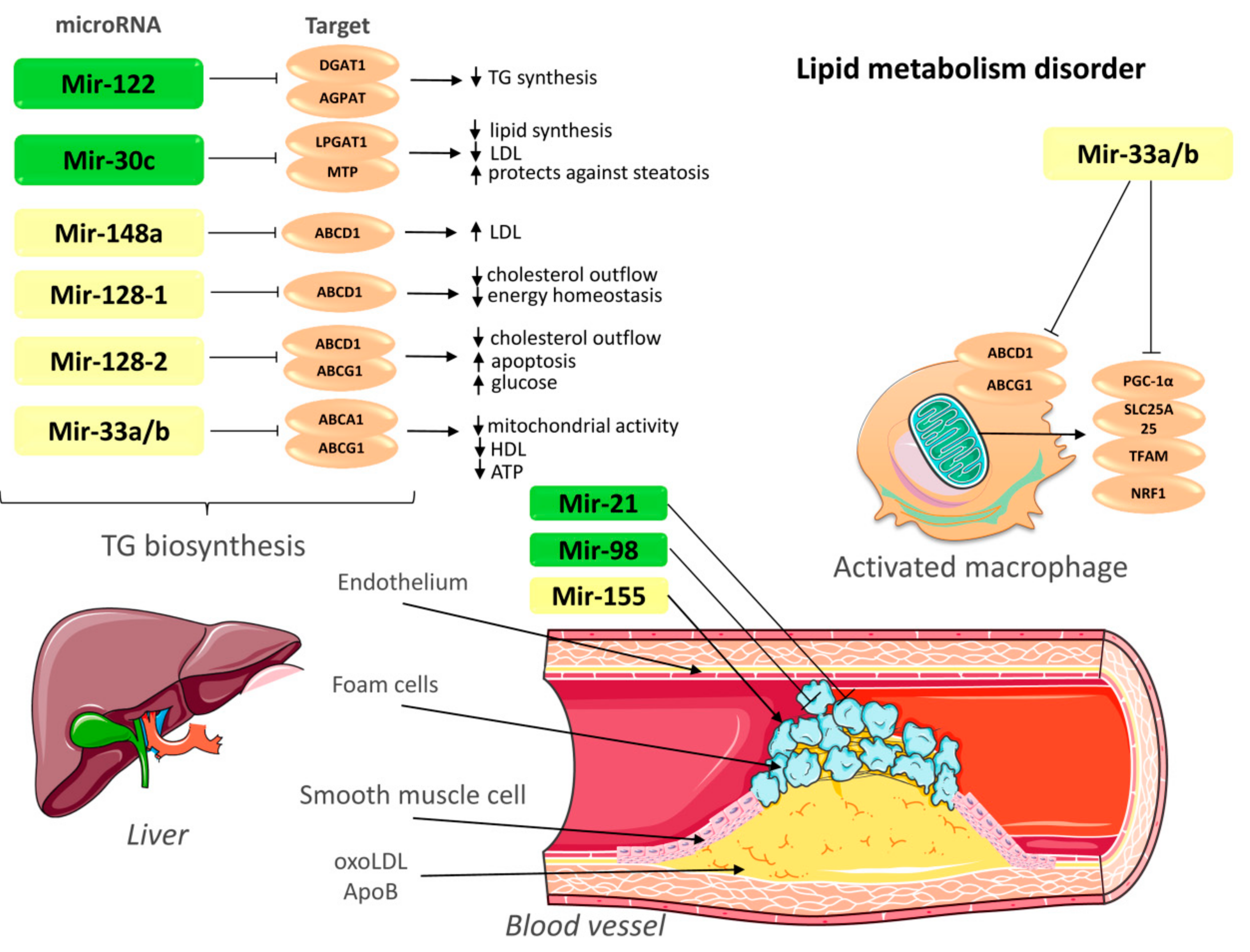
 ; upregulation ↑; downregulation ↓. TG—triglycerides; LDL—low-density lipoprotein; ABCA1—ATP-binding cassette A1; ABCG1—ATP-binding cassette subfamily G member 1; LPGAT1—lysophosphatidylglycerol acyltransferase-1; MTP—Microsomal triglyceride transfer protein; AGPAT—1-acylglycerol-3-phosphate-O-acyltransferase; DGAT1—Diacylglycerol O-acyltransferase 1; PGC-1α—peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SLC25A25—Solute Carrier Family 25 Member 25; NRF1—nuclear respiratory factor; TFAM: transcription factor A, mitochondrial; OxoLDL—oxidized low-density lipoprotein; ApoB—apolipoprotein B. This figure has been created by modifying the templates from Servier Medical Art (https://smart.servier.com).
; upregulation ↑; downregulation ↓. TG—triglycerides; LDL—low-density lipoprotein; ABCA1—ATP-binding cassette A1; ABCG1—ATP-binding cassette subfamily G member 1; LPGAT1—lysophosphatidylglycerol acyltransferase-1; MTP—Microsomal triglyceride transfer protein; AGPAT—1-acylglycerol-3-phosphate-O-acyltransferase; DGAT1—Diacylglycerol O-acyltransferase 1; PGC-1α—peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SLC25A25—Solute Carrier Family 25 Member 25; NRF1—nuclear respiratory factor; TFAM: transcription factor A, mitochondrial; OxoLDL—oxidized low-density lipoprotein; ApoB—apolipoprotein B. This figure has been created by modifying the templates from Servier Medical Art (https://smart.servier.com).
; upregulation ↑; downregulation ↓. TG—triglycerides; LDL—low-density lipoprotein; ABCA1—ATP-binding cassette A1; ABCG1—ATP-binding cassette subfamily G member 1; LPGAT1—lysophosphatidylglycerol acyltransferase-1; MTP—Microsomal triglyceride transfer protein; AGPAT—1-acylglycerol-3-phosphate-O-acyltransferase; DGAT1—Diacylglycerol O-acyltransferase 1; PGC-1α—peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SLC25A25—Solute Carrier Family 25 Member 25; NRF1—nuclear respiratory factor; TFAM: transcription factor A, mitochondrial; OxoLDL—oxidized low-density lipoprotein; ApoB—apolipoprotein B. This figure has been created by modifying the templates from Servier Medical Art (https://smart.servier.com).
; upregulation ↑; downregulation ↓. TG—triglycerides; LDL—low-density lipoprotein; ABCA1—ATP-binding cassette A1; ABCG1—ATP-binding cassette subfamily G member 1; LPGAT1—lysophosphatidylglycerol acyltransferase-1; MTP—Microsomal triglyceride transfer protein; AGPAT—1-acylglycerol-3-phosphate-O-acyltransferase; DGAT1—Diacylglycerol O-acyltransferase 1; PGC-1α—peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SLC25A25—Solute Carrier Family 25 Member 25; NRF1—nuclear respiratory factor; TFAM: transcription factor A, mitochondrial; OxoLDL—oxidized low-density lipoprotein; ApoB—apolipoprotein B. This figure has been created by modifying the templates from Servier Medical Art (https://smart.servier.com).

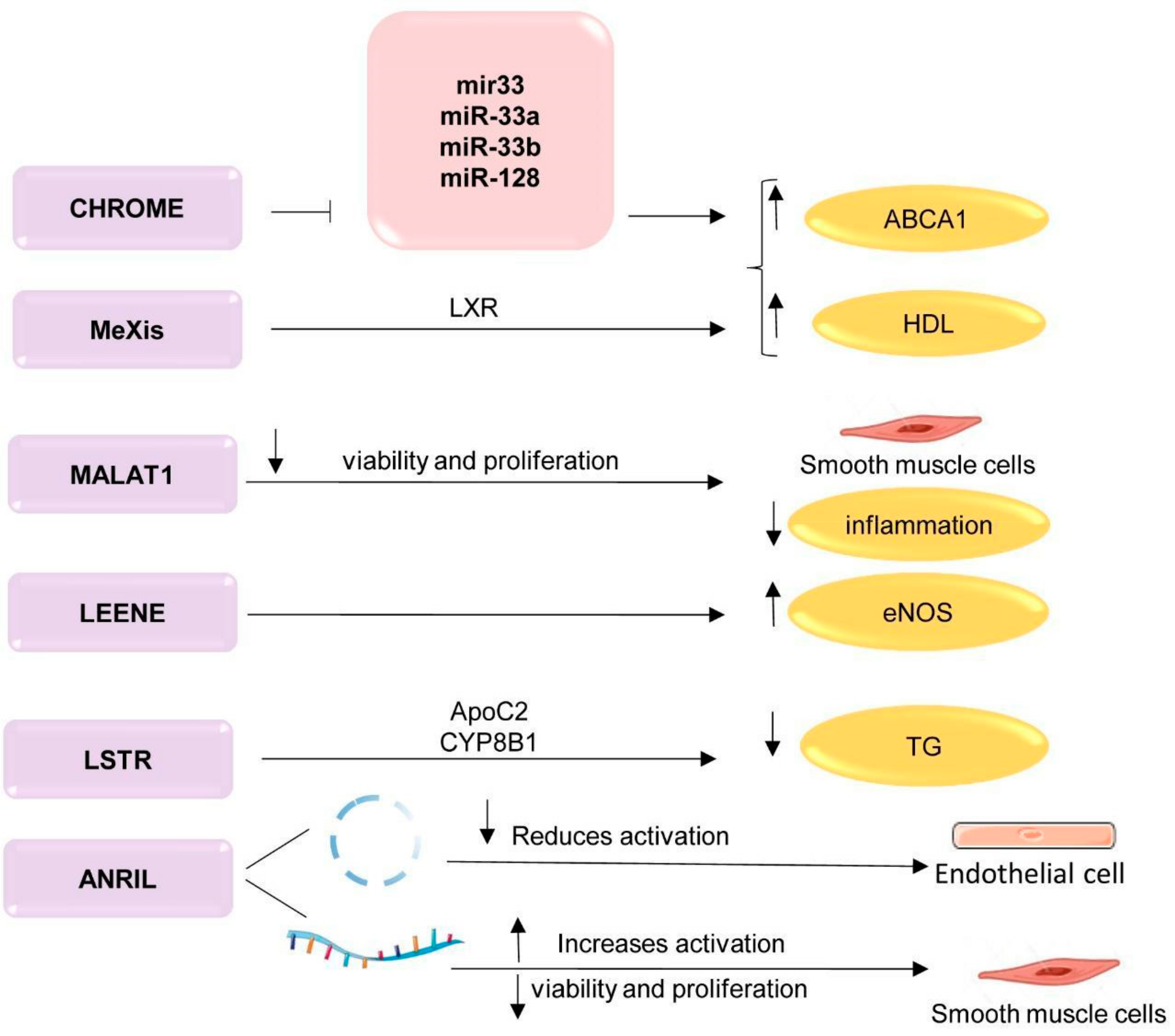
 ; upregulation ↑; downregulation ↓. TG—triglycerides; LXR—liver receptors; ApoC2—apolipoprotein C2; CYP8B1—cytochrome P450 family 8 subfamily B, polypeptide 1; ABCA1—ATP-binding cassette A; HDL—high-density lipoprotein; CHROME—cholesterol homeostasis regulator of miRNA expression; MeXis—Macrophage-expressed LXR-induced sequence; MALAT1—metastasis associated lung adenocarcinoma transcript 1; LSTR—liver specific hepatic triglyceride regulator; ANRIL antisense non-coding RNA in the INK4 locus. This figure has been created by modifying the templates from Servier Medical Art (https://smart.servier.com).
; upregulation ↑; downregulation ↓. TG—triglycerides; LXR—liver receptors; ApoC2—apolipoprotein C2; CYP8B1—cytochrome P450 family 8 subfamily B, polypeptide 1; ABCA1—ATP-binding cassette A; HDL—high-density lipoprotein; CHROME—cholesterol homeostasis regulator of miRNA expression; MeXis—Macrophage-expressed LXR-induced sequence; MALAT1—metastasis associated lung adenocarcinoma transcript 1; LSTR—liver specific hepatic triglyceride regulator; ANRIL antisense non-coding RNA in the INK4 locus. This figure has been created by modifying the templates from Servier Medical Art (https://smart.servier.com).
; upregulation ↑; downregulation ↓. TG—triglycerides; LXR—liver receptors; ApoC2—apolipoprotein C2; CYP8B1—cytochrome P450 family 8 subfamily B, polypeptide 1; ABCA1—ATP-binding cassette A; HDL—high-density lipoprotein; CHROME—cholesterol homeostasis regulator of miRNA expression; MeXis—Macrophage-expressed LXR-induced sequence; MALAT1—metastasis associated lung adenocarcinoma transcript 1; LSTR—liver specific hepatic triglyceride regulator; ANRIL antisense non-coding RNA in the INK4 locus. This figure has been created by modifying the templates from Servier Medical Art (https://smart.servier.com).
; upregulation ↑; downregulation ↓. TG—triglycerides; LXR—liver receptors; ApoC2—apolipoprotein C2; CYP8B1—cytochrome P450 family 8 subfamily B, polypeptide 1; ABCA1—ATP-binding cassette A; HDL—high-density lipoprotein; CHROME—cholesterol homeostasis regulator of miRNA expression; MeXis—Macrophage-expressed LXR-induced sequence; MALAT1—metastasis associated lung adenocarcinoma transcript 1; LSTR—liver specific hepatic triglyceride regulator; ANRIL antisense non-coding RNA in the INK4 locus. This figure has been created by modifying the templates from Servier Medical Art (https://smart.servier.com).

{kind=link}
{kind=link}
{kind=link}
| ncRNA | Impact Targets | Impact Level | Therapeutic Strategies Based on ncRNA | |
|---|---|---|---|---|
| Downregulation | Upregulation | |||
| miRNA | Regulate the expression of several genes | Repression translation degradation of mRNA | Anti-miRNA: Inhibition endogenous miRNA; miR sponges; Target site blocker; CRISPR | Mimics miRNA; re-introduction of miRNA ORN |
| siRNA | Highly specific, complementary to the target gene | Endonucleolytic cleavage of the target mRNA | – | – |
| lncRNA | Regulate the expression of several genes | Regulation of gene expression from the start of transcription to protein translation | Inhibition lncRNA: LNA-GapmeR; Short hairpin RNA CRISPR | – |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skuratovskaia, D.; Vulf, M.; Komar, A.; Kirienkova, E.; Litvinova, L. Promising Directions in Atherosclerosis Treatment Based on Epigenetic Regulation Using MicroRNAs and Long Noncoding RNAs. Biomolecules 2019, 9, 226. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9060226
Skuratovskaia D, Vulf M, Komar A, Kirienkova E, Litvinova L. Promising Directions in Atherosclerosis Treatment Based on Epigenetic Regulation Using MicroRNAs and Long Noncoding RNAs. Biomolecules. 2019; 9(6):226. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9060226
Chicago/Turabian StyleSkuratovskaia, Daria, Maria Vulf, Aleksandra Komar, Elena Kirienkova, and Larisa Litvinova. 2019. "Promising Directions in Atherosclerosis Treatment Based on Epigenetic Regulation Using MicroRNAs and Long Noncoding RNAs" Biomolecules 9, no. 6: 226. https://0-doi-org.brum.beds.ac.uk/10.3390/biom9060226