
Transcriptomic Analysis for the Identification of Metabolic Pathway Genes Related to Toluene Response in Ardisia pusilla

Abstract
:1. Introduction
2. Results
2.1. Illumina Sequencing and De Novo Assembly
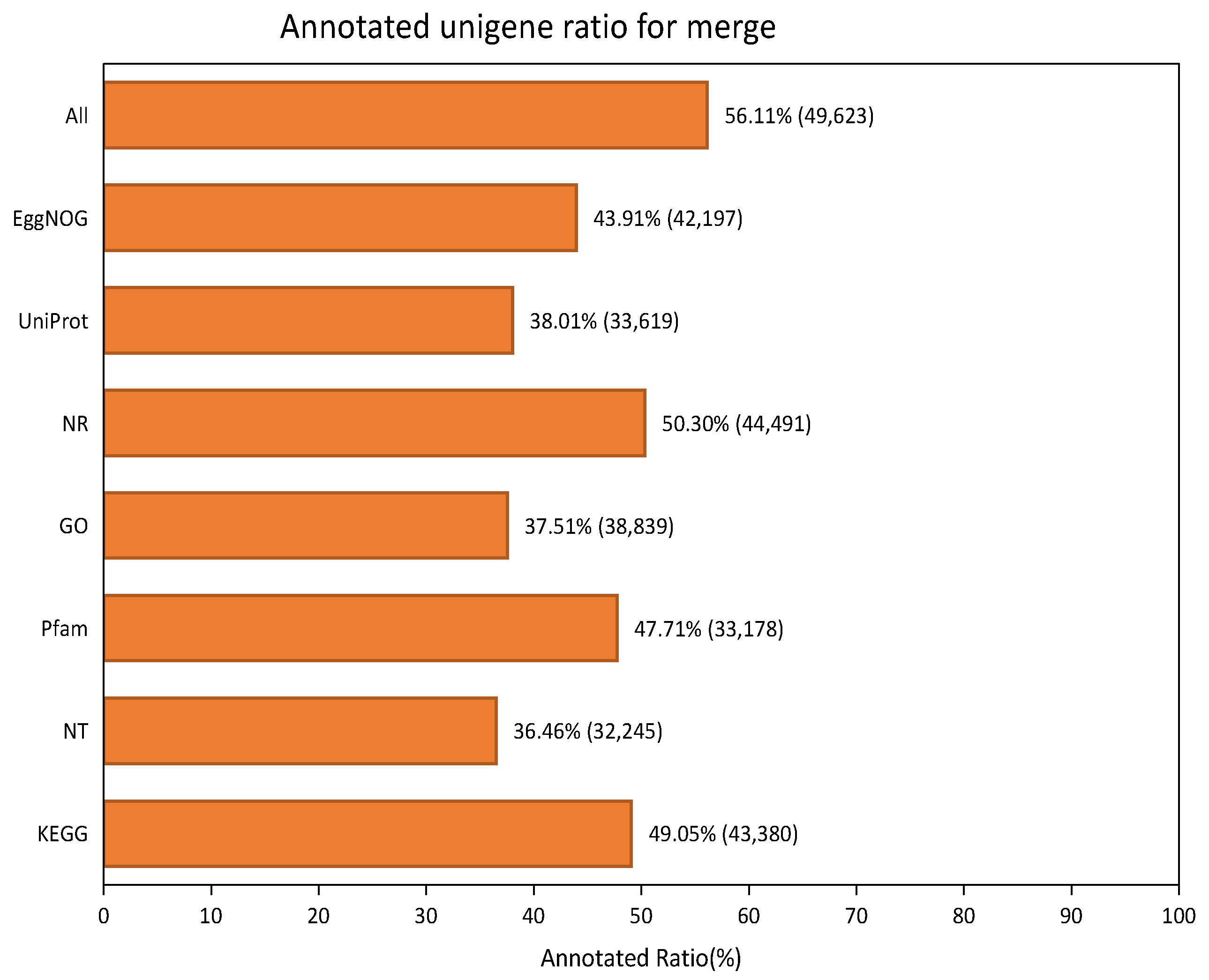
2.2. Annotation Results
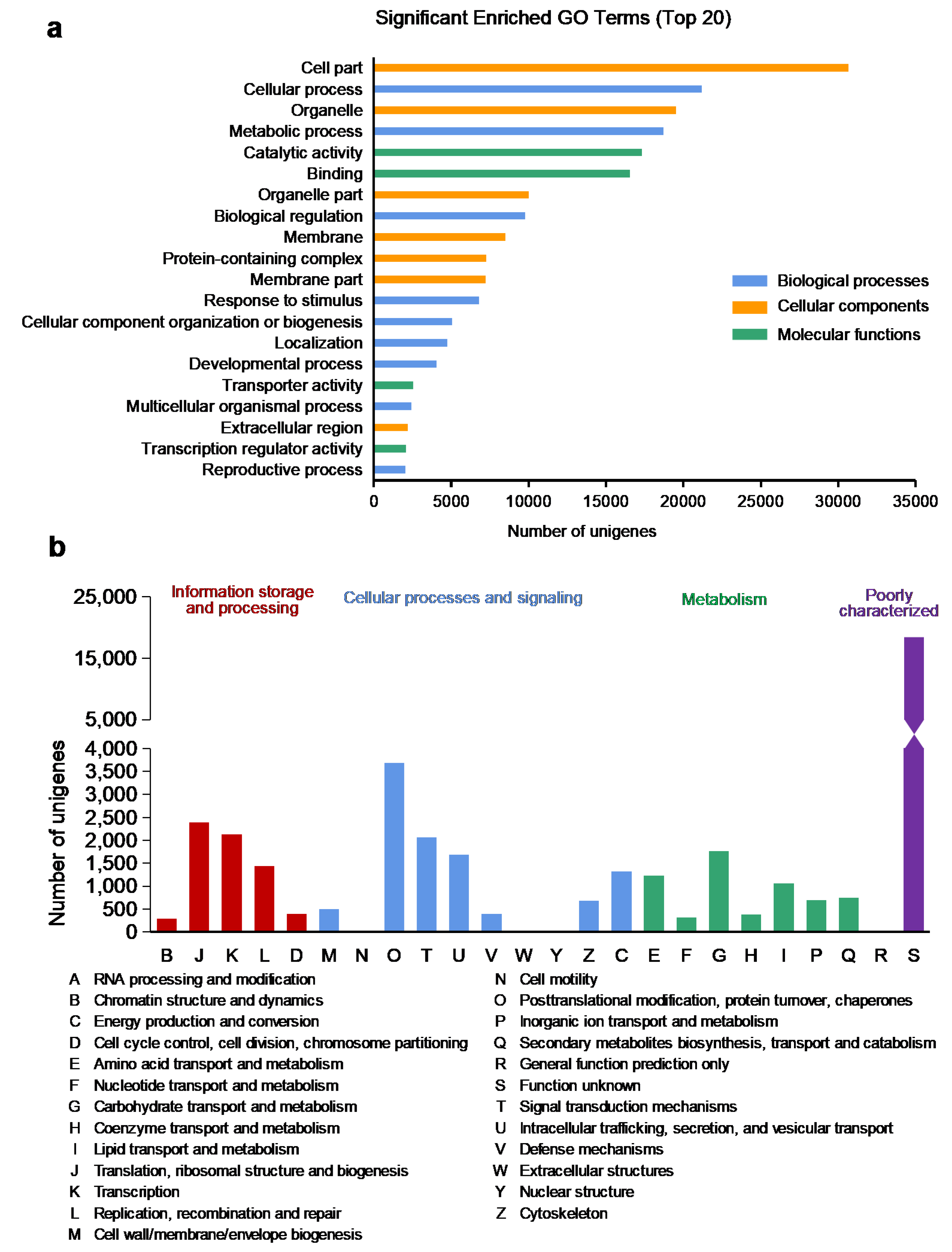
2.3. Annotation on GO Database
2.4. Annotation on EggNOG Database
2.5. DEG Analysis Results
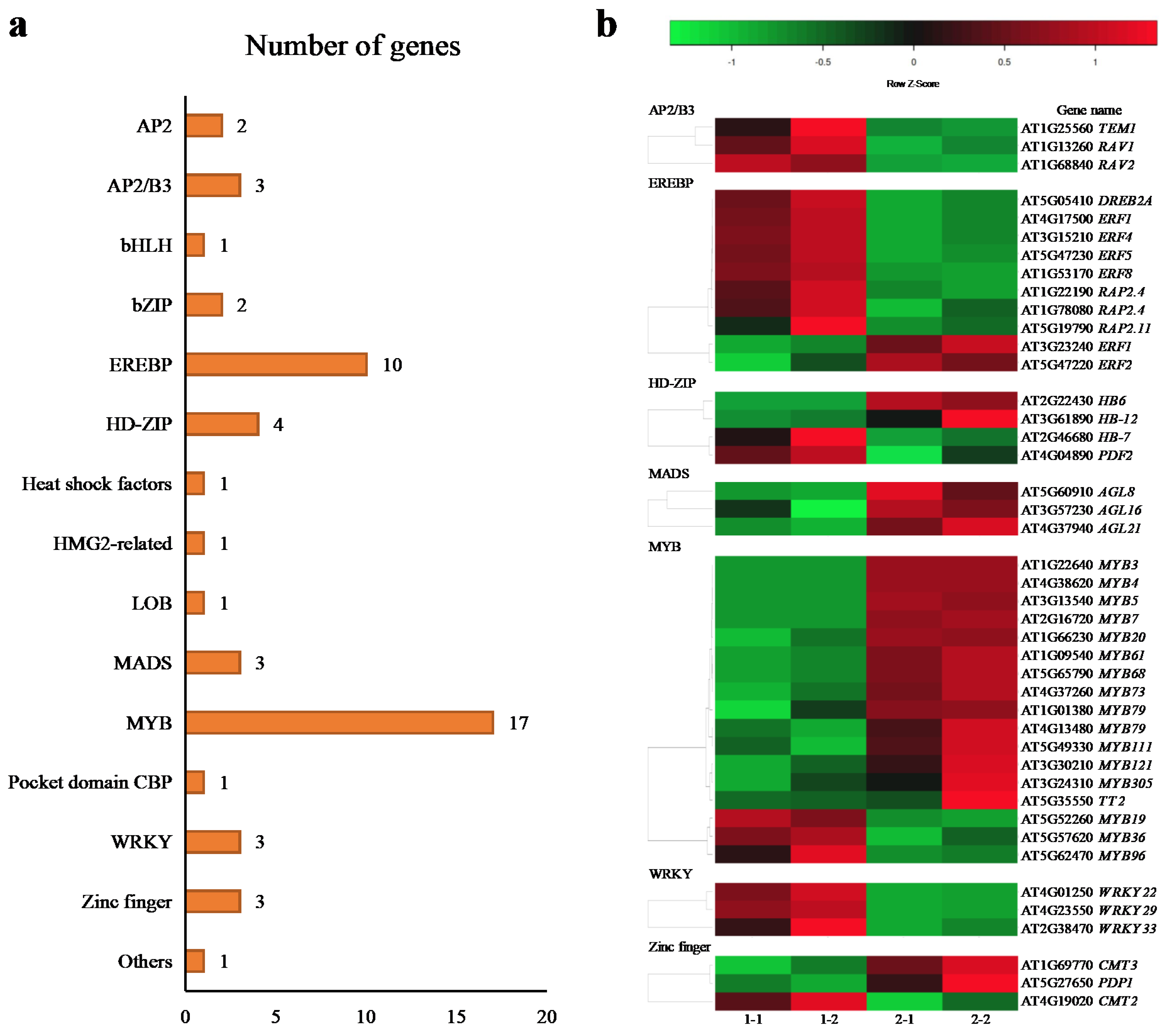
2.6. Transcriptional Factors (TFs)
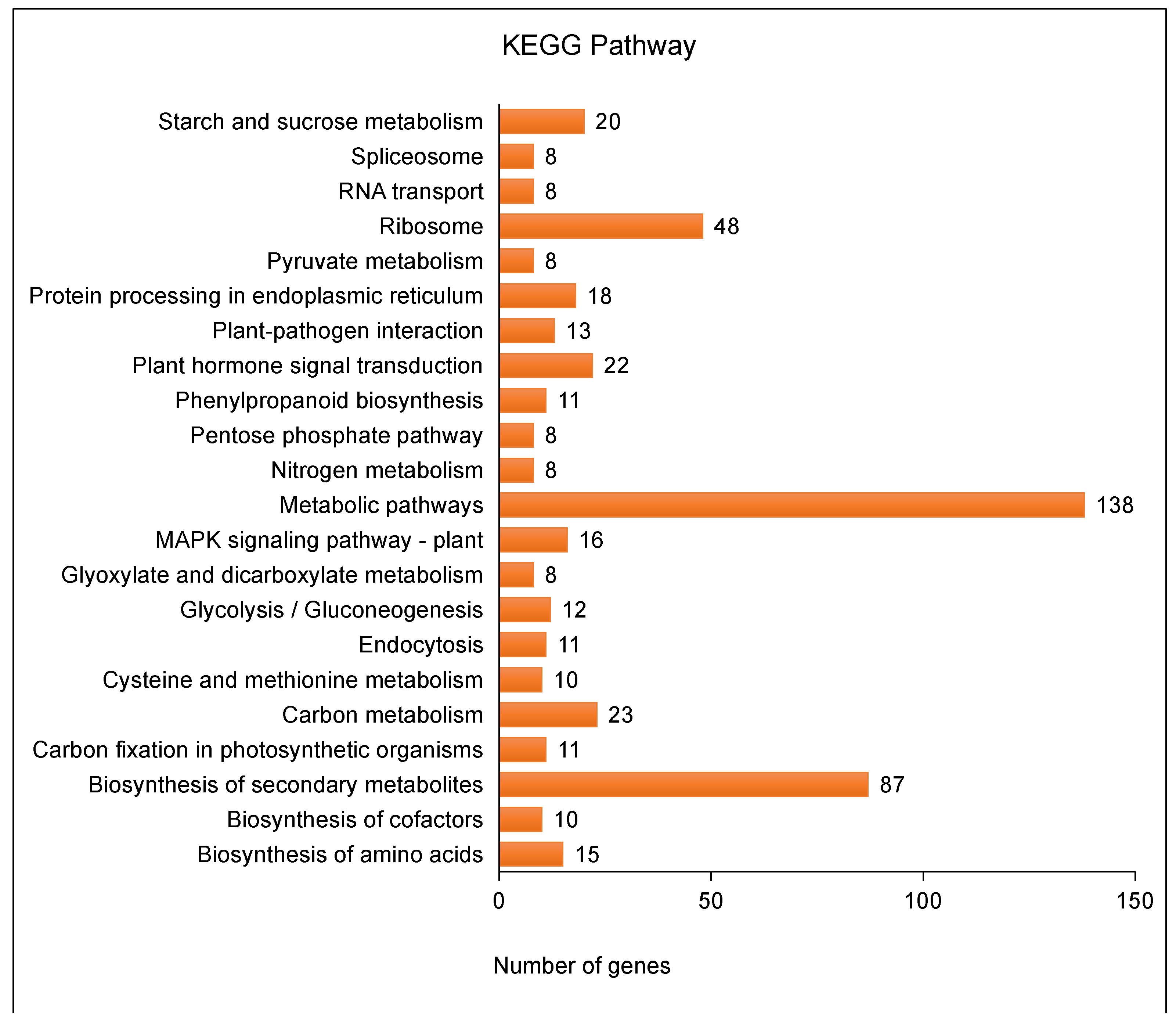
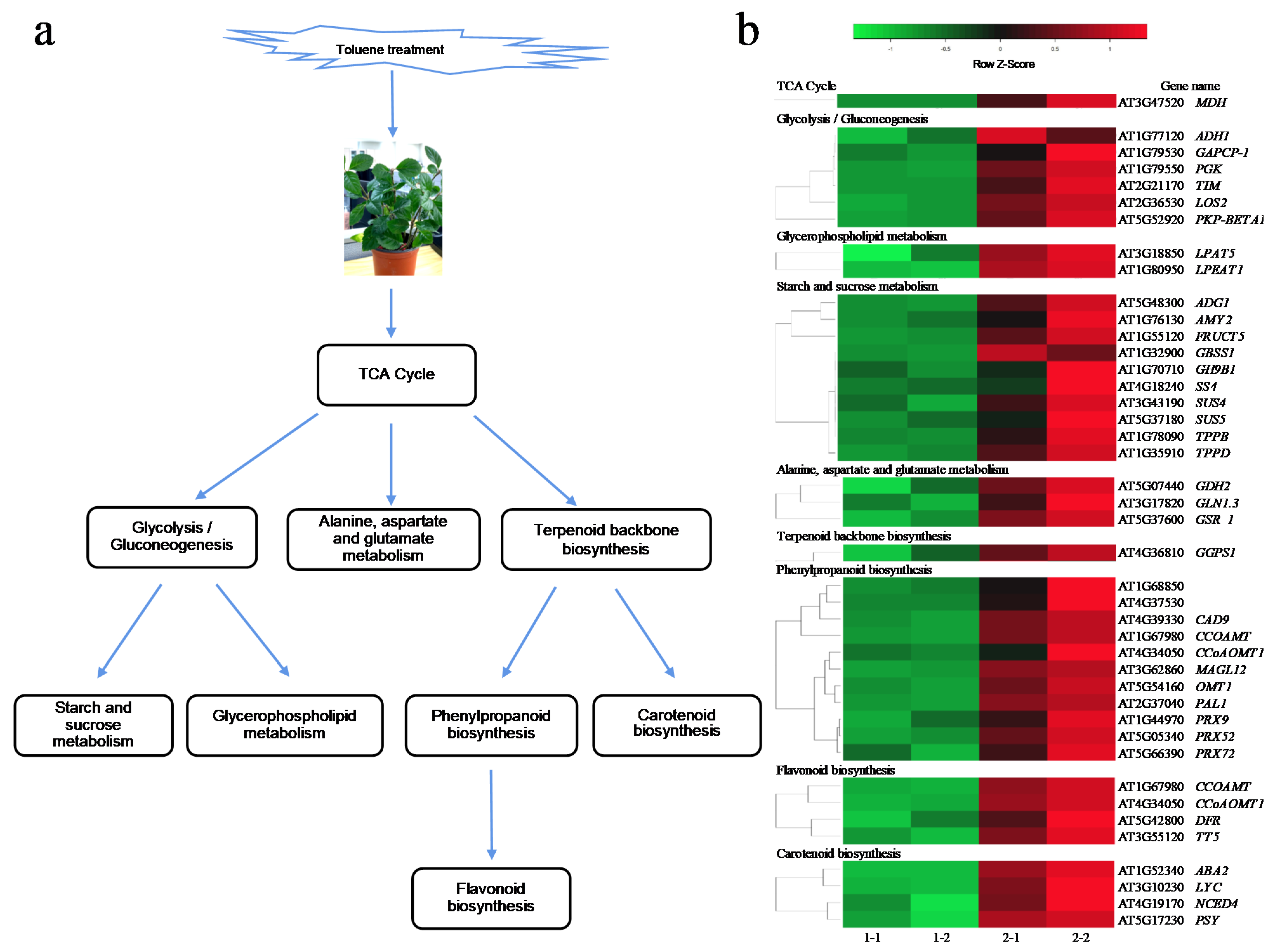
2.7. Toluene Metabolic Pathway
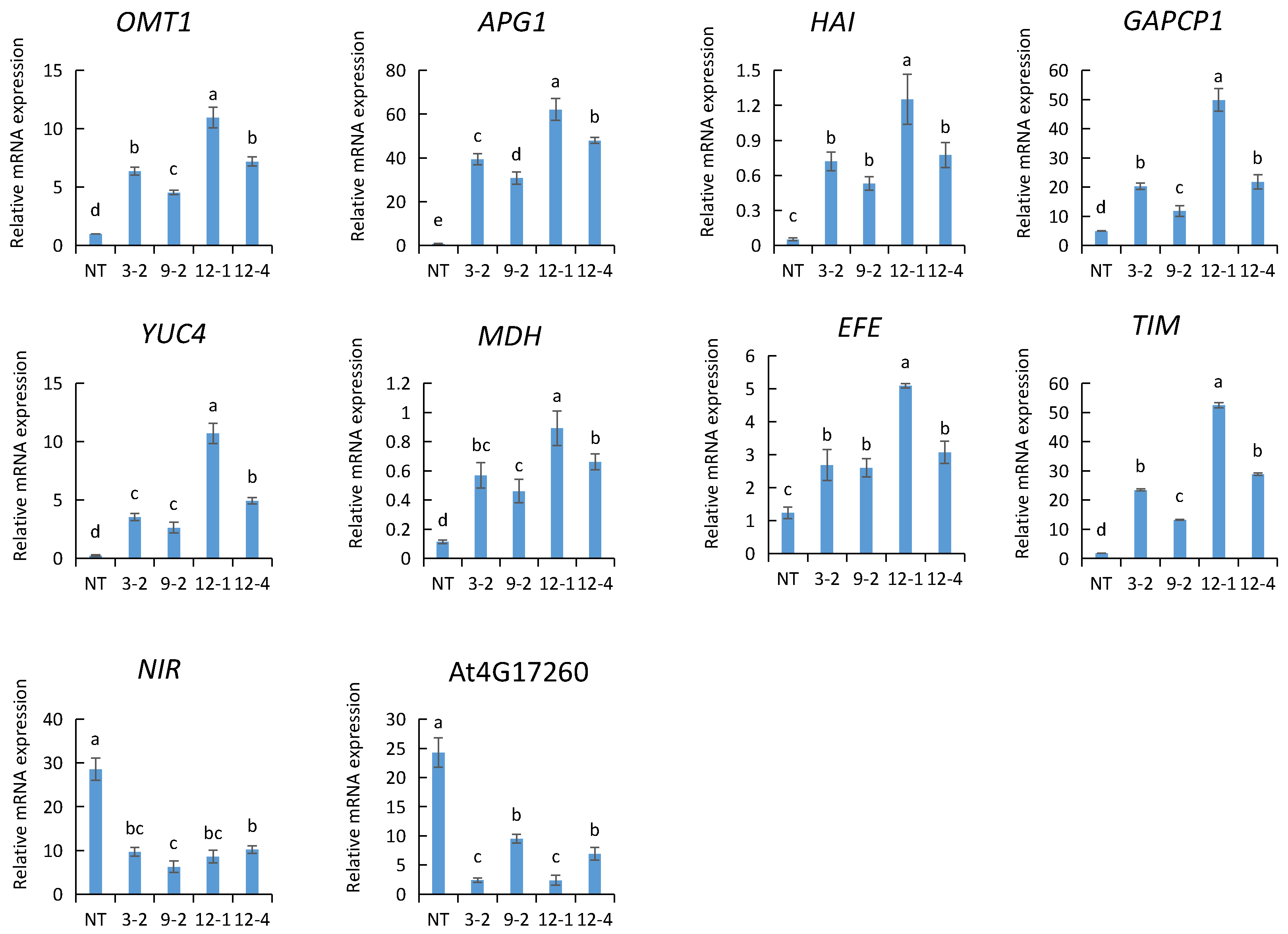
2.8. Validation of RNA-Seq Results by qRT-PCR
2.9. Gene Expression Analysis Related to Toluene Purification in AtNDPK2-Transgenic A. pusilla Plants
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Toluene Treatment Conditions
4.2. RNA Extraction
4.3. Deep Sequencing
4.4. Sequence Data Analysis and De Novo Assembly
4.5. Sequence Annotation and Classification
4.6. Identification of DEGs
4.7. Validation of RNA-Seq Analysis by qRT-PCR
4.8. Gene Expression Analysis Related to Toluene Purification in AtNDPK2-Transgenic A. pusilla Lines
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flowers, L.; Boyes, W.; Foster, S.; Gelhaus, M.; Hogan, K.; Marcus, A.; McClure, P.; Osier, M.; Ronney, A. Toxicological Review of Toluene (CAS No. 108–88-3); US Environmental Protection Agency: Washington, DC, USA, 2005.
- Muccee, F.; Ejaz, S.; Riaz, N. Toluene degradation via a unique metabolic route in indigenous bacterial species. Arch. Microbiol. 2019, 201, 1369–1383. [Google Scholar] [CrossRef]
- Becker, J.M.; Neal, M.W. Drinking Water Criteria Document for Toluene; Final Report (No. PB-91-143487/XAB); Syracuse Research Corp.: New York, NY, USA, 1990. [Google Scholar]
- De Bortoli, M.; Knöppel, H.; Pecchio, E.; Peil, A.; Rogora, L.; Schauenburg, H.; Schlitt, H.; Vissers, H. Concentrations of selected organic pollutants in indoor and outdoor air in northern Italy. Environ. Int. 1986, 12, 343–350. [Google Scholar] [CrossRef]
- Faradisha, J.; Tualeka, A.R.; Widajati, N. Analysis of Correlation between Toluene Exposure and Health Risk Characterization on Printing Worker of Plastic Bags Industry. Indian J. Public Health Res. Dev. 2019, 10, 351–355. [Google Scholar] [CrossRef]
- Shih, H.-T.; Yu, C.-L.; Wu, M.-T.; Liu, C.-S.; Tsai, C.-H.; Hung, D.-Z.; Wu, C.-S.; Kuo, H.-W. Subclinical abnormalities in workers with continuous low-level toluene exposure. Toxicol. Ind. Health 2011, 27, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Singhai, A. Toluene-induced acute lung injury. Med. J. DY Patil Univ. 2013, 6, 82. [Google Scholar] [CrossRef]
- Irga, P.J.; Pettit, T.; Irga, R.F.; Paull, N.J.; Douglas, A.N.; Torpy, F.R. Does plant species selection in functional active green walls influence VOC phytoremediation efficiency? Environ. Sci. Pollut. Res. 2019, 26, 12851–12858. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.J.; Khalekuzzaman, M.; Suh, J.N.; Kim, H.J.; Shagol, C.; Kim, H.-H.; Kim, H.J. Phytoremediation of volatile organic compounds by indoor plants: A review. Hortic. Environ. Biotechnol. 2018, 59, 143–157. [Google Scholar] [CrossRef]
- Hörmann, V.; Brenske, K.-R.; Ulrichs, C. Assessment of filtration efficiency and physiological responses of selected plant species to indoor air pollutants (toluene and 2-ethylhexanol) under chamber conditions. Environ. Sci. Pollut. Res. 2018, 25, 447–458. [Google Scholar] [CrossRef]
- Kim, K.J.; Yoo, E.H.; Jeong, M.I.; Song, J.S.; Lee, S.Y.; Kays, S.J. Changes in the phytoremediation potential of indoor plants with exposure to toluene. HortScience 2011, 46, 1646–1649. [Google Scholar] [CrossRef]
- Sriprapat, W.; Suksabye, P.; Areephak, S.; Klantup, P.; Waraha, A.; Sawattan, A.; Thiravetyan, P. Uptake of toluene and ethylbenzene by plants: Removal of volatile indoor air contaminants. Ecotoxicol. Environ. Saf. 2014, 102, 147–151. [Google Scholar] [CrossRef]
- Group, A.P. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar]
- Ahn, C.H.; Kim, N.-S.; Shin, J.Y.; Lee, Y.A.; Kim, K.J.; Kim, J.H.; Park, P.M.; An, H.R.; Kim, Y.-J.; Kim, W.H. Enhanced detoxification of exogenous toluene gas in transgenic Ardisia pusilla expressing AtNDPK2 gene. Hortic. Environ. Biotechnol. 2020, 61, 949–957. [Google Scholar] [CrossRef]
- Song, J.E. A study on the reduction of volatile organic compounds by Fatsia japonica and Ardisia pusilla. J. Korea Inst. Ecol. Archit. Environ. 2012, 12, 77–82. [Google Scholar]
- Kim, Y.-H.; Lim, S.; Yang, K.-S.; Kim, C.Y.; Kwon, S.-Y.; Lee, H.-S.; Wang, X.; Zhou, Z.; Ma, D.; Yun, D.-J. Expression of Arabidopsis NDPK2 increases antioxidant enzyme activities and enhances tolerance to multiple environmental stresses in transgenic sweetpotato plants. Mol. Breed. 2009, 24, 233–244. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Ke, Q.; Jeong, J.C.; Lee, H.-S.; Xu, B.; Deng, X.-P.; Lim, Y.P.; Kwak, S.-S. Transgenic alfalfa plants expressing AtNDPK2 exhibit increased growth and tolerance to abiotic stresses. Plant Physiol. Biochem. 2014, 84, 67–77. [Google Scholar] [CrossRef]
- Zhang, J.; Movahedi, A.; Sang, M.; Wei, Z.; Xu, J.; Wang, X.; Wu, X.; Wang, M.; Yin, T.; Zhuge, Q. Functional analyses of NDPK2 in Populus trichocarpa and overexpression of PtNDPK2 enhances growth and tolerance to abiotic stresses in transgenic poplar. Plant Physiol. Biochem. 2017, 117, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Ramya, M.; Park, P.H.; Chuang, Y.-C.; Kwon, O.K.; An, H.R.; Park, P.M.; Baek, Y.S.; Kang, B.-C.; Tsai, W.-C.; Chen, H.-H. RNA sequencing analysis of Cymbidium goeringii identifies floral scent biosynthesis related genes. BMC Plant Biol. 2019, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Le, X.C.; Zhu, L. Metabolomics and transcriptomics reveal defense mechanism of rice (Oryza sativa) grains under stress of 2, 2’, 4, 4’-tetrabromodiphenyl ether. Environ. Int. 2019, 133, 105154. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Xue, X.; Liu, Z.; Ye, Y.; Xiao, P.; Pu, Y.; Guan, W.; Mwacharo, J.M.; Ma, Y.; Zhao, Q. Expression Profiling and Functional Characterization of miR-26a and miR-130a in Regulating Zhongwei Goat Hair Development via the TGF-β/SMAD Pathway. Int. J. Mol. Sci. 2020, 21, 5076. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Xu, H.; Zhai, J.; Li, D.; Luo, H.; He, C.; Huang, X. RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis. Plant Mol. Biol. 2011, 77, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Altermann, E.; Klaenhammer, T.R. PathwayVoyager: Pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genom. 2005, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Durmishidze, S. Cleavage of the Aromatic Ring of Some Exogenous Compounds in Plants; Metsniereba: Tbilisi, GA, USA, 1975. [Google Scholar]
- Ugrekhelidze, D.S. Metabolizm Ekzogennykh Alkanov i Aromaticheskikh Uglevodorodov v Rasteniyakh (Metabolism of Exogenous Alkanes and Aromatic Hydrocarbons in Plants); Metsniereba: Tbilisi, Georgia, 1976; pp. 77–84. [Google Scholar]
- Ugrekhelidze, D.; Korte, F.; Kvesitadze, G. Uptake and transformation of benzene and toluene by plant leaves. Ecotoxicol. Environ. Saf. 1997, 37, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.B.; Foley, R.C.; Oñate-Sánchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef]
- Zhang, J.Z. Overexpression analysis of plant transcription factors. Curr. Opin. Plant Biol. 2003, 6, 430–440. [Google Scholar] [CrossRef]
- Ma, D.; Constabel, C.P. MYB repressors as regulators of phenylpropanoid metabolism in plants. Trends Plant Sci. 2019, 24, 275–289. [Google Scholar] [CrossRef]
- Ampomah-Dwamena, C.; Thrimawithana, A.H.; Dejnoprat, S.; Lewis, D.; Espley, R.V.; Allan, A.C. A kiwifruit (Actinidia deliciosa) R2R3-MYB transcription factor modulates chlorophyll and carotenoid accumulation. New Phytol. 2019, 221, 309–325. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Wang, Z.; Wang, Y.; Wang, C.; Zhu, B.; Liu, H.; Ji, W.; Wen, J.; Chu, C.; Tadege, M. The MYB activator WHITE PETAL1 associates with MtTT8 and MtWD40-1 to regulate carotenoid-derived flower pigmentation in Medicago truncatula. Plant Cell 2019, 31, 2751–2767. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Fan, R.; Guo, S.; Wang, P.; Zhu, X.; Fan, Y.; Chen, Y.; He, K.; Kumar, A.; Shi, J. The Arabidopsis MYB transcription factor, MYB111 modulates salt responses by regulating flavonoid biosynthesis. Environ. Exp. Bot. 2019, 166, 103807. [Google Scholar] [CrossRef]
- Wang, X.C.; Wu, J.; Guan, M.L.; Zhao, C.H.; Geng, P.; Zhao, Q. Arabidopsis MYB4 plays dual roles in flavonoid biosynthesis. Plant J. 2020, 101, 637–652. [Google Scholar] [CrossRef]
- Geng, P.; Zhang, S.; Liu, J.; Zhao, C.; Wu, J.; Cao, Y.; Fu, C.; Han, X.; He, H.; Zhao, Q. MYB20, MYB42, MYB43, and MYB85 regulate phenylalanine and lignin biosynthesis during secondary cell wall formation. Plant Physiol. 2020, 182, 1272–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.B.; Kim, H.U.; Suh, M.C. MYB94 and MYB96 additively activate cuticular wax biosynthesis in Arabidopsis. Plant Cell Physiol. 2016, 57, 2300–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-S.; Song, Z.-B.; Sun, Z.; Zhang, J.; Mei, Y.; Nian, H.-J.; Li, K.-Z.; Chen, L.-M. Physiological and transcriptional analysis of the effects of formaldehyde exposure on Arabidopsis thaliana. Acta Physiol. Plant. 2012, 34, 923–936. [Google Scholar] [CrossRef]
- Caarls, L.; Van der Does, D.; Hickman, R.; Jansen, W.; Verk, M.C.V.; Proietti, S.; Lorenzo, O.; Solano, R.; Pieterse, C.M.; Van Wees, S. Assessing the role of ETHYLENE RESPONSE FACTOR transcriptional repressors in salicylic acid-mediated suppression of jasmonic acid-responsive genes. Plant Cell Physiol. 2017, 58, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.Y.; Ohta, M.; Usui, A.; Shinshi, H.; Ohme-Takagi, M. Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box–mediated gene expression. Plant Cell 2000, 12, 393–404. [Google Scholar]
- Son, G.H.; Wan, J.; Kim, H.J.; Nguyen, X.C.; Chung, W.S.; Hong, J.C.; Stacey, G. Ethylene-responsive element-binding factor 5, ERF5, is involved in chitin-induced innate immunity response. Mol. Plant Microbe Interact. 2012, 25, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Wehner, N.; Hartmann, L.; Ehlert, A.; Böttner, S.; Oñate-Sánchez, L.; Dröge-Laser, W. High-throughput protoplast transactivation (PTA) system for the analysis of Arabidopsis transcription factor function. Plant J. 2011, 68, 560–569. [Google Scholar] [CrossRef]
- Yang, Z.; Tian, L.; Latoszek-Green, M.; Brown, D.; Wu, K. Arabidopsis ERF4 is a transcriptional repressor capable of modulating ethylene and abscisic acid responses. Plant Mol. Biol. 2005, 58, 585–596. [Google Scholar] [CrossRef]
- Chen, W.-H.; Li, P.-F.; Chen, M.-K.; Lee, Y.-I.; Yang, C.-H. FOREVER YOUNG FLOWER negatively regulates ethylene response DNA-binding factors by activating an ethylene-responsive factor to control Arabidopsis floral organ senescence and abscission. Plant Physiol. 2015, 168, 1666–1683. [Google Scholar] [CrossRef]
- Osnato, M.; Cereijo, U.; Sala, J.; Matías-Hernández, L.; Aguilar-Jaramillo, A.E.; Rodríguez-Goberna, M.R.; Riechmann, J.L.; Rodríguez-Concepción, M.; Pelaz, S. The floral repressors TEMPRANILLO1 and 2 modulate salt tolerance by regulating hormonal components and photo-protection in Arabidopsis. Plant J. 2021, 105, 7–21. [Google Scholar] [CrossRef]
- Fu, M.; Kang, H.K.; Son, S.-H.; Kim, S.-K.; Nam, K.H. A subset of Arabidopsis RAV transcription factors modulates drought and salt stress responses independent of ABA. Plant Cell Physiol. 2014, 55, 1892–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, F.-C.; Chou, M.-Y.; Chou, S.-J.; Li, Y.-R.; Peng, H.-P.; Shih, M.-C. Submergence confers immunity mediated by the WRKY22 transcription factor in Arabidopsis. Plant Cell 2013, 25, 2699–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, W.; Wang, X.; Chen, M.; Chai, T.; Wang, H. A WRKY transcription factor, PcWRKY33, from Polygonum cuspidatum reduces salt tolerance in transgenic Arabidopsis thaliana. Plant Cell Rep. 2018, 37, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Snider, N.T.; Altshuler, P.J.; Omary, M.B. Modulation of cytoskeletal dynamics by mammalian nucleoside diphosphate kinase (NDPK) proteins. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Weisman, D.; Tang, L.; Tan, L.; Zhang, W.-K.; Wang, Z.-H.; Huang, Y.-H.; Lin, W.-X.; Liu, X.-M.; Colón-Carmona, A. Stress signaling in response to polycyclic aromatic hydrocarbon exposure in Arabidopsis thaliana involves a nucleoside diphosphate kinase, NDPK-3. Planta 2015, 241, 95–107. [Google Scholar] [CrossRef]
- Luzarowski, M.; Kosmacz, M.; Sokolowska, E.; Jasińska, W.; Willmitzer, L.; Veyel, D.; Skirycz, A. Affinity purification with metabolomic and proteomic analysis unravels diverse roles of nucleoside diphosphate kinases. J. Exp. Bot. 2017, 68, 3487–3499. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The gene ontology project in 2008. Nucleic Acids Res. 2008, 36, D440–D444. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | 1-1 | 1-2 | 2-1 | 2-2 |
|---|---|---|---|---|
| Total raw reads | 137,518,750 | 165,188,286 | 144,957,778 | 138,077,056 |
| Total trimmed reads | 135,591,762 | 162,623,598 | 142,958,464 | 136,257,376 |
| Q20 (%) | 98.90 | 98.78 | 98.82 | 98.86 |
| Q30 (%) | 96.03 | 95.69 | 95.78 | 95.87 |
| GC (%) | 50.30 | 49.99 | 50.52 | 49.37 |
| Mapped reads | 100,780,248 (74.33%) | 117,220,734 (72.08%) | 110,070,246 (76.99%) | 97,991,310 (71.92%) |
| Unmapped reads | 34,811,514 (25.67%) | 45,402,864 (27.92%) | 32,888,218 (23.01%) | 38,266,066 (28.08%) |
| DEGs Set | Total DEGs | KEGG DEGs | GO DEGs | |||
|---|---|---|---|---|---|---|
| Total | Biological Processes | Cellular Components | Molecular Functions | |||
| All DEGs | 4101 | 3101 | 2854 | 2393 | 2525 | 2547 |
| Up regulation | 2100 (51.2%) | 1610 (51.9%) | 1456 (51.0%) | 1234 (51.6%) | 1262 (50.0%) | 1315 (51.6%) |
| Down regulation | 2001 (48.8%) | 1491 (48.1%) | 1398 (49.0%) | 1159 (48.4%) | 1263 (50.0%) | 1232 (48.4%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Ahn, C.H.; Shin, J.Y.; Park, P.M.; An, H.R.; Kim, Y.-J.; Lee, S.Y. Transcriptomic Analysis for the Identification of Metabolic Pathway Genes Related to Toluene Response in Ardisia pusilla. Plants 2021, 10, 1011. https://0-doi-org.brum.beds.ac.uk/10.3390/plants10051011
Xu J, Ahn CH, Shin JY, Park PM, An HR, Kim Y-J, Lee SY. Transcriptomic Analysis for the Identification of Metabolic Pathway Genes Related to Toluene Response in Ardisia pusilla. Plants. 2021; 10(5):1011. https://0-doi-org.brum.beds.ac.uk/10.3390/plants10051011
Chicago/Turabian StyleXu, Junping, Chang Ho Ahn, Ju Young Shin, Pil Man Park, Hye Ryun An, Yae-Jin Kim, and Su Young Lee. 2021. "Transcriptomic Analysis for the Identification of Metabolic Pathway Genes Related to Toluene Response in Ardisia pusilla" Plants 10, no. 5: 1011. https://0-doi-org.brum.beds.ac.uk/10.3390/plants10051011