
The Role of BMP Signaling in Osteoclast Regulation

Department of Biological Sciences, University of Delaware, Newark, DE 19716, USA
*
Authors to whom correspondence should be addressed.
J. Dev. Biol. 2021, 9(3), 24; https://0-doi-org.brum.beds.ac.uk/10.3390/jdb9030024
Submission received: 23 April 2021
/
Revised: 2 June 2021
/
Accepted: 18 June 2021
/
Published: 28 June 2021
(This article belongs to the Special Issue Feature Papers in Journal of Developmental Biology II)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The osteogenic effects of Bone Morphogenetic Proteins (BMPs) were delineated in 1965 when Urist et al. showed that BMPs could induce ectopic bone formation. In subsequent decades, the effects of BMPs on bone formation and maintenance were established. BMPs induce proliferation in osteoprogenitor cells and increase mineralization activity in osteoblasts. The role of BMPs in bone homeostasis and repair led to the approval of BMP2 by the Federal Drug Administration (FDA) for anterior lumbar interbody fusion (ALIF) to increase the bone formation in the treated area. However, the use of BMP2 for treatment of degenerative bone diseases such as osteoporosis is still uncertain as patients treated with BMP2 results in the stimulation of not only osteoblast mineralization, but also osteoclast absorption, leading to early bone graft subsidence. The increase in absorption activity is the result of direct stimulation of osteoclasts by BMP2 working synergistically with the RANK signaling pathway. The dual effect of BMPs on bone resorption and mineralization highlights the essential role of BMP-signaling in bone homeostasis, making it a putative therapeutic target for diseases like osteoporosis. Before the BMP pathway can be utilized in the treatment of osteoporosis a better understanding of how BMP-signaling regulates osteoclasts must be established.
1. Role of BMP in Bone Formation
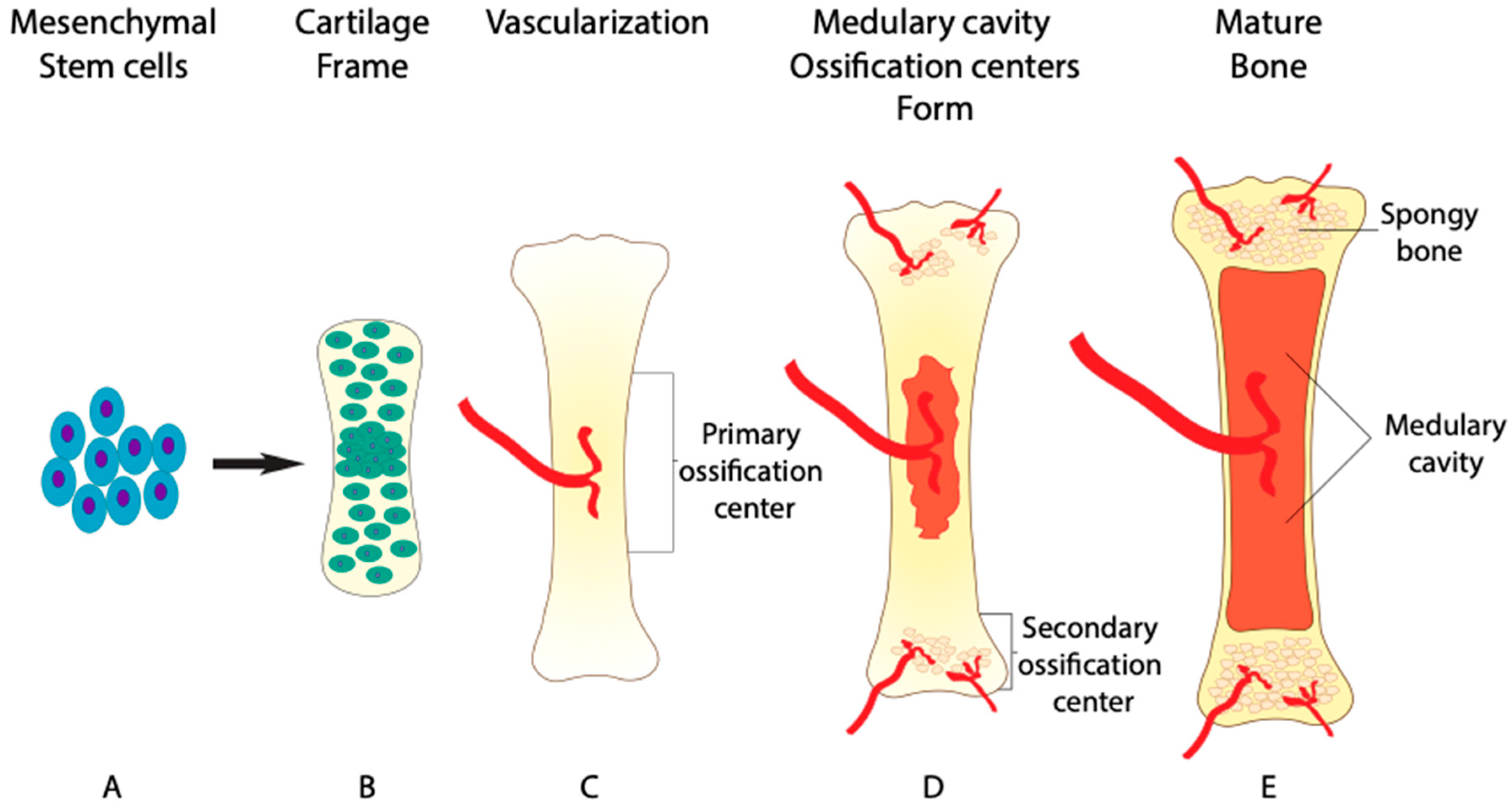
Bone is formed in two major ways: endochondral ossification and intramembranous ossification. The majority of the human skeleton is formed by endochondral ossification, excluding the flat bones of the skull, the mandible, and the clavicle [1]. Endochondral ossification begins during week six and seven of embryonic development with the differentiation and condensation of mesenchymal stem cells into chondrocytes [2]. Chondrocytes then lay down a framework for skeletal elements that is later mineralized by osteoblasts and remodeled by osteoclasts (Figure 1A,B). The initial mineralization takes place in the primary ossification center near the middle region of the skeletal element, known as the diaphysis (Figure 1C). The mineralization and elongation of the skeletal element remains active after birth until 25 years of age [2].
Each stage of endochondral bone formation is regulated by BMPs, including the activity of osteoclasts. In the initial stages of bone formation, BMP2 and BMP7 stimulate the proliferation and differentiation of mesenchymal stem cells into chondrocytes [3,4]. Later, the condensation process of chondrocytes is regulated by BMP-signaling transmitted through the phosphorylation and activation of SMADs [5,6]. Then, BMP-ERK1/2-signaling induces the proliferation and maturation of the chondrocyte condensation, encouraging these cells to grow into a proper framework for eventual bone formation through the regulation of Indian hedgehog (IHH) and parathyroid hormone (PTH) expression gradients [5,7,8]. Disruption in BMP-signaling during chondrocyte condensation results in deformed cartilage growth zones and short limb phenotypes [9].
After the framework of chondrocytes is formed, blood vessels invade the cartilage through a process called angiogenesis (Figure 1C). BMP-signaling further regulates this process, as the absence of the BMP receptor type 1a (BMPRIa) gene in vivo leads to impaired angiogenesis [10]. Once the blood vessels have penetrated the established cartilage, they bring in mature osteoblasts and osteoclasts, thus beginning the mineralization process [11,12,13]. Additionally, up to 60% of mature chondrocytes also transdifferentiate into osteoblasts themselves, adding to the mineralization of the extracellular matrix surrounding chondrocytes [14]. The transdifferentiation of chondrocytes is regulated by essential factor, Runt-related transcription factor 2 (RUNX2), that acts down stream of BMP-signaling [15]. Without proper of BMP-signaling chondrocyte transdifferentiation is lost in BMPRIa knockout mice [16].
While the mineralization process takes place, osteoclasts work to remodel the inner bone tissue to extend a network of blood vessels. (Figure 1D). The network of blood vessels within bone tissue is so extensive that it receives around 10% of the total cardiac output [17,18]. Due to this circulation of blood containing osteoclasts and osteoblasts, bone is capable of remodeling its structure and repairing damage, unlike avascular cartilage [19]. Once the primary ossification center has formed, secondary ossification centers develop at either end of long bones within the epiphysial plates, effectively shutting down the growth zones by age 25, preventing further elongation of skeletal elements [2,20]. The first remodeling process occurs at these ossification centers as osteoclasts resorb bone in the diaphysis. As a result, osteoclasts form a space for bone marrow to be stored, known as the medullary cavity (Figure 1E) [21]. The administration of BMP2 into the medullary cavity results in an initial increase in bone formation that is later lost [22]. The loss of bone density is attributed to the increase in absorption activity of osteoclast, indicating a dual role of BMP2 in bone homeostasis. With the emerging role of BMP-signaling in osteoclast differentiation, the same focus given to osteoprogenitors should be given to osteoclast progenitors. This is highlighted by the BMP-dependent ERK1/2 signaling in both osteoprogenitors and osteoclast progenitors.
Furthermore, it is has been established that ERK1/2 phosphorylation leads to an increase in mesenchymal stem cell proliferation [23,24,25]. However, activation of ERK1/2 also leads to an increase in proliferation of preosteoclasts [26,27,28]. Regulation of osteoclast differentiation then helps bone remolding at the secondary ossification centers. Osteoclast resorption at the epiphysis creates a complex microstructure important for support and shock absorption.
2. The Role of BMP and Osteoclasts in Maintaining Bone Microstructure
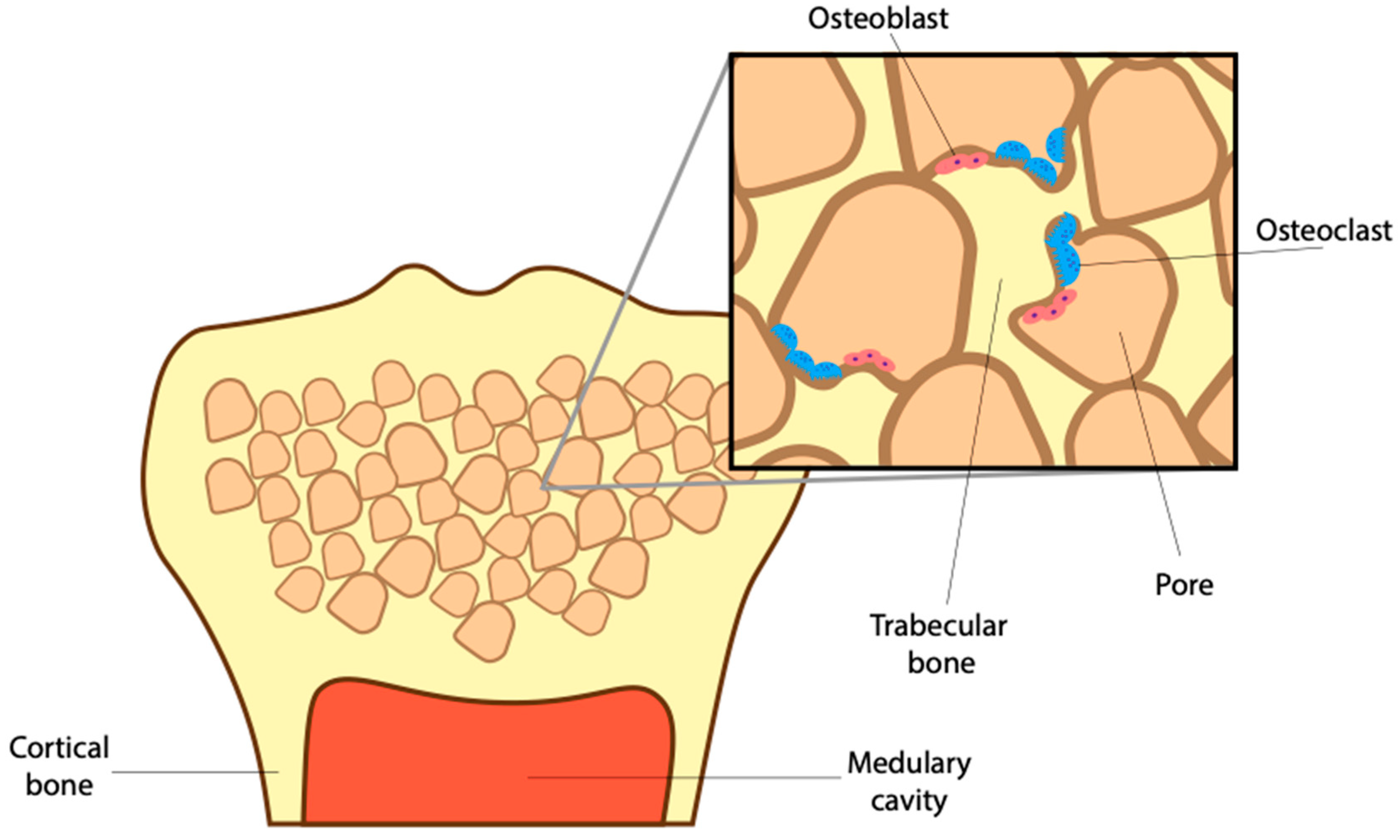
To avoid fractures, bone cells must maintain a healthy density while sustaining a complex supporting microstructure. That includes the trabecular network of spongy bone and surrounding cortical bone (Figure 2). To sustain a healthy bone structure, the mature skeleton is constantly remodeling, breaking down old worn-out bone and producing new bone in its place. The continued activity of osteoblasts and osteoclasts results in the complete turnover of an adult skeleton roughly every ten years [29]. This turnover is important, allowing for the release of growth factors, like BMPs, as well as calcium and phosphorus trapped in the mineralized matrix [30,31,32]. BMP-signaling helps maintain this healthy bone density by stimulating osteoblast mineralization, differentiation and survival [33]. However, it has become clear that BMPs not only aid in the anabolic process of bone turnover but also the catabolic processes [34]. To that end, the loss of the BMP receptor type 1 a (BMPRIa) increased trabecular bone thickness due to a decrease in osteoblast activity and a larger decrease in osteoclast activity [35]. Without proper BMP-signaling, the imbalance between osteoblast and osteoclasts results in reduced bone quality.
Healthy bone requires a suitable microstructure, organized to support weight and absorb shock. Beyond the formation of the medullary cavity, osteoclasts play a vital role in creating and retaining important porous structures in long bones (Figure 2) [21]. These structures make up trabecular bone, a weaving network of pores filled with red bone marrow and adipose tissue. The storage of marrow here is essential as it contains hematopoietic stem cells that give rise to many cell types, including red blood cells, immune cells, and osteoclasts [36]. Remarkably, the complex arrangement of trabecular bone that surrounds these pores provides structural integrity to withstand and transfer shock from falls and daily strain [37,38]. This means that the number of trabecular connections, as well as their thickness, needs to be tightly regulated to allow for the storage of marrow as well as shock absorption.
Surrounding the trabecular network is cortical bone or, the bone collar. Upon impact, trabecular bone transfers shock to the cortical bone, which is thick yet flexible, allowing for further shock absorption. To maintain proper trabecular connections and cortical thickness, osteoblasts create new bone and osteoclasts, in turn, resorb old bone (Figure 2). The balance between osteoclast and osteoblasts is regulated by BMPs. As a result, the loss of osteoblastic BMP2 causes a decrease in cortical bone thickness resulting in weaker long bones [39]. Beyond this, the effect of altered BMP-signaling in osteoblasts and the corresponding change in bone quality is well established [40,41,42]. While the effect of altered BMP-signaling in osteoclast differentiation and their impact on bone microstructure is underappreciated.
3. The Role of BMP in Stages of Osteoclast Differentiation
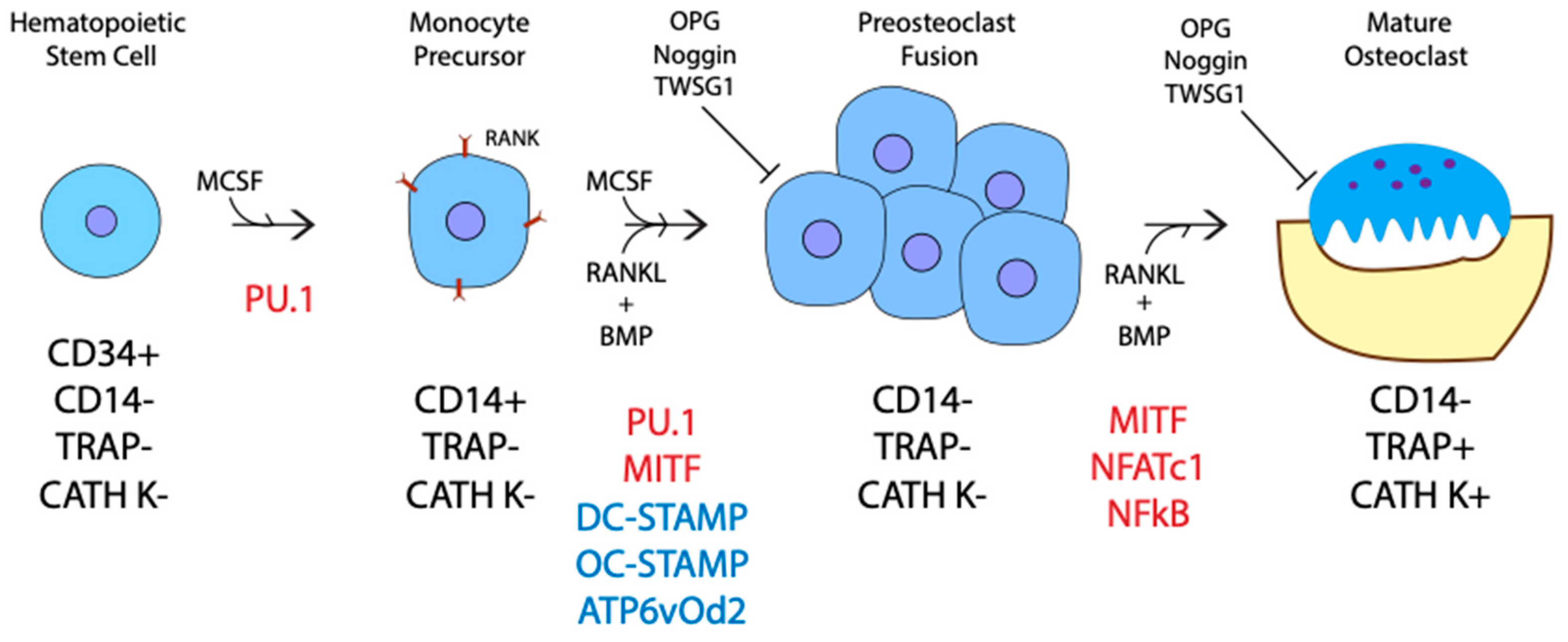
Originating from hematopoietic stem cells within the bone marrow, osteoclasts go through several stages of differentiation before becoming mature and active [43,44]. First, CD34+ hematopoietic stem cells differentiate into CD14+ monocytes [45]. Then, the monocytes differentiate into mononuclear preosteoclasts, which fuse to form polykarons or immature/inactive osteoclasts that are CD14- [46,47]. Lastly, the multinucleated polykaryons become active osteoclasts producing acid to break down calcified bone in absorption pits under their ruffled borders called Howship lacunae (Figure 3). These stages of differentiation are driven by a few critical factors: Macrophage Colony Stimulating Factor (M-CSF), Receptor Activator Nuclear Factor kb Ligand (RANKL), and BMPs. The roles of both RANKL and M-CSF in osteoclast differentiation were well studied, while the requirement of BMP-signaling is underappreciated [48,49,50,51,52]. Recent studies on the inhibition of BMP-signaling confirm that BMPs play a major role in RANKL-mediated osteoclast differentiation, proliferation, fusion, and survival [36,53,54,55,56,57,58,59,60,61]. The addition of M-CSF begins the initial differentiation of hematopoietic stem cells. M-CSF upregulates PU.1, a transcription factor, which in turn induces the expression of Receptor Activator of Nuclear Factor kappa-B (RANK) on the membrane of osteoclast precursors (Figure 3) [62,63]. In addition to M-CSF, BMP4 also helps to upregulate PU.1 transcription initiating differentiation [64]. A combined signaling pathway between M-CSF and RANK then upregulates genes required for preosteoclast fusion, including the master fusion regulators DC-STAMP, OC-STAMP, and ATP6v0d2 [65,66,67]. The expression of DC-STAMP and ATP6v02 are regulated by an essential transcription factor, NFATc1, which is activated downstream of RANKL and BMP-signaling [68,69]. BMP inhibition leads to a decrease in DC-STAMP, ATP6v02, and NFATc1 gene expression [55,61,70,71,72]. As a result, fewer, smaller, and less active osteoclasts form, showing the requirement of BMP-signaling in preosteoclast fusion [55,57,59,72]. Furthermore, BMP2 and NFATc1 are also required in immature osteoclasts for the proper induction of osteoclast specific genes as BMP2 promotes the nuclear translocation of NFATc1, which in turn upregulates the expression of TRAP and CATH K [69,73,74,75,76,77,78]. It is currently thought that BMPs only enhance RANKL mediated osteoclastogenesis. However, a study utilizing a soluble form of BMPRIa in bone marrow macrophages showed that in the absence of BMP-signaling, RANKL alone was not efficient in generating osteoclasts. Therefore, BMP-signaling may be required for RANKL-mediated osteoclastogenesis [54].
Several inhibitors are also known to regulate RANKL and BMP-driven osteoclastogenesis [55,61,72,79,80,81,82]. The most well-known inhibitor, osteoprotegerin (OPG), inhibits RANKL by binding to the ligand, preventing RANK-signaling. As such, an increase in OPG concentration may cancel out any increase in RANKL production. Like OPG, BMP inhibitors Twisted gastrulation 1 (TWSG1) and Noggin reduce BMP-mediated osteoclastogenesis [55,61,71,72]. This points to a more significant role for BMPs in osteoclast regulation and communication.
4. The Communication of Osteoclasts with Bone and Immune Cells
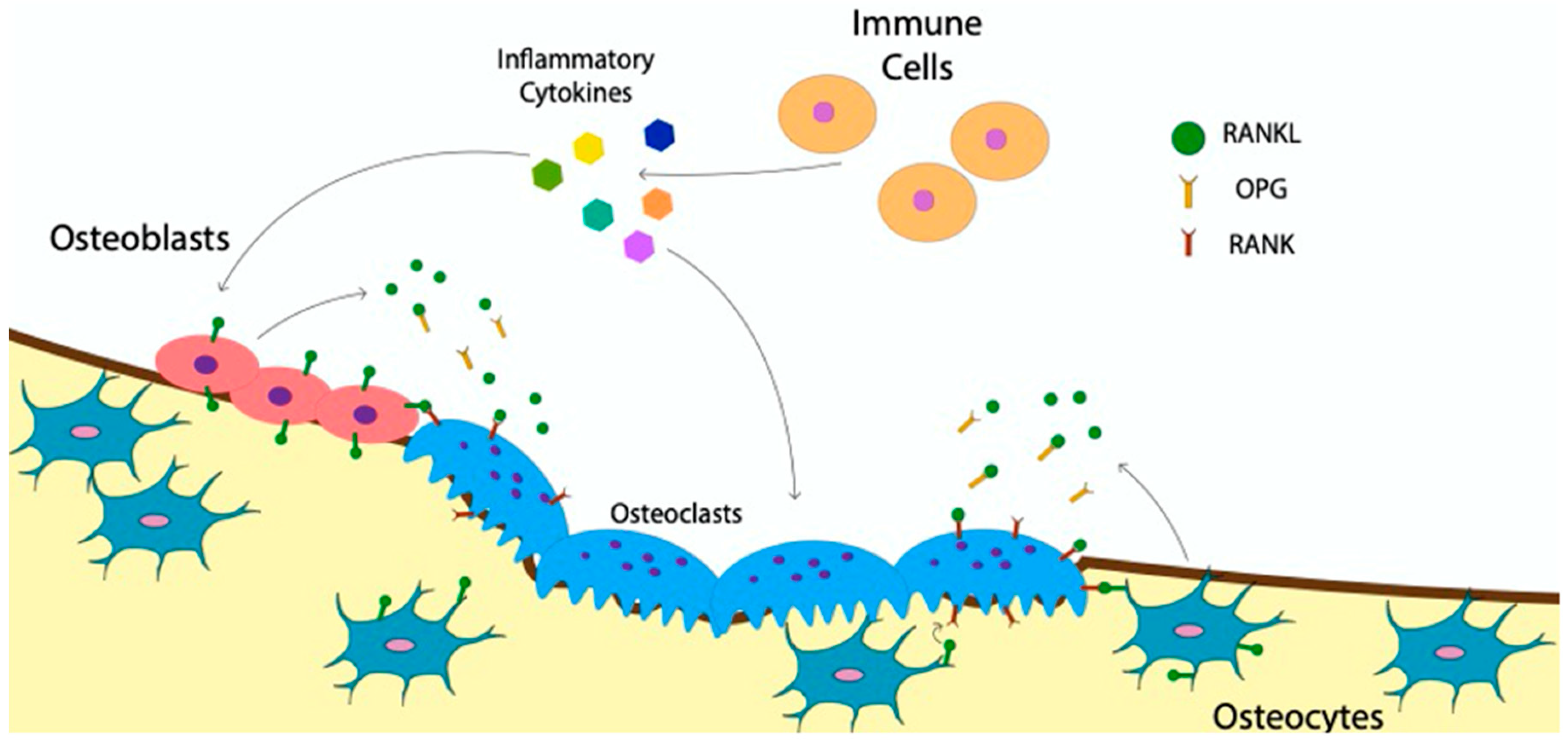
The catabolic activity of osteoclasts and the anabolic activity of osteoblasts are tightly regulated to ensure a healthy bone structure. For instance, in the bone disease, osteopetrosis, too little resorption reduces bone turnover, causing an accumulation of older more fragile bone [83]. While in the bone disease, osteoporosis, too much bone resorption leads to a decrease in bone density and higher risk of fractures [84]. To understand how osteoclast activity is regulated, the communication of immune cells and other bone cells with osteoclasts must be understood. The RANKL:OPG ratio is the most well-researched avenue utilized to regulate osteoclasts [85,86,87,88,89]. Many cell types, including immune cells, manipulate the RANKL:OPG ratio by stimulating osteoclast differentiation and or increasing their activity [90,91,92]. RANKL is an essential ligand in driving osteoclastogenesis that is inhibited when bound to OPG [81]. By producing RANKL, immune cells directly regulate the RANKL:OPG ratio and increase osteoclast activity [93]. In addition to this a growing body of research, immune cells also regulate osteoclasts through the release of inflammatory cytokines, both directly and indirectly, by altering the RANKL production in osteoblasts (Figure 4) [42,94,95,96]. As such, proinflammatory cytokines such as Tumor Necrosis Factor-alpha (TNF-α), Interleukin 6 (IL-6), Interleukin 23 (IL-23), and interleukin 1 beta (IL-1β) released by immune cells, including macrophages and dendritic cells, are major contributors to osteoclast activation and differentiation [64,83,94,95,97,98,99,100,101]. Immune cells also inhibit osteoclasts through the release of cytokines such as Interleukin 4 (IL-4), Interleukin 18 (IL-18), Interleukin 33 (IL-33) and Interferon (IFN) [99,100,101,102,103,104]. The impact of these inflammatory cytokines is crucial in regulating osteoclasts after a fracture [105].
Bone cells such as osteocytes and osteoblasts also communicate with osteoclasts to modulate their activity and differentiation. Osteocytes, which are mature osteoblasts that have become embedded within bone, signal osteoclasts through direct cell-to-cell contact. Osteocytes do so by extending a dendritic process with membrane-bound RANKL to the bone surface to interact with membrane bound RANK on osteoblasts (Figure 4) [106,107]. Osteocytes also secrete soluble RANKL to stimulate osteoclasts, but to a lesser extent (Figure 4) [107]. Additionally, osteoblasts communicate with osteoclasts through paracrine signaling or gap junctions, similar to osteocytes [80,108]. Whether osteocytes or osteoblasts are the main sources of RANKL for the regulation of osteoclast activity and differentiation is not yet established [109,110]. Nevertheless, both cell types play a critical role in communicating with osteoclasts to maintain bone homeostasis. The crosstalk between immune cells, osteoblasts and osteoclasts creates a network of communication. Ultimately resulting in the regulation of osteoclast differentiation and activity required to maintain bone homeostasis.
Indirect Effect of BMP-Signaling on Osteoclast Communication
Osteoblasts and osteoclasts are in constant communication, recruiting and stimulating each other to maintain proper bone homeostasis and repair bone damage. They do so by sending cytokines via paracrine signaling and contact with membrane-bound ligands [60,106,107]. The roles of RANKL, OPG, and inflammatory cytokines were well studied in their communication with osteoclasts [111,112,113]. However, the importance of BMP-signaling in the communication and regulation between osteoclasts and osteoblasts is underappreciated. Osteoclasts lacking BMPRIa caused an increase in osteoblast activity by increasing gap junction communication with osteoblasts [60]. Inversely, osteoblasts lacking BMPRIa caused a reduction in osteoclastogenesis due to a subsequent reduction in sclerostin, a glycoprotein that stimulates the release of RANKL in osteocytes [114,115,116]. The release of BMP6 by osteoclasts also recruits and stimulates osteoblasts, promoting mineralization [117,118,119,120]. Furthermore, a reservoir of growth factors including BMP2, BMP4, and BMP 7 are stored in bone tissue. Osteoclasts release these stored growth factors via bone resorption [121,122,123]. The release of these factors thereby stimulates osteoblasts. Stimulated osteoblasts in turn release factors that regulate osteoclast differentiation and function, including M-CSF, which is required to drive hematopoietic stem cells into the osteoclast lineage [76,124]. Furthermore, BMP2 and 4 alter the RANKL:OPG ratio by modifying the levels of either RANKL or OPG released from osteoblasts [54,70,86,88]. The conditional knockout of BMPRIa in osteoblasts decreased the RANKL:OPG ratio [114]. This establishes the indirect effect of BMP-signaling in regulating the RANKL:OPG ratio. The recruitment of osteoblasts to a fracture site highlights the significance of this communication. When a fracture occurs, osteoclasts come in to resorb the damaged bone and then recruit osteoblasts to lay down new bone at the site of injury through the release of growth factors including, BMP2, BMP4, BMP6 and 7 [69,121,125]. In the absence of BMP2 fracture healing does not begin, highlighting the requirement for its release during bone resorption [126]. Beyond indirect effects of BMPs, a growing body of studies indicates that BMPs have more than just an indirect effect on osteoclasts [53,54,55,56].
5. Direct Effect of BMP-Signaling in Osteoclasts
First, it was determined that purified osteoclasts and osteoclast precursors express BMP receptors. Then, it was established that BMP2 directly stimulates osteoclast precursors with the help of M-CSF by Kanatani et al., 1995, thus suggesting a greater role for BMPs in bone homeostasis. This was also supported by the increased expression of the preosteoclast fusion protein, DC-STAMP, upon BMP2 stimulation [55]. In addition to BMP2, BMP4 and BMP6 also stimulate osteoclasts, but to a lesser degree [56,57,59,127]. To elucidate the potential direct effect of BMPs on osteoclasts, the BMP pathway was disrupted by Twisted gastrulation 1 (TWSG1) and Noggin, a BMP antagonist, resulting in the inhibition of osteoclast differentiation [55,61,70,71,72]. Later, the conditional knockout of the BMPRIa receptor resulted in a decrease in osteoclastogenesis and DC-STAMP expression [58]. Furthermore, the deletion of BMPRIa in vivo led to an increase in trabecular bone and bone density, along with a decrease in osteoclast activity and osteoclast differentiation [35,58,128]. A deletion of BMPRIb also showed an increase in proliferation of preosteoclasts and increase differentiation while decreasing resorption [129]. Additionally, the deletion of BMPR2 decreased osteoclast differentiation and activity [53]. Taken together, BMP-signaling plays a critical role in not only osteoblasts but also the proper differentiation of osteoclasts, making it an ideal target for future therapeutic treatments.
6. BMP-Signaling Pathway in Osteoclasts
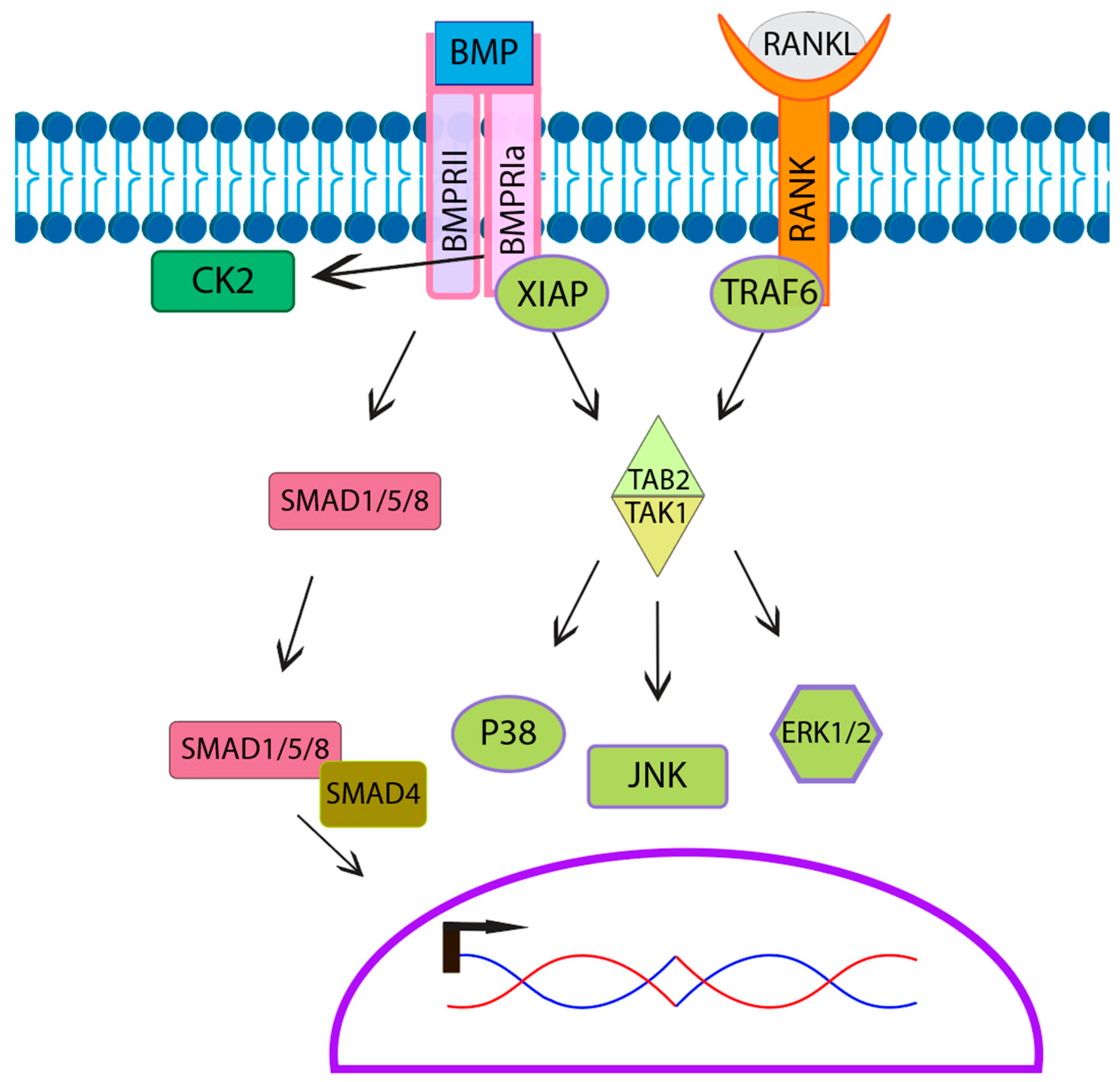
BMPs signal either through the canonical or noncanonical pathways. The canonical pathway is also known as the SMAD signaling pathway. The SMAD pathway involves a few different types of SMADs including R-SMADs (SMAD1/5/8), which are receptor-regulated SMADs, and Co-SMADs such as SMAD 4 that bind to R-SMADs, facilitating nuclear translocation for signal transduction (Figure 5) [130]. Multiple studies established that osteoclasts express SMADs as well as phospho-active SMADs [53,54,55,131,132]. Furthermore, research suggests that BMP-mediated SMAD signaling may play a role in osteoclast fusion and activation, as inhibition of SMAD leads to smaller and less active osteoclasts [53,69,132,133]. However, SMAD signaling is unaffected in BMPRII knockout in osteoclasts, suggesting that BMPs primarily signal through the noncanonical pathway in osteoclasts [53,131]. The noncanonical signaling pathway consists of mitogen-activated protein kinase MAPK downstream signaling molecules including c-Jun N-terminal kinase (JNK), mitogen-activated protein kinase 38 alpha (p38α), and extracellular regulated kinases (ERK), all of which are activated by BMP2 in osteoclasts [53,131]. TGF-β Activated Kinase 1 (TAK1), an upstream signaling molecule of the noncanonical signaling pathway, is required for osteoclast differentiation. The specific knockout of TAK1 in osteoclasts leads to an osteopetrosis-like phenotype with decreased resorptive activity [134]. Furthermore, while not shown in osteoclasts, X-linked inhibitor of apoptosis protein (XIAP) associates with both BMPRIa and TGF-β Activated Kinase 1 binding protein (TAB1) [135]. TAB1 then forms a complex with TGF-β Activated Kinase 1 binding protein 2 (TAB2) associating with and activating TAK1. This indicates that the BMP/BMPR-XIAP-TAB1/TAB2-TAK1 pathway allows for BMPs to stimulate the noncanonical pathway including MAPK’s JNK, p38, and ERK in osteoclasts. Further, this overlaps with the RANK signal transduction, which also uses the MAPK pathway, activating downstream the molecules JNK, p38, and ERK [27,136]. The difference in RANK signaling is that Tumor Necrosis Factor Receptor-associated Factor Protein 6 (TRAF6) rather than XIAP recruits TAB1/TAB2 to the RANK receptor, activating TAK1. MAPK signaling converges to increase the expression and translocation of NFATc1a, an essential transcription factor that leads to the upregulation of osteoclast genes for fusion and activation [69,73,137,138,139,140] This overlap in MAPK signaling between the RANKL pathway and the BMP pathway may contribute to the enhancement of osteoclastogenesis (Figure 5).
7. The Role of Osteoclasts in Bone Disease
Diseases impact the health of bone by altering the microstructure and overall bone mineral density. These diseases primarily alter the function and balance of osteoblasts and osteoclasts. Two major diseases that impact bone density are osteopetrosis and osteoporosis. These diseases are two sides of the same coin, as they both impact osteoblasts and osteoclasts inversely. Osteopetrosis occurs when either osteoblast activity is increased or osteoclast activity is decreased, causing an imbalance in bone maintenance and an increase in bone mineral density. Osteopetrosis causes an increased risk of fracture as the trabecular network is disrupted [141]. Further ossification also leads to the shrinking of the medullary cavity reducing stem cell storage [142]. This leads to immune disruption and inhibition of healing [143]. Conversely, in osteoporosis, osteoblast activity is decreased, and osteoclast activity is increased. This imbalance favors bone resorption, causing a decrease in bone mineral density. The trabecular number and thickness decreases within spongy bone, resulting in a high risk of fracture [84,144,145]. Osteoporosis is one of the most prevalent bone diseases in the world, affecting 37% of women and 20% of men over the age of 50 [146]. The most common cause of osteoporosis is the onset of menopause, brought about primarily by a drop in estrogen in women [21,147]. The decrease in estrogen expression leads to an increase in the RANKL:OPG ratio osteoclastogenesis, promoting bone resorption [148]. Additionally, the onset of menopause decreases the BMP antagonist, inhibin A, stimulating both osteoblastogenesis and osteoclastogenesis. This suggests a dual role for BMP-signaling in bone metabolism of post-menopausal women [149].
As the aging population increases, a higher proportion of people are affected by osteoporosis for a greater portion of their lives [150,151]. The longer an individual suffers from osteoporosis, the more likely they are to experience fractures. Over 50% of the people that experience osteoporotic fractures cannot continue to live independently and more than 25% will die within 12 months after their injury [152,153,154]. Furthermore, the International Osteoporosis Foundation found that the risk of fracture from osteoporosis is such that one in every three women and one in every five men across the globe will experience an osteoporotic-related fracture. This results in a substantial annual medical cost with an estimate of 16 billion dollars spent in the United States in 2002 alone [155]. Moreover, the European Union spent more than 37 billion euros on osteoporosis-related treatment in 2010 [156]. Clearly, an effective long-term treatment for this disease is desperately needed worldwide.
8. Treatments for Osteoporosis
One of the early treatments developed for osteoporosis was hormone replacement therapy [157,158]. As the most common cause of osteoporosis is the post-menopausal loss of estrogen, the hope for this treatment was that, by replacing the lost hormones, they could restore homeostasis of bone regulation. Unfortunately, this treatment showed an increased risk of heart attacks, breast cancer, and strokes, causing it to discontinue [159,160]. The use of BMP2 has also been considered for treatment of osteoporosis due to its osteogenic effects post ALIF surgery [161,162,163,164]. However, increased resorption activity has been associated with this procedure, [165,166]. A total of 12 months after treatment, ALIF patients showed increased graft subsidence over patients that did not receive treatment with BMP 2 [166]. Additionally, treatment of osteoblasts isolated from patients with osteoporosis with BMP 2 showed no osteogenic response compared to osteoblasts derived from osteoarthritic patients [167]. As a result, the use of BMP2 for the treatment of osteoporosis is still under consideration. However, the dual impact on bone formation and bone resorption makes the BMP pathway a potential therapeutic target for osteoporosis.
Currently, the most popular treatment for osteoporosis is bisphosphonates. Bisphosphonates increase bone mineral density by inhibiting the mevalonate pathway, inducing apoptosis, and reducing osteoclast resorption [168,169,170,171]. Despite their popularity, they come with various side-effects, such as the increased risk of esophagus cancer, intestinal irritation, and osteonecrosis of the jaw [172,173,174]. Doctors may recommend taking a break from bisphosphonates after five years to reduce the risk of these side effects. Another antiresorptive drug called Denosumab is an alternative for bisphosphonates, acting as a RANKL inhibitor [175]. Denosumab blocks RANK signaling by binding to its ligand and preventing it from binding to the receptor on osteoclasts; therefore, reducing resorptive activity and increasing bone mineral density [175,176]. In opposition to antiresorptive drugs, other treatments have sought to increase the anabolic process of bone homeostasis by stimulating osteoblasts. One such new anabolic peptide, called p-PTH 1-34 (Teriparatide), stimulates osteoblast activity to increase bone mineralization, was recently approved for the treatment of osteoporosis [177,178,179]. This is a peptide derived from PTH, a known anabolic regulator of bone formation [180,181]. However, Teriparatide increased the risk of hypercalcemia [180,182]. Furthermore, long-term treatment causes an increase in RANKL production within osteoblasts, causing an indirect increase in bone resorption by osteoclasts [177].
Two potential therapeutics that have utilized the dual anabolic and antiresorptive properties include a sclerostin antibody, namely Romosozumab, and a Casein Kinase 2 (CK2), inhibitor (CK2.3). Sclerostin is a known inhibitor of osteoblast activity, released by osteoclasts to reduce anabolic activity [115,183,184]. Romosozumab is an antibody inhibiting sclerostin from binding to its Wnt receptor, thereby removing sclerostin’s negative effect on osteoblasts. Remarkably, Romosozumab not only stimulates osteoblasts, but it also inhibits osteoclasts [185]. This antiresorptive effect is likely due to increased OPG production, produced downstream of the Wnt pathway. However, OPG is not always increased after treatment with other sclerostin inhibitors, thus a separate mechanism may be responsible for the antiresorptive effect of Romosozumab [186]. Furthermore, the effect of sclerostin inhibitors is attenuated in osteoblasts, likely due to decreased proliferation in osteoprogenitor cells [187]. Furthermore, Romosozumab increased the risk of cardiac events and should be closely monitored for extended treatment [188]. Overall, Romosozumab is a potentially valuable alternative to bisphosphonates.
The use of BMP2 has also been considered for the treatment of osteoporosis due to its osteogenic effects post ALIF surgery [48,161,163]. However, increased resorption activity has been associated with this procedure, [165,166]. A year after BMP2 treatment ALIF patients showed increased graft subsidence over patients that did not receive surgery with BMP2 treatment [166]. Additionally, BMP2 stimulated osteoblasts isolated from patients with osteoporosis showed no osteogenic response when compared to osteoblasts derived from osteoarthritic patients [167]. As a result, the use of BMP 2 for the treatment of osteoporosis is still under investigation. Nevertheless, the dual impact on bone formation and bone resorption makes the BMP pathway a potential therapeutic target for osteoporosis. The peptide CK2.3 utilizes the BMP pathway in the absence of a BMP ligand to stimulate osteoblast mineralization and inhibit osteoclast resorption [140,189,190,191,192]. Furthermore, CK2.3 increases mineralization in osteoporotic patients where BMP2 is not [167]. While the effects of CK2.3 are still being analyzed, it offers a new approach to the use of BMP-signaling in the treatment of osteoporosis.
9. Conclusions and Future Directions
BMP-signaling is essential for proper bone formation and maintenance. BMPs regulate the condensation of chondrocytes in endochondral ossification as well as the transdifferentiation and activation of osteoblastic bone mineralization. Improper BMP-signaling results in short limb phenotypes, highlighting its importance during skeletal development. Mature bone is also regulated by BMP-signaling, as it maintains a balance of anabolic and catabolic activity between osteoblasts and osteoclasts. The requirement for BMP-signaling in osteoblasts has been established as a potent driver of bone mineralization. However, increasing evidence points to an additional role BMP-signaling in bone resorption. This is supported by an increase in trabecular bone and bone density in BMPR1a knockout models, attributed to a decrease in osteoblast activity and a more significant decrease in osteoclast differentiation and activity [35,58,128]. Despite this, the research on BMPs has primarily focused on osteoblasts.
The dual role of BMP-signaling in bone homeostasis presents it as a potential treatment for Osteoporosis. As osteoporosis is the result of increased resorption and decreased mineralization of bone, a treatment that can impact both osteoblasts and osteoclasts is imperative. However, the majority of currently available treatments focus on just one side of bone metabolism. This presents a problem for long-term treatment as osteoblasts and osteoclasts are in constant communication, regulating one another to maintain suitable bone mineral density. A problem that BMP2 treatment itself faces. Treatment with BMP2 in spinal fusion surgeries results in increased bone mineralization followed by an increase in osteoclast absorption. To utilize the dual effect of the BMP-signaling pathway a treatment called, CK2.3 was created. CK2.3 stimulates the BMP pathway in the absence of BMP2, resulting in the stimulation of osteoblast and inhibition of osteoclasts. However, more research is still needed to determine its effectiveness for the treatment of osteoporotic patients. With the emerging role of BMP-signaling in bone resorption, a greater focus should be given to its impact on osteoclast regulation.
Funding
This research was funded NIH NIAMS grant 1R21AR076689—01A1.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank Daniel Halloran for advice edits and discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goldring, M.B.; Tsuchimochi, K.; Ijiri, K. The control of chondrogenesis. J. Cell. Biochem. 2006, 97, 33–44. [Google Scholar] [CrossRef]
- Breeland, G.; Sinkler, M.A.; Menezes, R.G. Embryology, Bone Ossification. In StatPearls; StatPearls Publishing Copyright ©: Treasure Island, FL, USA, 2021. [Google Scholar]
- Cicione, C.; Muiños-López, E.; Hermida-Gómez, T.; Fuentes-Boquete, I.; Díaz-Prado, S.; Blanco, F.J. Alternative protocols to induce chondrogenic differentiation: Transforming growth factor-β superfamily. Cell Tissue Bank. 2015, 16, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Pountos, I.; Georgouli, T.; Henshaw, K.; Bird, H.; Jones, E.; Giannoudis, P.V. The effect of bone morphogenetic protein-2, bone morphogenetic protein-7, parathyroid hormone, and platelet-derived growth factor on the proliferation and osteogenic differentiation of mesenchymal stem cells derived from osteoporotic bone. J. Orthop. Trauma 2010, 24, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Li, T.F.; Yukata, K.; Yin, G.; Sheu, T.; Maruyama, T.; Jonason, J.H.; Hsu, W.; Zhang, X.; Xiao, G.; Konttinen, Y.T.; et al. BMP-2 induces ATF4 phosphorylation in chondrocytes through a COX-2/PGE2 dependent signaling pathway. Osteoarthr. Cartil. 2014, 22, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Tu, X.; Choi, K.; Akiyama, H.; Mishina, Y.; Long, F. BMP-Smad4 signaling is required for precartilaginous mesenchymal condensation independent of Sox9 in the mouse. Dev. Biol. 2015, 400, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Grimsrud, C.D.; Romano, P.R.; D’Souza, M.; Puzas, J.E.; Schwarz, E.M.; Reynolds, P.R.; Roiser, R.N.; O’Keefe, R.J. BMP signaling stimulates chondrocyte maturation and the expression of Indian hedgehog. J. Orthop. Res. 2001, 19, 18–25. [Google Scholar] [CrossRef]
- Shu, B.; Zhang, M.; Xie, R.; Wang, M.; Jin, H.; Hou, W.; Tang, D.; Harris, S.E.; Mishina, Y.; O’Keefe, R.J.; et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci. 2011, 124, 3428–3440. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Lian, N.; Li, L.; Moss, H.E.; Wang, W.; Perrien, D.S.; Elefteriou, F.; Yang, X. Atf4 regulates chondrocyte proliferation and differentiation during endochondral ossification by activating Ihh transcription. Development 2009, 136, 4143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, B.S.; Pogue, R.; Ovchinnikov, D.A.; Yoshii, I.; Mishina, Y.; Behringer, R.R.; Lyons, K.M. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 2006, 133, 4667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipowska, J.; Tomaszewski, K.A.; Niedźwiedzki, Ł.; Walocha, J.A.; Niedźwiedzki, T. The role of vasculature in bone development, regeneration and proper systemic functioning. Angiogenesis 2017, 20, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Olsen, B.R.; Reginato, A.M.; Wang, W.F. Bone development. Annu. Rev. Cell Dev. Biol. 2000, 16, 191–220. [Google Scholar] [CrossRef] [PubMed]
- Sivaraj, K.K.; Adams, R.H. Blood vessel formation and function in bone. Development 2016, 143, 2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; von der Mark, K.; Henry, S.; Norton, W.; Adams, H.; de Crombrugghe, B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014, 10, e1004820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Jiang, Q.; Nagano, K.; Moriishi, T.; Miyazaki, T.; Komori, H.; Ito, K.; Mark, K.V.D.; Sakane, C.; Kaneko, H.; et al. Runx2 is essential for the transdifferentiation of chondrocytes into osteoblasts. PLoS Genet. 2020, 16, e1009169. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Wang, Z.; Li, H.; Ma, C.; Feng, J. Chondrogenesis Defines Future Skeletal Patterns Via Cell Transdifferentiation from Chondrocytes to Bone Cells. Curr. Osteoporos. Rep. 2020, 18, 199–209. [Google Scholar] [CrossRef]
- Gross, P.M.; Marcus, M.L.; Heistad, D.D. Measurement of blood flow to bone and marrow in experimental animals by means of the microsphere technique. J. Bone Jt. Surg. Am. 1981, 63, 1028–1031. [Google Scholar] [CrossRef]
- Ray, R.D.; Kawabata, M.; Galante, J. Experimental study of peripheral circulation and bone growth. An experimental method for the quantitative determination of bone blood flow. 3. Clin. Orthop. Relat. Res. 1967, 54, 175–185. [Google Scholar] [CrossRef]
- Mishbak, H.; Vyas, C.; Cooper, G.; Peach, C.; Pereira, R.F.; Bártolo, P.J. Engineering Natural-Based Photocrosslinkable Hydrogels for Cartilage Applications. In Bio-Materials and Prototyping Applications in Medicine; Bártolo, P.J., Bidanda, B., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 111–138. [Google Scholar]
- Turner, C.H. Biomechanical Aspects of Bone Formation. In Bone Formation; Bronner, F., Farach-Carson, M.C., Eds.; Springer: London, UK, 2004; pp. 79–105. [Google Scholar]
- Chow, J.; Tobias, J.H.; Colston, K.W.; Chambers, T.J. Estrogen maintains trabecular bone volume in rats not only by suppression of bone resorption but also by stimulation of bone formation. J. Clin. Investig. 1992, 89, 74–78. [Google Scholar] [CrossRef]
- Ueyama, H.; Ohta, Y.; Imai, Y.; Suzuki, A.; Sugama, R.; Minoda, Y.; Takaoka, K.; Nakamura, H. Topical co--administration of zoledronate with recombinant human bone morphogenetic protein-2 can induce and maintain bone formation in the bone marrow environment. BMC Musculoskelet. Disord. 2021, 22, 94. [Google Scholar] [CrossRef]
- Lou, J.; Tu, Y.; Li, S.; Manske, P.R. Involvement of ERK in BMP-2 Induced Osteoblastic Differentiation of Mesenchymal Progenitor Cell Line C3H10T1/2. Biochem. Biophys. Res. Commun. 2000, 268, 757–762. [Google Scholar] [CrossRef]
- Lysdahl, H.; Baatrup, A.; Foldager, C.B.; Bünger, C. Preconditioning Human Mesenchymal Stem Cells with a Low Concentration of BMP2 Stimulates Proliferation and Osteogenic Differentiation In Vitro. BioRes. Open Access 2014, 3, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Griffith, L.G.; Wells, A. Growth factor regulation of proliferation and survival of multipotential stromal cells. Stem Cell Res. Ther. 2010, 1, 32. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-R.; Plotkin, L.I.; Aguirre, J.I.; Han, L.; Jilka, R.L.; Kousteni, S.; Bellido, T.; Manolagas, S.C. Transient Versus Sustained Phosphorylation and Nuclear Accumulation of ERKs Underlie Anti-Versus Pro-apoptotic Effects of Estrogens. J. Biol. Chem. 2005, 280, 4632–4638. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Seo, I.; Choi, M.H.; Jeong, D. Roles of Mitogen-Activated Protein Kinases in Osteoclast Biology. Int. J. Mol. Sci. 2018, 19, 3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhao, D.; Wang, S.; Ding, J.; Zhao, L. Bone morphogenetic protein-9 promotes the differentiation of mouse spleen macrophages into osteoclasts via the ALK1 receptor and ERK 1/2 pathways in vitro. Mol. Med. Rep. 2016, 14, 4545–4550. [Google Scholar] [CrossRef] [Green Version]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [PubMed] [Green Version]
- Baylink, D.J.; Finkelman, R.D.; Mohan, S. Growth factors to stimulate bone formation. J. Bone Miner. Res. 1993, 8, S565–S572. [Google Scholar] [CrossRef]
- Schlesinger, P.H.; Blair, H.C.; Teitelbaum, S.L.; Edwards, J.C. Characterization of the Osteoclast Ruffled Border Chloride Channel and Its Role in Bone Resorption *. J. Biol. Chem. 1997, 272, 18636–18643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzawa, M.; Takeuchi, Y.; Fukumoto, S.; Kato, S.; Ueno, N.; Miyazono, K.; Matsumoto, T.; Fujita, T. Extracellular Matrix-Associated Bone Morphogenetic Proteins Are Essential for Differentiation of Murine Osteoblastic Cells in Vitro. Endocrinology 1999, 140, 2125–2133. [Google Scholar] [CrossRef]
- Wang, C.-Y. Bone Morphogenic Proteins, Osteoblast Differentiation, and Cell Survival during Osteogenesis. In The Skeleton: Biochemical, Genetic, and Molecular Interactions in Development and Homeostasis; Massaro, E.J., Rogers, J.M., Eds.; Humana Press: Totowa, NJ, USA, 2004; pp. 185–193. [Google Scholar]
- Halloran, D.; Durbano, H.W.; Nohe, A. Bone Morphogenetic Protein-2 in Development and Bone Homeostasis. J. Dev. Biol. 2020, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Mishina, Y.; Starbuck, M.W.; Gentile, M.A.; Fukuda, T.; Kasparcova, V.; Seedor, J.G.; Hanks, M.C.; Amling, M.; Pinero, G.J.; Harada, S.; et al. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J. Biol. Chem. 2004, 279, 27560–27566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef] [PubMed]
- McGrath, C.; Wu, Z.Y.; Sankaran, J.S.; Sen, B.; Xie, Z.H.; Styner, M.; Zong, X.P.; Chen, W.Q.; Rubin, J.; Coleman, R.A. Exercise Effect on Marrow Adipose Tissue, Metabolic and Skeletal Health in Lipodystrophic SEIPIN Deficient Mice. J. Bone Miner. Res. 2017, 32, S204. [Google Scholar]
- Mosekilde, L.; Ebbesen, E.N.; Tornvig, L.; Thomsen, J.S. Trabecular bone structure and strength—Remodelling and repair. J. Musculoskelet. Neuronal Interact. 2000, 1, 25–30. [Google Scholar] [PubMed]
- McBride, S.H.; McKenzie, J.A.; Bedrick, B.S.; Kuhlmann, P.; Pasteris, J.D.; Rosen, V.; Silva, M.J. Long bone structure and strength depend on BMP2 from osteoblasts and osteocytes, but not vascular endothelial cells. PLoS ONE 2014, 9, e96862. [Google Scholar] [CrossRef]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; Hiepen, C.; Knaus, P.; ten Dijke, P. Bone morphogenetic protein signaling in bone homeostasis. Bone 2015, 80, 43–59. [Google Scholar] [CrossRef]
- Wu, M.; Chen, G.; Li, Y.-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef]
- Kahn, A.J.; Simmons, D.J. Investigation of cell lineage in bone using a chimaera of chick and quial embryonic tissue. Nature 1975, 258, 325–327. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Montanari, M.; Gemelli, C.; Tenedini, E.; Zanocco Marani, T.; Vignudelli, T.; Siena, M.; Zini, R.; Salati, S.; Chiossi, G.; Tagliafico, E.; et al. Correlation between differentiation plasticity and mRNA expression profiling of CD34+-derived CD14− and CD14+ human normal myeloid precursors. Cell Death Differ. 2005, 12, 1588–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massey, H.M.; Flanagan, A.M. Human osteoclasts derive from CD14-positive monocytes. Br. J. Haematol. 1999, 106, 167–170. [Google Scholar] [CrossRef]
- Jin, X.; Kruth, H.S. Culture of Macrophage Colony-stimulating Factor Differentiated Human Monocyte-derived Macrophages. J. Vis. Exp. 2016, 112. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Mossadegh-Keller, N.; Sarrazin, S.; Kandalla, P.K.; Espinosa, L.; Stanley, E.R.; Nutt, S.L.; Moore, J.; Sieweke, M.H. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 2013, 497, 239–243. [Google Scholar] [CrossRef]
- Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; Yasuda, H.; Yano, K.; Morinaga, T.; Higashio, K. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 1998, 253, 395–400. [Google Scholar] [CrossRef]
- Quinn, J.M.; Elliott, J.; Gillespie, M.T.; Martin, T.J. A combination of osteoclast differentiation factor and macrophage-colony stimulating factor is sufficient for both human and mouse osteoclast formation in vitro. Endocrinology 1998, 139, 4424–4427. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Hayashi, S.I.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S.I. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Broege, A.; Pham, L.; Jensen, E.D.; Emery, A.; Huang, T.H.; Stemig, M.; Beppu, H.; Petryk, A.; O’Connor, M.; Mansky, K.; et al. Bone morphogenetic proteins signal via SMAD and mitogen-activated protein (MAP) kinase pathways at distinct times during osteoclastogenesis. J. Biol. Chem. 2013, 288, 37230–37240. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Udagawa, N.; Katagiri, T.; Iemura, S.; Ueno, N.; Yasuda, H.; Higashio, K.; Quinn, J.M.; Gillespie, M.T.; Martin, T.J.; et al. Bone morphogenetic protein 2 stimulates osteoclast differentiation and survival supported by receptor activator of nuclear factor-kappaB ligand. Endocrinology 2001, 142, 3656–3662. [Google Scholar] [CrossRef]
- Jensen, E.D.; Pham, L.; Billington, C.J., Jr.; Espe, K.; Carlson, A.E.; Westendorf, J.J.; Petryk, A.; Gopalakrishnan, R.; Mansky, K. Bone morphogenic protein 2 directly enhances differentiation of murine osteoclast precursors. J. Cell. Biochem. 2010, 109, 672–682. [Google Scholar] [CrossRef] [Green Version]
- Kanatani, M.; Sugimoto, T.; Kaji, H.; Kobayashi, T.; Nishiyama, K.; Fukase, M.; Kumegawa, M.; Chihara, K. Stimulatory effect of bone morphogenetic protein-2 on osteoclast-like cell formation and bone-resorbing activity. J. Bone Miner. Res. 1995, 10, 1681–1690. [Google Scholar] [CrossRef]
- Kaneko, H.; Arakawa, T.; Mano, H.; Kaneda, T.; Ogasawara, A.; Nakagawa, M.; Toyama, Y.; Yabe, Y.; Kumegawa, M.; Hakeda, Y. Direct stimulation of osteoclastic bone resorption by bone morphogenetic protein (BMP)-2 and expression of BMP receptors in mature osteoclasts. Bone 2000, 27, 479–486. [Google Scholar] [CrossRef]
- Li, A.; Cong, Q.; Xia, X.; Leong, W.F.; Yeh, J.; Miao, D.; Mishina, Y.; Liu, H.; Li, B. Pharmacologic Calcitriol Inhibits Osteoclast Lineage Commitment via the BMP-Smad1 and IκB-NF-κB Pathways. J. Bone Miner. Res. 2017, 32, 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Murai, J.; Yoshikawa, H.; Tsumaki, N. Bone Morphogenetic Proteins in Bone Stimulate Osteoclasts and Osteoblasts During Bone Development. J. Bone Miner. Res. 2006, 21, 1022–1033. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, H.; Louie, K.A.; Mishina, Y.; Sun, H. BMP Signaling Mediated by BMPR1A in Osteoclasts Negatively Regulates Osteoblast Mineralization Through Suppression of Cx43. J. Cell. Biochem. 2017, 118, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Sotillo Rodriguez, J.E.; Mansky, K.C.; Jensen, E.D.; Carlson, A.E.; Schwarz, T.; Pham, L.; MacKenzie, B.; Prasad, H.; Rohrer, M.D.; Petryk, A.; et al. Enhanced Osteoclastogenesis Causes Osteopenia in Twisted Gastrulation-Deficient Mice Through Increased BMP Signaling. J. Bone Miner. Res. 2009, 24, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.H.; Lee, C.K.; Lee, Y.I.; Paik, S.G.; Lee, H.J. The hematopoietic transcription factor PU.1 regulates RANK gene expression in myeloid progenitors. Biochem. Biophys. Res. Commun. 2005, 335, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Tondravi, M.M.; McKercher, S.R.; Anderson, K.; Erdmann, J.M.; Quiroz, M.; Maki, R.; Teitelbaum, S.L. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature 1997, 386, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Buckley, S.; Schouteden, S.; Ekker, S.; Petryk, A.; Delforge, M.; Zwijsen, A.; Verfaillie, C.M. A novel role of BMP4 in adult hematopoietic stem and progenitor cell homing via Smad independent regulation of integrin-α4 expression. Blood 2013, 121, 781–790. [Google Scholar] [CrossRef] [Green Version]
- Courtial, N.; Smink, J.J.; Kuvardina, O.N.; Leutz, A.; Göthert, J.R.; Lausen, J. Tal1 regulates osteoclast differentiation through suppression of the master regulator of cell fusion DC-STAMP. FASEB J. 2012, 26, 523–532. [Google Scholar] [CrossRef]
- Lee, S.H.; Rho, J.; Jeong, D.; Sul, J.Y.; Kim, T.; Kim, N.; Kang, J.S.; Miyamoto, T.; Suda, T.; Lee, S.K.; et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 2006, 12, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Piemontese, M.; Onal, M.; Campbell, J.; Goellner, J.J.; Dusevich, V.; Bonewald, L.; Manolagas, S.C.; O’Brien, C.A. Osteocytes, not Osteoblasts or Lining Cells, are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PLoS ONE 2015, 10, e0138189. [Google Scholar] [CrossRef]
- Mandal, C.C.; Das, F.; Ganapathy, S.; Harris, S.E.; Choudhury, G.G.; Ghosh-Choudhury, N. Bone Morphogenetic Protein-2 (BMP-2) Activates NFATc1 Transcription Factor via an Autoregulatory Loop Involving Smad/Akt/Ca2+ Signaling. J. Biol. Chem. 2016, 291, 1148–1161. [Google Scholar] [CrossRef] [Green Version]
- Omi, M.; Kaartinen, V.; Mishina, Y. Activin A receptor type 1-mediated BMP signaling regulates RANKL-induced osteoclastogenesis via canonical SMAD-signaling pathway. J. Biol. Chem. 2019, 294, 17818–17836. [Google Scholar] [CrossRef] [PubMed]
- Abe, E.; Yamamoto, M.; Taguchi, Y.; Lecka-Czernik, B.; O’Brien, C.A.; Economides, A.N.; Stahl, N.; Jilka, R.L.; Manolagas, S.C. Essential Requirement of BMPs-2/4 for Both Osteoblast and Osteoclast Formation in Murine Bone Marrow Cultures from Adult Mice: Antagonism by Noggin. J. Bone Miner. Res. 2000, 15, 663–673. [Google Scholar] [CrossRef]
- Huntley, R.; Davydova, J.; Petryk, A.; Billington, C.J., Jr.; Jensen, E.D.; Mansky, K.C.; Gopalakrishnan, R. The Function of Twisted Gastrulation in Regulating Osteoclast Differentiation is Dependent on BMP Binding. J. Cell. Biochem. 2015, 116, 2239–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, L.; Beyer, K.; Jensen, E.D.; Rodriguez, J.S.; Davydova, J.; Yamamoto, M.; Petryk, A.; Gopalakrishnan, R.; Mansky, K.C. Bone morphogenetic protein 2 signaling in osteoclasts is negatively regulated by the BMP antagonist, twisted gastrulation. J. Cell. Biochem. 2011, 112, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [Green Version]
- Hollberg, K.; Nordahl, J.; Hultenby, K.; Mengarelli-Widholm, S.; Andersson, G.; Reinholt, F.P. Polarization and secretion of cathepsin K precede tartrate-resistant acid phosphatase secretion to the ruffled border area during the activation of matrix-resorbing clasts. J. Bone Min. Metab. 2005, 23, 441–449. [Google Scholar] [CrossRef]
- Jarrar, H.; Çetin Altındal, D.; Gümüşderelioğlu, M. Effect of melatonin/BMP-2 co-delivery scaffolds on the osteoclast activity. J. Mater. Sci. Mater. Med. 2021, 32, 32. [Google Scholar] [CrossRef] [PubMed]
- Mandal, C.C.; Ghosh Choudhury, G.; Ghosh-Choudhury, N. Phosphatidylinositol 3 Kinase/Akt Signal Relay Cooperates with Smad in Bone Morphogenetic Protein-2-Induced Colony Stimulating Factor-1 (CSF-1) Expression and Osteoclast Differentiation. Endocrinology 2009, 150, 4989–4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Udagawa, N.; Kobayashi, Y.; Suda, T. Generation of Osteoclasts In Vitro, and Assay of Osteoclast Activity. In Arthritis Research: Methods and Protocols; Cope, A.P., Ed.; Humana Press: Totowa, NJ, USA, 2007; Volume 1, pp. 285–301. [Google Scholar]
- Udagawa, N.; Takito, J.; Suda, T. [Mechanism of acid production and secretion by osteoclasts]. Nihon Rinsho Jpn. J. Clin. Med. 1992, 50, 2133–2138. [Google Scholar]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.L.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Lüthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Goto, M.; Mochizuki, S.I.; Tsuda, E.; Morinaga, T.; Udagawa, N.; et al. A novel molecular mechanism modulating osteoclast differentiation and function. Bone 1999, 25, 109–113. [Google Scholar] [CrossRef]
- Bhargava, A.; Vagela, M.; Lennox, C.M. “Challenges in the management of fractures in osteopetrosis”! Review of literature and technical tips learned from long-term management of seven patients. Injury 2009, 40, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Graeff, C.; Marin, F.; Petto, H.; Kayser, O.; Reisinger, A.; Peña, J.; Zysset, P.; Glüer, C.-C. High resolution quantitative computed tomography-based assessment of trabecular microstructure and strength estimates by finite-element analysis of the spine, but not DXA, reflects vertebral fracture status in men with glucocorticoid-induced osteoporosis. Bone 2013, 52, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Granholm, S.; Henning, P.; Lindholm, C.; Lerner, U.H. Osteoclast progenitor cells present in significant amounts in mouse calvarial osteoblast isolations and osteoclastogenesis increased by BMP-2. Bone 2013, 52, 83–92. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Dunstan, C.R.; Spelsberg, T.C.; Riggs, B.L.; Khosla, S. Osteoprotegerin production by human osteoblast lineage cells is stimulated by vitamin D, bone morphogenetic protein-2, and cytokines. Biochem. Biophys. Res. Commun. 1998, 250, 776–781. [Google Scholar] [CrossRef]
- Krajewski, A.C.; Ghuman, M.; Reddi, D.; McKay, I.; Hughes, F.; Belibasakis, G. Influence of BMP-2 on RANKL/ OPG production in W-20-17 cells. In Proceedings of the IADR General Session, Barcelona, Spain, 14–17 July 2010. [Google Scholar]
- Tazoe, M.; Mogi, M.; Goto, S.; Togari, A. Involvement of p38MAP kinase in bone morphogenetic protein-4-induced osteoprotegerin in mouse bone-marrow-derived stromal cells. Arch. Oral Biol. 2003, 48, 615–619. [Google Scholar] [CrossRef]
- Theoleyre, S.; Wittrant, Y.; Tat, S.K.; Fortun, Y.; Redini, F.; Heymann, D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004, 15, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Matsuyama, T.; Hosokawa, Y.; Makihira, S.; Seki, M.; Karimbux, N.Y.; Goncalves, R.B.; Valverde, P.; Dibart, S.; Li, Y.-P.; et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am. J. Pathol. 2006, 169, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Valverde, P.; Kawai, T.; Taubman, M.A. Selective blockade of voltage-gated potassium channels reduces inflammatory bone resorption in experimental periodontal disease. J. Bone Miner. Res. 2004, 19, 155–164. [Google Scholar] [CrossRef]
- Yun, T.J.; Chaudhary, P.M.; Shu, G.L.; Frazer, J.K.; Ewings, M.K.; Schwartz, S.M.; Pascual, V.; Hood, L.E.; Clark, E.A. OPG/FDCR-1, a TNF receptor family member, is expressed in lymphoid cells and is up-regulated by ligating CD40. J. Immunol. 1998, 161, 6113–6121. [Google Scholar]
- Leibbrandt, A.; Penninger, J.M. Novel functions of RANK(L) signaling in the immune system. Adv. Exp. Med. Biol. 2010, 658, 77–94. [Google Scholar] [PubMed]
- Chen, L.; Wei, X.Q.; Evans, B.; Jiang, W.; Aeschlimann, D. IL-23 promotes osteoclast formation by up-regulation of receptor activator of NF-kappaB (RANK) expression in myeloid precursor cells. Eur. J. Immunol. 2008, 38, 2845–2854. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Hayakawa, N.; Mihara, M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-α and IL-17. Rheumatology 2008, 47, 1635–1640. [Google Scholar] [CrossRef] [Green Version]
- Komine, M.; Kukita, A.; Kukita, T.; Ogata, Y.; Hotokebuchi, T.; Kohashi, O. Tumor necrosis factor-alpha cooperates with receptor activator of nuclear factor kappaB ligand in generation of osteoclasts in stromal cell-depleted rat bone marrow cell culture. Bone 2001, 28, 474–483. [Google Scholar] [CrossRef]
- Azuma, Y.; Kaji, K.; Katogi, R.; Takeshita, S.; Kudo, A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 2000, 275, 4858–4864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiratori, T.; Kyumoto-Nakamura, Y.; Kukita, A.; Uehara, N.; Zhang, J.; Koda, K.; Kamiya, M.; Badawy, T.; Tomoda, E.; Xu, X.; et al. IL-1β Induces Pathologically Activated Osteoclasts Bearing Extremely High Levels of Resorbing Activity: A Possible Pathological Subpopulation of Osteoclasts, Accompanied by Suppressed Expression of Kindlin-3 and Talin-1. J. Immunol. 2018, 200, 218. [Google Scholar] [CrossRef]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, F.; Duplomb, L.; Baud’huin, M.; Brounais, B. The dual role of IL-6-type cytokines on bone remodeling and bone tumors. Cytokine Growth Factor Rev. 2009, 20, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The Mechanism of Osteoclast Differentiation Induced by IL-1. J. Immunol. 2009, 183, 1862. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Grassi, F.; Ryan, M.R.; Terauchi, M.; Page, K.; Yang, X.; Weitzmann, M.N.; Pacifici, R. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J. Clin. Investig. 2007, 117, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Kitaura, H.; Yoshimatsu, M.; Fujimura, Y.; Kohara, H.; Eguchi, T.; Yoshida, N. IL-18 inhibits TNF-alpha-induced osteoclastogenesis possibly via a T cell-independent mechanism in synergy with IL-12 in vivo. Calcif. Tissue Int. 2010, 86, 242–248. [Google Scholar] [CrossRef]
- Saleh, H.; Eeles, D.; Hodge, J.M.; Nicholson, G.C.; Gu, R.; Pompolo, S.; Gillespie, M.T.; Quinn, J.M.W. Interleukin-33, a Target of Parathyroid Hormone and Oncostatin M, Increases Osteoblastic Matrix Mineral Deposition and Inhibits Osteoclast Formation in Vitro. Endocrinology 2011, 152, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Saribal, D.; Hocaoglu-Emre, F.S.; Erdogan, S.; Bahtiyar, N.; Caglar Okur, S.; Mert, M. Inflammatory cytokines IL-6 and TNF-α in patients with hip fracture. Osteoporos. Int. 2019, 30, 1025–1031. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Honma, M.; Ikebuchi, Y.; Kariya, Y.; Hayashi, M.; Hayashi, N.; Aoki, S.; Suzuki, H. RANKL subcellular trafficking and regulatory mechanisms in osteocytes. J. Bone Miner. Res. 2013, 28, 1936–1949. [Google Scholar] [CrossRef]
- Gu, G.; Nars, M.; Hentunen, T.A.; Metsikkö, K.; Väänänen, H.K. Isolated primary osteocytes express functional gap junctions in vitro. Cell Tissue Res. 2006, 323, 263–271. [Google Scholar] [CrossRef]
- Fumoto, T.; Takeshita, S.; Ito, M.; Ikeda, K. Physiological functions of osteoblast lineage and T cell-derived RANKL in bone homeostasis. J. Bone Min. Res. 2014, 29, 830–842. [Google Scholar] [CrossRef]
- Wutzl, A.; Brozek, W.; Lernbass, I.; Rauner, M.; Hofbauer, G.; Schopper, C.; Watzinger, F.; Peterlik, M.; Pietschmann, P. Bone morphogenetic proteins 5 and 6 stimulate osteoclast generation. J. Biomed. Mater. Res. A 2006, 77, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. The RANKL/RANK/OPG pathway. Curr. Osteoporos. Rep. 2007, 5, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Del Fattore, A.; Teti, A. The tight relationship between osteoclasts and the immune system. Inflamm. Allergy Drug Targets 2012, 11, 181–187. [Google Scholar] [CrossRef]
- Infante, M.; Fabi, A.; Cognetti, F.; Gorini, S.; Caprio, M.; Fabbri, A. RANKL/RANK/OPG system beyond bone remodeling: Involvement in breast cancer and clinical perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 12. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, N.; Ye, L.; Kobayashi, T.; Lucas, D.J.; Mochida, Y.; Yamauchi, M.; Kronenberg, H.M.; Feng, J.Q.; Mishina, Y. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J. Bone Min. Res. 2008, 23, 2007–2017. [Google Scholar] [CrossRef] [Green Version]
- Kusu, N.; Laurikkala, J.; Imanishi, M.; Usui, H.; Konishi, M.; Miyake, A.; Thesleff, I.; Itoh, N. Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J. Biol. Chem. 2003, 278, 24113–24117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, X.; Delgado-Calle, J.; Condon, K.W.; Maycas, M.; Zhang, H.; Carlesso, N.; Taketo, M.M.; Burr, D.B.; Plotkin, L.I.; Bellido, T. Osteocytes mediate the anabolic actions of canonical Wnt/β-catenin signaling in bone. Proc. Natl. Acad. Sci. USA 2015, 112, E478–E486. [Google Scholar] [CrossRef] [Green Version]
- Garimella, R.; Tague, S.E.; Zhang, J.; Belibi, F.; Nahar, N.; Sun, B.H.; Insogna, K.; Wang, J.; Anderson, H.C. Expression and synthesis of bone morphogenetic proteins by osteoclasts: A possible path to anabolic bone remodeling. J. Histochem. Cytochem. 2008, 56, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Hayden, J.M.; Mohan, S.; Baylink, D.J. The insulin-like growth factor system and the coupling of formation to resorption. Bone 1995, 17 (Suppl. 2), 93s–98s. [Google Scholar] [CrossRef]
- Ota, K.; Quint, P.; Weivoda, M.M.; Ruan, M.; Pederson, L.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Transforming growth factor beta 1 induces CXCL16 and leukemia inhibitory factor expression in osteoclasts to modulate migration of osteoblast progenitors. Bone 2013, 57, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Pederson, L.; Ruan, M.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc. Natl. Acad. Sci. USA 2008, 105, 20764–20769. [Google Scholar] [CrossRef] [Green Version]
- Aluganti Narasimhulu, C.; Singla, D.K. The Role of Bone Morphogenetic Protein 7 (BMP-7) in Inflammation in Heart Diseases. Cells 2020, 9, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliorini, E.; Guevara-Garcia, A.; Albiges-Rizo, C.; Picart, C. Learning from BMPs and their biophysical extracellular matrix microenvironment for biomaterial design. Bone 2020, 141, 115540. [Google Scholar] [CrossRef]
- Pang, D.-D.; Cai, L.; Zhang, J.-R.; Dai, S.-M. IL-23 induziert die Expression pro-osteogener Faktoren in Osteoklasten. Aktuelle Rheumatol. 2020, 45, 467–474. [Google Scholar] [CrossRef]
- Ghosh-Choudhury, N.; Singha, P.K.; Woodruff, K.; Clair, P.S.; Bsoul, S.; Werner, S.L.; Choudhury, G.G. Concerted Action of Smad and CREB-binding Protein Regulates Bone Morphogenetic Protein-2-stimulated Osteoblastic Colony-stimulating Factor-1 Expression. J. Biol. Chem. 2006, 281, 20160–20170. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, G.; Xiao, Y.; Luo, G.; Li, G.; Li, Z. Friend or Foe? Essential Roles of Osteoclast in Maintaining Skeletal Health. BioMed Res. Int. 2020, 2020, 4791786. [Google Scholar] [CrossRef]
- Tsuji, K.; Bandyopadhyay, A.; Harfe, B.D.; Cox, K.; Kakar, S.; Gerstenfeld, L.; Einhorn, T.; Tabin, C.J.; Rosen, V. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet. 2006, 38, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhou, X.; Huang, D.; Ji, Y.; Kang, F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell. Physiol. Biochem. 2017, 41, 1360–1369. [Google Scholar] [CrossRef]
- Okamoto, M.; Murai, J.; Imai, Y.; Ikegami, D.; Kamiya, N.; Kato, S.; Mishina, Y.; Yoshikawa, H.; Tsumaki, N. Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J. Bone Miner. Res. 2011, 26, 2511–2522. [Google Scholar] [CrossRef]
- Shi, C.; Iura, A.; Terajima, M.; Liu, F.; Lyons, K.; Pan, H.; Zhang, H.; Yamauchi, M.; Mishina, Y.; Sun, H. Deletion of BMP receptor type IB decreased bone mass in association with compromised osteoblastic differentiation of bone marrow mesenchymal progenitors. Sci. Rep. 2016, 6, 24256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attisano, L.; Lee-Hoeflich, S.T. The Smads. Genome Biol. 2001, 2. [Google Scholar] [CrossRef]
- Fong, D.; Bisson, M.; Laberge, G.; McManus, S.; Grenier, G.; Faucheux, N.; Roux, S. Bone morphogenetic protein-9 activates Smad and ERK pathways and supports human osteoclast function and survival in vitro. Cell. Signal. 2013, 25, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Tasca, A.; Astleford, K.; Blixt, N.C.; Jensen, E.D.; Gopalakrishnan, R.; Mansky, K.C. SMAD1/5 signaling in osteoclasts regulates bone formation via coupling factors. PLoS ONE 2018, 13, e0203404. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Yuan, J.; Wu, J.; Zheng, J.; Zheng, W.; Wang, F.; Wang, C.; Li, X.; Liu, S.; Shi, Z.; et al. Bone Morphogenetic Protein-2 Promotes Osteoclasts-mediated Osteolysis via Smad1 and p65 Signaling Pathways. Spine 2021, 46. [Google Scholar] [CrossRef]
- Qi, B.; Cong, Q.; Li, P.; Ma, G.; Guo, X.; Yeh, J.; Xie, M.; Schneider, M.D.; Liu, H.; Li, B. Ablation of Tak1 in osteoclast progenitor leads to defects in skeletal growth and bone remodeling in mice. Sci. Rep. 2014, 4, 7158. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Nagai, S.-I.; Ninomiya-Tsuji, J.; Nishita, M.; Tamai, K.; Irie, K.; Ueno, N.; Nishida, E.; Shibuya, H.; Matsumoto, K. XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1–TAK1 in the BMP signaling pathway. EMBO J. 1999, 18, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Feng, X. RANKing Intracellular Signaling in Osteoclasts. IUBMB Life 2005, 57, 389–395. [Google Scholar] [CrossRef]
- Guo, Q.; Cao, Z.; Wu, B.; Chen, F.; Tickner, J.; Wang, Z.; Qiu, H.; Wang, C.; Chen, K.; Tan, R.; et al. Modulating calcium-mediated NFATc1 and mitogen-activated protein kinase deactivation underlies the inhibitory effects of kavain on osteoclastogenesis and bone resorption. J. Cell. Physiol. 2019, 234, 789–801. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Lee, S.-H.; Ha Kim, J.; Choi, Y.; Kim, N. NFATc1 Induces Osteoclast Fusion Via Up-Regulation of Atp6v0d2 and the Dendritic Cell-Specific Transmembrane Protein (DC-STAMP). Mol. Endocrinol. 2008, 22, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Wang, B.; Yang, X.; Guo, H.; Zhang, K.; Zhu, Z.; Liu, J.; Hao, D. Picrasidine I from Picrasma Quassioides Suppresses Osteoclastogenesis via Inhibition of RANKL Induced Signaling Pathways and Attenuation of ROS Production. Cell. Physiol. Biochem. 2017, 43, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.; Kelly, S.; Wood, R.; Heubel, B.; Nohe, A. A Synthetic Peptide, CK2.3, Inhibits RANKL-Induced Osteoclastogenesis through BMPRIa and ERK Signaling Pathway. J. Dev. Biol. 2020, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Bollerslev, J.; Andersen, P.E. Fracture patterns in two types of autosomal-dominant osteopetrosis. Acta Orthop. Scand. 1989, 60, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Sreehari, S.; Naik, D.R.; Eapen, M. Osteopetrosis: A rare cause of anemia. Hematol. Rep. 2011, 3, e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, E.; Xu, H.; Wang, L.; Kryczek, I.; Wu, K.; Hu, Y.; Wang, G.; Zou, W. Bone marrow and the control of immunity. Cell. Mol. Immunol. 2012, 9, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Lane, N.E.; Thompson, J.M.; Haupt, D.; Kimmel, D.B.; Modin, G.; Kinney, J.H. Acute Changes in Trabecular Bone Connectivity and Osteoclast Activity in the Ovariectomized Rat In Vivo. J. Bone Miner. Res. 1998, 13, 229–236. [Google Scholar] [CrossRef]
- Legrand, E.; Chappard, D.; Pascaretti, C.; Duquenne, M.; Krebs, S.; Rohmer, V.; Basle, M.-F.; Audran, M. Trabecular Bone Microarchitecture, Bone Mineral Density, and Vertebral Fractures in Male Osteoporosis. J. Bone Miner. Res. 2000, 15, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Dawson-Hughes, B.; Looker, A.C.; Tosteson, A.N.; Johansson, H.; Kanis, J.A.; Melton, L.J., 3rd. The potential impact of new National Osteoporosis Foundation guidance on treatment patterns. Osteoporos. Int. 2010, 21, 41–52. [Google Scholar] [CrossRef]
- Compston, J.; Cooper, A.; Cooper, C.; Gittoes, N.; Gregson, C.; Harvey, N.; Hope, S.; Kanis, J.A.; McCloskey, E.V.; Poole, K.E.S.; et al. UK clinical guideline for the prevention and treatment of osteoporosis. Arch. Osteoporos. 2017, 12, 24. [Google Scholar] [CrossRef]
- Streicher, C.; Heyny, A.; Andrukhova, O.; Haigl, B.; Slavic, S.; Schüler, C.; Kollmann, K.; Kantner, I.; Sexl, V.; Kleiter, M.; et al. Estrogen Regulates Bone Turnover by Targeting RANKL Expression in Bone Lining Cells. Sci. Rep. 2017, 7, 6460. [Google Scholar] [CrossRef] [Green Version]
- Nicks, K.M.; Fowler, T.W.; Akel, N.S.; Perrien, D.S.; Suva, L.J.; Gaddy, D. Bone turnover across the menopause transition: The role of gonadal inhibins. Ann. N. Y. Acad. Sci. 2010, 1192, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Reginster, J.-Y.; Burlet, N. Osteoporosis: A still increasing prevalence. Bone 2006, 38 (Suppl. 1), 4–9. [Google Scholar] [CrossRef]
- Sözen, T.; Özışık, L.; Başaran, N.Ç. An overview and management of osteoporosis. Eur. J. Rheumatol. 2017, 4, 46–56. [Google Scholar] [CrossRef]
- Johnell, O.; Kanis, J.A. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos. Int. 2006, 17, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Johnell, O.; Kanis, J.A.; Odén, A.; Sernbo, I.; Redlund-Johnell, I.; Petterson, C.; De Laet, C.; Jönsson, B. Mortality after osteoporotic fractures. Osteoporos. Int. 2004, 15, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Neuburger, J.; Currie, C.; Wakeman, R.; Tsang, C.; Plant, F.; De Stavola, B.; Cromwell, D.A.; van der Meulen, J. The impact of a national clinician-led audit initiative on care and mortality after hip fracture in England: An external evaluation using time trends in non-audit data. Med. Care 2015, 53, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Blume, S.W.; Curtis, J.R. Medical costs of osteoporosis in the elderly Medicare population. Osteoporos. Int. 2011, 22, 1835–1844. [Google Scholar] [CrossRef] [Green Version]
- Hernlund, E.; Svedbom, A.; Ivergård, M.; Compston, J.; Cooper, C.; Stenmark, J.; McCloskey, E.V.; Jönsson, B.; Kanis, J.A. Osteoporosis in the European Union: Medical management, epidemiology and economic burden. A report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch. Osteoporos. 2013, 8, 136. [Google Scholar] [CrossRef] [Green Version]
- Draper, J.; Roland, M. Perimenopausal women’s views on taking hormone replacement therapy to prevent osteoporosis. Br. Med. J. 1990, 300, 786. [Google Scholar] [CrossRef] [Green Version]
- Grady, D.; Rubin, S.M.; Petitti, D.B.; Fox, C.S.; Black, D.; Ettinger, B.; Ernster, V.L.; Cummings, S.R. Hormone therapy to prevent disease and prolong life in postmenopausal women. Ann. Intern. Med. 1992, 117, 1016–1037. [Google Scholar] [CrossRef]
- Colditz, G.A. Relationship Between Estrogen Levels, Use of Hormone Replacement Therapy, and Breast Cancer. JNCI J. Natl. Cancer Inst. 1998, 90, 814–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grodstein, F.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C.; Manson, J.E.; Joffe, M.; Rosner, B.; Fuchs, C.; Hankinson, S.E.; Hunter, D.J.; et al. Postmenopausal hormone therapy and mortality. N. Engl. J. Med. 1997, 336, 1769–1775. [Google Scholar] [CrossRef]
- Bodalia, P.N.; Balaji, V.; Kaila, R.; Wilson, L. Effectiveness and safety of recombinant human bone morphogenetic protein-2 for adults with lumbar spine pseudarthrosis following spinal fusion surgery. Bone Jt. Res. 2016, 5, 145–152. [Google Scholar] [CrossRef]
- Deyo, R.A.; Ching, A.; Matsen, L.; Martin, B.I.; Kreuter, W.; Jarvik, J.G.; Angier, H.; Mirza, S.K. Use of bone morphogenetic proteins in spinal fusion surgery for older adults with lumbar stenosis: Trends, complications, repeat surgery, and charges. Spine 2012, 37, 222–230. [Google Scholar] [CrossRef]
- Huang, K.; Wu, G.; Zou, J.; Peng, S. Combination therapy with BMP-2 and psoralen enhances fracture healing in ovariectomized mice. Exp. Ther. Med. 2018, 16, 1655–1662. [Google Scholar] [CrossRef] [Green Version]
- Tannoury, C.A.; An, H.S. Complications with the use of bone morphogenetic protein 2 (BMP-2) in spine surgery. Spine J. 2014, 14, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.M.; Sasso, R.C. Resorptive response of rhBMP2 simulating infection in an anterior lumbar interbody fusion with a femoral ring. Clin. Spine Surg. 2006, 19, 130–134. [Google Scholar] [CrossRef]
- Vaidya, R.; Weir, R.; Sethi, A.; Meisterling, S.; Hakeos, W.; Wybo, C.D. Interbody fusion with allograft and rhBMP-2 leads to consistent fusion but early subsidence. J. Bone Jt. Surg. Br. Vol. 2007, 89, 342–345. [Google Scholar] [CrossRef] [Green Version]
- Durbano, H.W.; Halloran, D.; Nguyen, J.; Stone, V.; McTague, S.; Eskander, M.; Nohe, A. Aberrant BMP2 Signaling in Patients Diagnosed with Osteoporosis. Int. J. Mol. Sci. 2020, 21, 6909. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, A.M.; Chambers, T.J. Inhibition of bone resorption by bisphosphonates: Interactions between bisphosphonates, osteoclasts, and bone. Calcif. Tissue Int. 1991, 49, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Hadji, P.; Coleman, R.; Gnant, M.; Green, J. The impact of menopause on bone, zoledronic acid, and implications for breast cancer growth and metastasis. Ann. Oncol. 2012, 23, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.E.; Wright, K.R.; Uy, H.L.; Sasaki, A.; Yoneda, T.; Roodman, D.G.; Mundy, G.R.; Boyce, B.F. Bisphosphonates promote apoptosis in murine osteoclasts in vitro and in vivo. J. Bone Miner. Res. 1995, 10, 1478–1487. [Google Scholar] [CrossRef]
- Rogers, M.J.; Mönkkönen, J.; Munoz, M.A. Molecular mechanisms of action of bisphosphonates and new insights into their effects outside the skeleton. Bone 2020, 139, 115493. [Google Scholar] [CrossRef]
- Marx, R.E.; Sawatari, Y.; Fortin, M.; Broumand, V. Bisphosphonate-induced exposed bone (osteonecrosis/osteopetrosis) of the jaws: Risk factors, recognition, prevention, and treatment. J. Oral Maxillofac. Surg. 2005, 63, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Watts, N.B.; Diab, D.L. Long-term use of bisphosphonates in osteoporosis. J. Clin. Endocrinol. Metab. 2010, 95, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-B.; Hellstein, J.W.; Kalmar, J.R. Systematic Review: Bisphosphonates and Osteonecrosis of the Jaws. Ann. Intern. Med. 2006, 144, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Hamdy, N.A. Denosumab: RANKL inhibition in the management of bone loss. Drugs Today 2008, 44, 7–21. [Google Scholar] [CrossRef]
- Törring, O. Effects of denosumab on bone density, mass and strength in women with postmenopausal osteoporosis. Ther. Adv. Musculoskelet. Dis. 2015, 7, 88–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, M.; Horwitz, M.J. Parathyroid hormone and parathyroid hormone-related protein analogs as therapies for osteoporosis. Curr. Osteoporos. Rep. 2013, 11, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.; Han, S.; Kim, H.; Kim, K.S.; Hahn, S.K. Hyaluronate—Parathyroid hormone peptide conjugate for transdermal treatment of osteoporosis. J. Biomater. Sci. Polym. Ed. 2018, 29, 793–804. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Johnson, J.; Le Henaff, C.; Bitel, C.L.; Tamasi, J.A.; Partridge, N.C. Catabolic Effects of Human PTH (1–34) on Bone: Requirement of Monocyte Chemoattractant Protein-1 in Murine Model of Hyperparathyroidism. Sci. Rep. 2017, 7, 15300. [Google Scholar] [CrossRef] [Green Version]
- Camirand, A.; Goltzman, D.; Gupta, A.; Kaouass, M.; Panda, D.; Karaplis, A. The Role of Parathyroid Hormone-Related Protein (PTHrP) in Osteoblast Response to Microgravity: Mechanistic Implications for Osteoporosis Development. PLoS ONE 2016, 11, e0160034. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J. Osteoblast-derived PTHrP is a physiological regulator of bone formation. J. Clin. Investig. 2005, 115, 2322–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, M.J.; Tedesco, M.B.; Garcia-Ocaña, A.; Sereika, S.M.; Prebehala, L.; Bisello, A.; Hollis, B.W.; Gundberg, C.M.; Stewart, A.F. Parathyroid Hormone-Related Protein for the Treatment of Postmenopausal Osteoporosis: Defining the Maximal Tolerable Dose. J. Clin. Endocrinol. Metab. 2010, 95, 1279–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.D.; Shoback, D.; Lewiecki, E.M. Sclerostin inhibition: A novel therapeutic approach in the treatment of osteoporosis. Int. J. Womens Health 2015, 7, 565–580. [Google Scholar]
- Suen, P.K.; Qin, L. Sclerostin, an emerging therapeutic target for treating osteoporosis and osteoporotic fracture: A general review. J. Orthop. Transl. 2016, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef]
- Stolina, M.; Dwyer, D.; Niu, Q.T.; Villasenor, K.S.; Kurimoto, P.; Grisanti, M.; Han, C.Y.; Liu, M.; Li, X.; Ominsky, M.S.; et al. Temporal changes in systemic and local expression of bone turnover markers during six months of sclerostin antibody administration to ovariectomized rats. Bone 2014, 67, 305–313. [Google Scholar] [CrossRef]
- Boyce, R.W.; Brown, D.; Felx, M.; Mellal, N.; Locher, K.; Pyrah, I.; Ominsky, M.S.; Taylor, S. Decreased osteoprogenitor proliferation precedes attenuation of cancellous bone formation in ovariectomized rats treated with sclerostin antibody. Bone Rep. 2018, 8, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Lewiecki, E.M.; Blicharski, T.; Goemaere, S.; Lippuner, K.; Meisner, P.D.; Miller, P.D.; Miyauchi, A.; Maddox, J.; Chen, L.; Horlait, S. A Phase III Randomized Placebo-Controlled Trial to Evaluate Efficacy and Safety of Romosozumab in Men With Osteoporosis. J. Clin. Endocrinol. Metab. 2018, 103, 3183–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkiraju, H.; Bonor, J.; Olli, K.; Bowen, C.; Bragdon, B.; Coombs, H.; Donahue, L.R.; Duncan, R.; Nohe, A. Systemic Injection of CK2.3, a Novel Peptide Acting Downstream of Bone Morphogenetic Protein Receptor BMPRIa, Leads to Increased Trabecular Bone Mass. J. Orthop. Res. 2015, 33, 208–215. [Google Scholar] [CrossRef]
- Bragdon, B.; Thinakaran, S.; Moseychuk, O.; King, D.; Young, K.; Litchfield, D.W.; Petersen, N.O.; Nohe, A. Casein Kinase 2 β-Subunit Is a Regulator of Bone Morphogenetic Protein 2 Signaling. Biophys. J. 2010, 99, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Vrathasha, V.; Weidner, H.; Nohe, A. Mechanism of CK2.3, a Novel Mimetic Peptide of Bone Morphogenetic Protein Receptor Type IA, Mediated Osteogenesis. Int. J. Mol. Sci. 2019, 20, 2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, H.; Yuan Gao, V.; Dibert, D.; McTague, S.; Eskander, M.; Duncan, R.; Wang, L.; Nohe, A. CK2.3, a Mimetic Peptide of the BMP Type I Receptor, Increases Activity in Osteoblasts over BMP2. Int. J. Mol. Sci. 2019, 20, 5877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Endochondral bone formation. The phases of bone maturation and remodeling. (A) condensation of mesenchymal stem cells differentiating into chondrocytes. (B) Chondrocyte cells organize into a framework for the maturing skeletal element. (C) Initial vascularization beginning primary ossification at a long bone diaphysis. (D) Formation of secondary ossification centers and initiation of osteoclast-driven remodeling of medullary cavity and spongy bone. (E) Mature skeletal element with fully formed trabecular bone structure and medullary cavity filled with bone marrow.
Figure 1.
Endochondral bone formation. The phases of bone maturation and remodeling. (A) condensation of mesenchymal stem cells differentiating into chondrocytes. (B) Chondrocyte cells organize into a framework for the maturing skeletal element. (C) Initial vascularization beginning primary ossification at a long bone diaphysis. (D) Formation of secondary ossification centers and initiation of osteoclast-driven remodeling of medullary cavity and spongy bone. (E) Mature skeletal element with fully formed trabecular bone structure and medullary cavity filled with bone marrow.

Figure 2.
Maintenance of spongy bone microstructure. The epiphysial head of a long bone containing pores of adipocytes and stem cells (in beige). Surrounded by trabecular bone (in yellow). Osteoclasts (in blue) absorb bone and expand pores while osteoblasts (in pink) mineralize new bone closing pores and thickening trabecular bone. Cortical bone and medullary cavity run down to the diaphysis of the skeletal element (not shown in this image).
Figure 2.
Maintenance of spongy bone microstructure. The epiphysial head of a long bone containing pores of adipocytes and stem cells (in beige). Surrounded by trabecular bone (in yellow). Osteoclasts (in blue) absorb bone and expand pores while osteoblasts (in pink) mineralize new bone closing pores and thickening trabecular bone. Cortical bone and medullary cavity run down to the diaphysis of the skeletal element (not shown in this image).

Figure 3.
Osteoclast development. Hematopoietic precursor cells stimulated by mammalian colony stimulating factor (M-CSF) are driven toward the osteoclast lineage. M-CSF stimulates transcription factor PU.1 to express RANK on progenitor cells. CD14+ monocytes stimulated by RANKL, along with BMP4, help to upregulate the MITF and PU.1 transcription factors to increase expression of essential fusion proteins DC-STAMP, OC-STAMP and ATP6vOd2. Preosteoclasts fuse to form a polykaron. BMP2 and RANKL further the activation of mature osteoclasts and the transcription factors Microphthalmia-associated transcription factor (MITF), Nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) and nuclear factor kappa light chain enhancer of activated B cells (NFKB) upregulate osteoclast specific markers tartrate-resistant acid phosphatase (TRAP) and cathepsin k (CATH K). Mature osteoclasts form resorptive pits, or Howship lacunae, by pumping CATH K and TRAP against the bone surface, breaking down the mineralized bone. OPG, Noggin and Twisted gastrulation 1 (TWSG1) inhibit RANKL and BMP mediate osteoclast fusion and activation.
Figure 3.
Osteoclast development. Hematopoietic precursor cells stimulated by mammalian colony stimulating factor (M-CSF) are driven toward the osteoclast lineage. M-CSF stimulates transcription factor PU.1 to express RANK on progenitor cells. CD14+ monocytes stimulated by RANKL, along with BMP4, help to upregulate the MITF and PU.1 transcription factors to increase expression of essential fusion proteins DC-STAMP, OC-STAMP and ATP6vOd2. Preosteoclasts fuse to form a polykaron. BMP2 and RANKL further the activation of mature osteoclasts and the transcription factors Microphthalmia-associated transcription factor (MITF), Nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) and nuclear factor kappa light chain enhancer of activated B cells (NFKB) upregulate osteoclast specific markers tartrate-resistant acid phosphatase (TRAP) and cathepsin k (CATH K). Mature osteoclasts form resorptive pits, or Howship lacunae, by pumping CATH K and TRAP against the bone surface, breaking down the mineralized bone. OPG, Noggin and Twisted gastrulation 1 (TWSG1) inhibit RANKL and BMP mediate osteoclast fusion and activation.

Figure 4.
Osteoclasts communicate with immune cells and other bone cells. Osteoclasts communicate with immune cells through the releases of inflammatory cytokines. Osteoblast and osteocytes release soluble RANKL as well as osteoprotegerin (OPG) to communicate with osteoclasts. Osteocytes and osteoblasts also communicate with Osteoclasts via direct cell–cell contact utilizing membrane-bound RANKL.
Figure 4.
Osteoclasts communicate with immune cells and other bone cells. Osteoclasts communicate with immune cells through the releases of inflammatory cytokines. Osteoblast and osteocytes release soluble RANKL as well as osteoprotegerin (OPG) to communicate with osteoclasts. Osteocytes and osteoblasts also communicate with Osteoclasts via direct cell–cell contact utilizing membrane-bound RANKL.

Figure 5.
The BMP-signaling pathway and RANKL signaling overlap. BMP stimulates the downstream signaling pathways by activating BMP receptors BMPRIa and BMPRII. The canonical SMAD pathway transduces the signal into the nucleus with the help of co-SMADs (SMAD4). The noncanonical BMP-signal is transduced by the recruitment of TAB2/TAK1 complex the BMP receptor through XIAP. Thus, activating the same downstream effector proteins as the RANKL signaling pathway. These proteins can then modulate the expression of osteoclast genes.
Figure 5.
The BMP-signaling pathway and RANKL signaling overlap. BMP stimulates the downstream signaling pathways by activating BMP receptors BMPRIa and BMPRII. The canonical SMAD pathway transduces the signal into the nucleus with the help of co-SMADs (SMAD4). The noncanonical BMP-signal is transduced by the recruitment of TAB2/TAK1 complex the BMP receptor through XIAP. Thus, activating the same downstream effector proteins as the RANKL signaling pathway. These proteins can then modulate the expression of osteoclast genes.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Heubel, B.; Nohe, A. The Role of BMP Signaling in Osteoclast Regulation. J. Dev. Biol. 2021, 9, 24. https://0-doi-org.brum.beds.ac.uk/10.3390/jdb9030024
AMA Style
Heubel B, Nohe A. The Role of BMP Signaling in Osteoclast Regulation. Journal of Developmental Biology. 2021; 9(3):24. https://0-doi-org.brum.beds.ac.uk/10.3390/jdb9030024
Chicago/Turabian StyleHeubel, Brian, and Anja Nohe. 2021. "The Role of BMP Signaling in Osteoclast Regulation" Journal of Developmental Biology 9, no. 3: 24. https://0-doi-org.brum.beds.ac.uk/10.3390/jdb9030024
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.