
Detection of QTLs for Plant Height Architecture Traits in Rice (Oryza sativa L.) by Association Mapping and the RSTEP-LRT Method



 ,
,
Abstract
:1. Introduction
2. Results
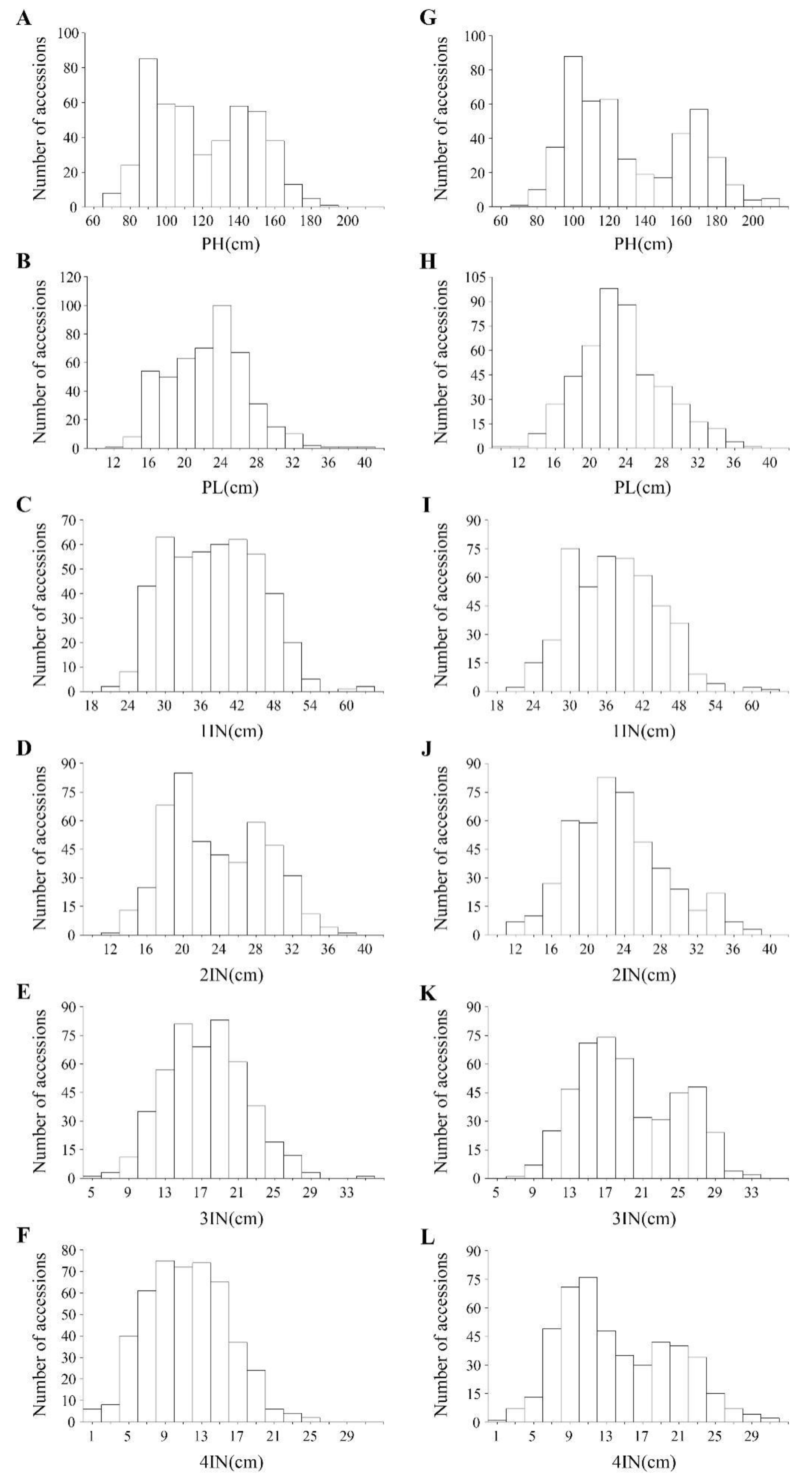
2.1. Phenotypic Evaluations of the Six Traits in the Natural Population Composed of 504 Rice Accessions
2.2. Genetic Diversity Revealed by SSR Molecular Markers
2.3. Population Structure and Genetic Relatedness
2.4. Genetic Differentiation among Subpopulations
2.5. Phenotypic Variation of the Six Traits in the XC-CSSL Population and Its Parents
2.6. Association Mapping and Discovery of Favorable Alleles for PH and Its Component Traits in the Natural Population
2.6.1. SSR Association Loci and Favorable Alleles for PH in the Natural Population
2.6.2. SSR Association Loci and Favorable Alleles for PL in the Natural Population
2.6.3. SSR Association Loci and Favorable Alleles for 1IN in the Natural Population
2.6.4. SSR Association Loci and Favorable Alleles for 2IN in the Natural Population
2.6.5. SSR Association Loci and Favorable Alleles for 3IN in the Natural Population
2.6.6. SSR Association Loci and Favorable Alleles for 4IN in the Natural Population
2.7. QTL Identification for PH and Its Component Traits in XC-CSSL Population
2.8. Comparison of Mapping Results in the Natural and XC-CSSL Populations
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Field Planting and Trait Measurement
4.3. Phenotypic Data Analysis and Heritability in a Broad Sense
4.4. DNA Extraction and SSR Marker Genotyping
4.5. Population Structure Analysis, Genetic Diversity and Phylogenesis
4.6. Linkage Disequilibrium Analysis
4.7. Association Mapping
4.8. QTL Detection for PH and Its Component Traits in the XC-CSSL Population
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sasaki, T.; Burr, B. International Rice Genome Sequencing Project: The effort to completely sequence the rice genome. Curr. Opin. Plant Biol. 2000, 3, 138–142. [Google Scholar] [CrossRef]
- Muthayya, S.; Sugimoto, J.D.; Montgomery, S.; Maberly, G.F. An overview of global rice production, supply, trade, and consumption. Ann. N. Y. Acad. Sci. 2014, 1324, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Statista. Leading Countries Based on the Production of Milled Rice in 2018/2019. 2020. Available online: https://0-www-statista-com.brum.beds.ac.uk/statistics/255945/top-countries-of-destination-for-us-rice-exports-2011/ (accessed on 12 October 2020).
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [Green Version]
- Khush, G.S. Green revolution: Preparing for the 21st century. Genome 1999, 42, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Ashikari, M.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Swapan, D.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.S. A mutant gibberellin-synthesis gene in rice. Nature 2002, 416, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zheng, S.; Gui, J.; Fu, C.; Yu, H.; Song, D.; Shen, J.; Qin, P.; Liu, X.; Han, B. Shortened basal internodes encodes a gibberellin 2-oxidase and contributes to lodging resistance in rice. Mol. Plant 2018, 11, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-H.; Zhuang, J.-Y.; Fan, Y.-Y.; Du, J.-H.; Cao, L.-Y. Progress in research and development on hybrid rice: A super-domesticate in China. Ann. Bot. 2007, 100, 959–966. [Google Scholar] [CrossRef]
- Rout, D.; Jena, D.; Singh, V.; Kumar, M.; Arsode, P.; Singh, P.; Katara, J.L.; Samantaray, S.; Verma, R.L. Hybrid Rice Research: Current Status and Prospects. In Recent Advances in Rice Research; IntechOpen: Rijeka, Croatia, 2020. [Google Scholar] [CrossRef]
- Sabouri, A.; Toorchi, M.; Rabiei, B.; Aharizad, S.; Moumeni, A.; Singh, R. Identification and mapping of QTLs for agronomic traits in indica—indica cross of rice (Oryza sativa L.). Cereal Res. Commun. 2010, 38, 317–326. [Google Scholar] [CrossRef]
- Sowadan, O.; Li, D.; Zhang, Y.; Zhu, S.; Hu, X.; Bhanbhro, L.B.; Edzesi, W.M.; Dang, X.; Hong, D. Mining of favorable alleles for lodging resistance traits in rice (Oryza sativa) through association mapping. Planta 2018, 248, 155–169. [Google Scholar] [CrossRef]
- Srividhya, A.; Vemireddy, L.R.; Sridhar, S.; Jayaprada, M.; Ramanarao, P.V.; Hariprasad, A.S.; Reddy, H.K.; Anuradha, G.; Siddiq, E. Molecular mapping of QTLs for yield and its components under two water supply conditions in rice (Oryza sativa L.). J. Crop Sci. Biotechnol. 2011, 14, 45–56. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, X.; Tong, H.; Lu, Y.; Li, J. Association mapping for important agronomic traits in core collection of rice (Oryza sativa L.) with SSR markers. PLoS ONE 2014, 9, e111508. [Google Scholar] [CrossRef] [Green Version]
- Kurotani, K.-I.; Hattori, T.; Takeda, S. Overexpression of a CYP94 family gene CYP94C2b increases internode length and plant height in rice. Plant Signal. Behav. 2015, 10, e1046667. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yu, C.; Lin, J.; Liu, J.; Liu, B.; Wang, J.; Huang, A.; Li, H.; Zhao, T. OsMPH1 regulates plant height and improves grain yield in rice. PLoS ONE 2017, 12, e0180825. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Li, L.; Zhang, Y.; Xu, C.; Li, X.; Xing, Y. Comparison of quantitative trait loci for rice yield, panicle length and spikelet density across three connected populations. J. Genet. 2011, 90, 377–382. [Google Scholar] [CrossRef]
- Cho, Y.G.; Kang, H.J.; Lee, J.S.; Lee, Y.T.; Lim, S.J.; Gauch, H.; Eun, M.Y.; McCouch, S.R. Identification of quantitative trait loci in rice for yield, yield components, and agronomic traits across years and locations. Crop Sci. 2007, 47, 2403–2417. [Google Scholar] [CrossRef]
- Marathi, B.; Guleria, S.; Mohapatra, T.; Parsad, R.; Mariappan, N.; Kurungara, V.K.; Atwal, S.S.; Prabhu, K.V.; Singh, N.K.; Singh, A.K. QTL analysis of novel genomic regions associated with yield and yield related traits in new plant type based recombinant inbred lines of rice (Oryza sativa L.). BMC Plant Biol. 2012, 12, 137. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, J.; Wang, J.; Wang, L.; Ma, B.; Zeng, L.; Qi, Y.; Li, Q.; He, Z. Quantitative trait locus analysis and fine mapping of the qPL6 locus for panicle length in rice. Theor. Appl. Genet. 2015, 128, 1151–1161. [Google Scholar] [CrossRef]
- Liu, E.; Liu, Y.; Wu, G.; Zeng, S.; Tran Thi, T.G.; Liang, L.; Liang, Y.; Dong, Z.; She, D.; Wang, H. Identification of a candidate gene for panicle length in rice (Oryza sativa L.) via association and linkage analysis. Front. Plant Sci. 2016, 7, 596. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Liu, W.; Tang, J.; Chen, J.; Tong, H.; Hu, B.; Li, C.; Fang, J.; Chen, M.; Chu, C. Rice DENSE AND ERECT PANICLE 2 is essential for determining panicle outgrowth and elongation. Cell Res. 2010, 20, 838–849. [Google Scholar] [CrossRef]
- Qiao, Y.; Piao, R.; Shi, J.; Lee, S.-I.; Jiang, W.; Kim, B.-K.; Lee, J.; Han, L.; Ma, W.; Koh, H.-J. Fine mapping and candidate gene analysis of dense and erect panicle 3, DEP3, which confers high grain yield in rice (Oryza sativa L.). Theor. Appl. Genet. 2011, 122, 1439–1449. [Google Scholar] [CrossRef]
- Bai, J.; Zhu, X.; Wang, Q.; Zhang, J.; Chen, H.; Dong, G.; Zhu, L.; Zheng, H.; Xie, Q.; Nian, J. Rice TUTOU1 encodes a suppressor of cAMP receptor-like protein that is important for actin organization and panicle development. Plant Physiol. 2015, 169, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.H.; Zhong, D.B.; Xu, J.L.; Yu, S.B.; Li, Z.K. Differential expression of lodging resistance related QTLs in rice (Oryza sativa L.). Plant Sci. 2008, 175, 898–905. [Google Scholar] [CrossRef]
- Dang, X.; Fang, B.; Chen, X.; Li, D.; Sowadan, O.; Dong, Z.; Liu, E.; She, D.; Wu, G.; Liang, Y.; et al. Favorable marker alleles for panicle exsertion length in rice (Oryza sativa L.) mined by association mapping and the RSTEP-LRT method. Front. Plant Sci. 2017, 8, 2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, A.; Qian, Q.; Yin, H.; Liu, X.; Yin, C.; Lan, Y.; Tang, J.; Tang, Z.; Cao, S.; Wang, X. EUI1, encoding a putative cytochrome P450 monooxygenase, regulates internode elongation by modulating gibberellin responses in rice. Plant Cell Physiol. 2006, 47, 181–191. [Google Scholar] [CrossRef]
- Zhu, Y.; Nomura, T.; Xu, Y.; Zhang, Y.; Peng, Y.; Mao, B.; Hanada, A.; Zhou, H.; Wang, R.; Li, P. Elongated uppermost internode encodes a cytochrome P450 monooxygenase that epoxidizes gibberellins in a novel deactivation reaction in rice. Plant Cell 2006, 18, 442–456. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Saeda, T.; Takeda, K.; Kitano, H. Dominant gene, Ssi1 expresses semidwarfism by inhibiting the second internode elongation in rice. Breed. Sci. 2000, 50, 17–22. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Sasaki, H.; Ishimaru, K. Factors responsible for decreasing sturdiness of the lower part in lodging of rice (Oryza sativa L.). Plant Prod. Sci. 2005, 8, 166–172. [Google Scholar] [CrossRef]
- Donde, R.; Mohapatra, S.; Baksh, S.K.Y.; Padhy, B.; Mukherjee, M.; Roy, S.; Chattopadhyay, K.; Anandan, A.; Swain, P.; Sahoo, K.K. Identification of QTLs for high grain yield and component traits in new plant types of rice. PLoS ONE 2020, 15, e0227785. [Google Scholar] [CrossRef]
- Swamy, B.M.; Shamsudin, N.A.A.; Abd Rahman, S.N.; Mauleon, R.; Ratnam, W.; Cruz, M.T.S.; Kumar, A.J.R. Association mapping of yield and yield-related traits under reproductive stage drought stress in rice (Oryza sativa L.). Rice 2017, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Chen, Y.; Ji, Z.; Liang, Y.; Zheng, A.; Wen, Z.; Yang, C. Control of plant height by 24 alleles at 12 quantitative trait loci in rice. Crop Breed. Appl. Biotechnol. 2019, 19, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, S.; Wu, W.; Chen, D.; Zhan, X.; Zhu, A.; Zhang, Y.; Cheng, S.; Cao, L.; Lou, X. Dissection of genetic architecture of rice plant height and heading date by multiple-strategy-based association studies. Sci. Rep. 2016, 6, 29718. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Liu, Q.; Lu, C.; Zhang, H.; Liu, X.; Dang, X.; Niu, F.; Breria, M.C.; Zhao, K.; Hong, D. Genetic basis dissection of heterosis in japonica rice (Oryza sativa L.). Acta Agron. Sin. 2012, 38, 2147–2161. [Google Scholar] [CrossRef]
- Bai, X.F.; Luo, L.J.; Yan, W.H.; Kovi, M.R.; Xing, Y.Z. Quantitative trait loci for rice yield-related traits using recombinant inbred lines derived from two diverse cultivars. J. Genet. 2011, 90, 209–215. [Google Scholar] [CrossRef]
- Hong, D.L.; Masahiko, I. Studies on Agronomic Characters of Short Root Mutants in Rice. Chin. J. Rice Sci. 1996, 10, 57–61. [Google Scholar]
- Wang, J.; Gang, S.; Yang, L.; Zheng, H.; Sun, J.; Liu, H.; Zhao, H.; Zou, D. Markers Associated with Culm Length and Elongated Internode Length in Japonica Rice. Crop Sci. 2017, 57, 2329–2344. [Google Scholar] [CrossRef]
- Yamamoto, T.; Taguchi-Shiobara, F.; Ukai, Y.; Sasaki, T.; Yano, M. Mapping quantitative trait loci for days-to-heading, and culm, panicle and internode lengths in a BC1F3 population using an elite rice variety, Koshihikari, as the recurrent parent. Breed. Sci. 2001, 51, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Andrew-Peter-Leon, M.T.; Selvaraj, R.; Kumar, K.K.; Muthamilarasan, M.; Yasin, J.K.; Pillai, M.A. Loss of function of OsFBX267 and OsGA20ox2 in rice promotes early maturing and semi-dwarfism in γ-irradiated IWP and genome-edited Pusa Basmati-1. Front. Plant Sci. 2021, 1968. [Google Scholar] [CrossRef]
- Bhuvaneswari, S.; Gopala Krishnan, S.; Ellur, R.K.; Vinod, K.K.; Bollinedi, H.; Bhowmick, P.K.; Bansal, V.P.; Nagarajan, M.; Singh, A.K. Discovery of a novel induced polymorphism in SD1 gene governing semi-dwarfism in rice and development of a functional marker for marker-assisted selection. Plants 2020, 9, 1198. [Google Scholar] [CrossRef]
- Meng, B.; Wang, T.; Luo, Y.; Xu, D.; Li, L.; Diao, Y.; Gao, Z.; Hu, Z.; Zheng, X. Genome-Wide Association Study Identified Novel Candidate Loci/Genes Affecting Lodging Resistance in Rice. Genes 2021, 12, 718. [Google Scholar] [CrossRef]
- Jiang, C.-J.; Shimono, M.; Maeda, S.; Inoue, H.; Mori, M.; Hasegawa, M.; Sugano, S.; Takatsuji, H. Suppression of the rice fatty-acid desaturase gene OsSSI2 enhances resistance to blast and leaf blight diseases in rice. Mol. Plant-Microbe Interact. 2009, 22, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Peng, S.; Visperas, R.M.; Ereful, N.; Bhuiya, M.S.U.; Julfiquar, A.W. Lodging-related morphological traits of hybrid rice in a tropical irrigated ecosystem. Field Crops Res. 2007, 101, 240–248. [Google Scholar] [CrossRef]
- Ookawa, T.; Ishihara, K. Varietal difference of physical characteristics of the culm related to lodging resistance in paddy rice. Jpn. J. Crop Sci. 1992, 61, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Karutz, C.; Schmid, J.E.; Stamp, P.; Winzeler, M.; Keller, B.; Messmer, M.M. Quantitative trait loci for lodging resistance in a segregating wheat× spelt population. Theor. Appl. Genet. 1999, 98, 1171–1182. [Google Scholar] [CrossRef]
- Monna, L.; Kitazawa, N.; Yoshino, R.; Suzuki, J.; Masuda, H.; Maehara, Y.; Tanji, M.; Sato, M.; Nasu, S.; Minobe, Y. Positional cloning of rice semidwarfing gene, sd-1: Rice “green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 2002, 9, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Spielmeyer, W.; Ellis, M.H.; Chandler, P.M. Semidwarf (sd-1),“green revolution” rice, contains a defective gibberellin 20-oxidase gene. Proc. Natl. Acad. Sci. USA 2002, 99, 9043–9048. [Google Scholar] [CrossRef] [Green Version]
- Pearce, S.; Saville, R.; Vaughan, S.P.; Chandler, P.M.; Wilhelm, E.P.; Sparks, C.A.; Al-Kaff, N.; Korolev, A.; Boulton, M.I.; Phillips, A.L. Molecular characterization of Rht-1 dwarfing genes in hexaploid wheat. Plant Physiol. 2011, 157, 1820–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Huang, J.; Wang, Y.; Xu, R.; Yang, Z.; Zhao, Z.; Liu, S.; Tian, Y.; Zheng, X.; Li, F. Identification and genetic analysis of qCL1. 2, a novel allele of the “green revolution” gene SD1 from wild rice (Oryza rufipogon) that enhances plant height. BMC Genet. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Peng, Y.; Hu, Y.; Qian, Q.; Ren, D. Progress and prospect of breeding utilization of green revolution gene SD1 in rice. Agriculture 2021, 11, 611. [Google Scholar] [CrossRef]
- Hirano, K.; Ordonio, R.L.; Matsuoka, M. Engineering the lodging resistance mechanism of post-Green Revolution rice to meet future demands. Proc. Jpn. Acad. Ser. B 2017, 93, 220–233. [Google Scholar] [CrossRef] [Green Version]
- Bux, L.; Li, D.; Faheem, M.; Sowadan, O.; Dong, Z.; Liu, E.; Ali, M.; Li, Y.; Sitoe, H.M.; Mirani, A.A. Detection of QTLs for outcrossing-related traits in rice (Oryza sativa L.) by association mapping and the RSTEP-LRT method. Euphytica 2019, 215, 204. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [Green Version]
- McCouch, S.R.; Teytelman, L.; Xu, Y.; Lobos, K.B.; Clare, K.; Walton, M.; Fu, B.; Maghirang, R.; Li, Z.; Xing, Y. Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res. 2002, 9, 199–207. [Google Scholar] [CrossRef]
- Temnykh, S.; Park, W.D.; Ayres, N.; Cartinhour, S.; Hauck, N.; Lipovich, L.; Cho, Y.G.; Ishii, T.; McCouch, S.R. Mapping and genome organization of microsatellite sequences in rice (Oryza sativa L.). Theor. Appl. Genet. 2000, 100, 697–712. [Google Scholar] [CrossRef]
- Creste, S.; Neto, A.T.; Figueira, A. Detection of single sequence repeat polymorphisms in denaturing polyacrylamide sequencing gels by silver staining. Plant Mol. Biol. Report. 2001, 19, 299–306. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Nei, M.; Tajima, F.; Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H. Arlequin (version 3.0): An Integrated Software Package for Population Genetics Data Analysis; Computational and Molecular Population Genetics Lab (CMPG), Zoological Institute, University of Berne: Berne, Switzerland, 2006. [Google Scholar]
- Weir, B.S.; Hill, W.G. Estimating F-statistics. Annu. Rev. Genet. 2002, 36, 721–750. [Google Scholar] [CrossRef] [Green Version]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of Linkage Disequilibrium in Plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [Green Version]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, S.; Dang, X.; Liu, E.; Hu, X.; Eltahawy, M.S.; Zaid, I.U.; Hong, D. Favorable alleles mining for gelatinization temperature, gel consistency and amylose content in Oryza sativa by association mapping. BMC Genet. 2019, 20, 34. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Breseghello, F.; Sorrells, M.E. Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 2006, 172, 1165–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wan, X.; Crossa, J.; Crouch, J.; Weng, J.; Zhai, H.; Wan, J. QTL mapping of grain length in rice (Oryza sativa L.) using chromosome segment substitution lines. Genet. Res. 2006, 88, 93–104. [Google Scholar] [CrossRef] [PubMed]
- McCouch, S.R. Gene nomenclature system for rice. Rice 2008, 1, 72–84. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Trait (cm) | Year | Mean ± SD | Min | Max | Skew | Kurtosis | CV (%) | |
|---|---|---|---|---|---|---|---|---|
| PH | 2018 | 120.07 ± 27.24 | 68.78 | 193.82 | 0.22 | −1.13 | 22.68 | 92.59 |
| 2019 | 128.78 ± 29.90 | 75.73 | 230.27 | 0.42 | −0.93 | 23.21 | 99.55 | |
| PL | 2018 | 22.82 ± 4.60 | 7.02 | 40.38 | 0.35 | 0.45 | 20.16 | 45.27 |
| 2019 | 23.32 ± 4.66 | 9.87 | 37.16 | 0.38 | 0.01 | 19.99 | 94.01 | |
| 1IN | 2018 | 38.04 ± 7.51 | 21.86 | 63.57 | 0.21 | −0.57 | 19.75 | 93.00 |
| 2019 | 37.30 ± 7.21 | 12.47 | 62.28 | 0.17 | −0.05 | 19.32 | 95.03 | |
| 2IN | 2018 | 23.30 ± 5.10 | 11.02 | 35.74 | 0.30 | −0.80 | 21.91 | 82.27 |
| 2019 | 23.33 ± 5.22 | 11.48 | 37.63 | 0.45 | −0.07 | 22.36 | 92.33 | |
| 3IN | 2018 | 17.76 ± 4.32 | 6.27 | 29.14 | 0.06 | −0.52 | 24.33 | 77.20 |
| 2019 | 19.30 ± 5.45 | 6.82 | 32.93 | 0.28 | −0.87 | 28.26 | 94.55 | |
| 4IN | 2018 | 12.81 ± 5.37 | 2.42 | 27.16 | 0.45 | −0.49 | 41.88 | 76.31 |
| 2019 | 14.19 ± 6.03 | 1.96 | 31.44 | 0.41 | −0.75 | 42.49 | 93.27 |
| Trait (cm) | Year | Xiushui79 | C-bao | XC-CSSL Population | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Mean ± SD | Mean ± SD | Min | Max | Skew | Kurtosis | CV (%) | |||
| PH | 2018 | 86.04 ± 0.54 | 94.48 ± 1.11 | 90.64 ± 12.19 | 72.91 | 123.73 | 0.93 | 0.43 | 13.45 | 96.97 |
| 2019 | 87.08 ± 0.70 | 95.84 ± 1.58 | 108.68 ± 12.79 | 86.2 | 141.96 | 1.08 | 0.67 | 11.76 | 94.93 | |
| PL | 2018 | 15.03 ± 0.74 | 25.32 ± 0.57 | 16.91 ± 2.56 | 13.46 | 23.72 | 1.1 | 0.66 | 15.14 | 83.75 |
| 2019 | 15.92 ± 0.63 | 26.38 ± 0.41 | 18.37 ± 2.62 | 15.5 | 26.82 | 1.35 | 1.18 | 14.25 | 89.71 | |
| 1IN | 2018 | 24.81 ± 1.02 | 34.06 ± 0.64 | 28.54 ± 5.21 | 22.72 | 44.64 | 1.55 | 1.48 | 18.26 | 93.95 |
| 2019 | 26.43 ± 0.18 | 36.28 ± 0.73 | 29.20 ± 5.29 | 23.76 | 45.36 | 1.72 | 1.96 | 18.13 | 89.22 | |
| 2IN | 2018 | 17.30 ± 0.35 | 15.99 ± 0.26 | 18.07 ± 2.84 | 12.79 | 24.78 | 0.43 | −0.2 | 15.71 | 94.92 |
| 2019 | 18.31 ± 1.07 | 18.01 ± 0.80 | 21.99 ± 2.44 | 18.04 | 30.12 | 1.27 | 2.65 | 11.11 | 71.39 | |
| 3IN | 2018 | 16.30 ± 0.61 | 10.10 ± 0.27 | 15.97 ± 2.69 | 8.78 | 20.76 | −0.49 | −0.15 | 16.86 | 91.66 |
| 2019 | 17.5 ± 0.06 | 12.23 ± 0.98 | 19.22 ± 2.41 | 12.94 | 26.14 | −0.11 | 0.76 | 12.55 | 89.61 | |
| 4IN | 2018 | 7.04 ± 0.78 | 3.77 ± 0.82 | 9.12 ± 2.57 | 4.32 | 15.13 | 0.23 | −0.31 | 28.2 | 79.83 |
| 2019 | 8.40 ± 0.32 | 6.61 ± 0.76 | 12.87 ± 2.76 | 5.88 | 19.32 | −0.22 | 0.46 | 21.49 | 81.09 | |
| Traits | SSR | Chr | Position | Position (bp) b | 2018 | 2019 | Average | QTL Reported in the Previous Studies | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Marker | (cM) a | Start Pos | End Pos | p-Value | PVE (%) | FDR | p-Value | PVE (%) | FDR | PVE (%) | References | ||
| PH | RM301 | 2 | 53.5 | 12,216,463 | 12,216,613 | 1.38 × 10−3 | 5.46 | 6.25 × 10−3 | 2.29 × 10−3 | 5.17 | 5.88 × 10−3 | 5.31 | |
| RM348 | 4 | 160.8 | 32,650,358 | 32,650,527 | 4.55 × 10−2 | 2.41 | 5.00 × 10−2 | 8.04 × 10−3 | 3.34 | 1.47 × 10−2 | 2.88 | ||
| RM305 | 5 | 96.9 | 12,216,463 | 12,216,613 | 4.80 × 10−3 | 4.38 | 9.38 × 10−3 | 1.70 × 10−2 | 3.f65 | 1.76 × 10−2 | 4.01 | [35] | |
| RM264 | 8 | 138.2 | 27,926,633 | 27,926,652 | 2.00 × 10−2 | 5.16 | 2.81 × 10−2 | 1.38 × 10−3 | 6.92 | 2.94 × 10−3 | 6.04 | [36] | |
| RM512 | 12 | 39.4 | 5,104,270 | 5,104,483 | 1.55 × 10−2 | 3.00 | 1.25 × 10−2 | 2.72 × 10−3 | 3.90 | 8.82 × 10−3 | 3.45 | ||
| PL | RM1 | 1 | 25.4 | 4,635,793 | 4,635,870 | 3.30 × 10−2 | 4.50 | 4.71 × 10−2 | 8.97 × 10−3 | 5.39 | 1.76 × 10−2 | 4.94 | [37] |
| RM583 | 1 | 38.8 | 8,328,958 | 8,329,017 | 6.29 × 10−3 | 3.86 | 1.47 × 10−2 | 4.12 × 10−3 | 4.09 | 1.18 × 10−2 | 3.97 | ||
| RM338 | 3 | 61.9 | 13,221,482 | 13,221,665 | 3.17 × 10−2 | 3.60 | 4.12 × 10−2 | 2.92 × 10−2 | 3.66 | 3.82 × 10−2 | 3.63 | ||
| RM122 | 5 | 3 | 311,055 | 311,218 | 1.17 × 10−2 | 3.15 | 2.06 × 10−2 | 4.52 × 10−4 | 4.82 | 2.94 × 10−3 | 3.98 | ||
| RM3330 | 6 | 61.6 | 11,064,125 | 11,064,294 | 5.54 × 10−3 | 6.69 | 8.82 × 10−3 | 4.39 × 10−3 | 6.85 | 1.47 × 10−2 | 6.77 | ||
| RM5380 | 7 | 67 | 19,361,277 | 19,361,383 | 3.54 × 10−3 | 5.64 | 5.88 × 10−3 | 7.54 × 10−4 | 6.57 | 5.88 × 10−3 | 6.10 | ||
| RM3533 | 9 | 65.1 | 17,833,841 | 17,833,864 | 1.03 × 10−3 | 7.82 | 2.94 × 10−3 | 1.31 × 10−3 | 7.66 | 8.82 × 10−3 | 7.74 | ||
| RM3600 | 9 | 62.7 | 17,054,142 | 17,054,167 | 2.72 × 10−2 | 4.94 | 2.94 × 10−2 | 2.66 × 10−2 | 4.96 | 2.94 × 10−2 | 4.95 | [19] | |
| RM6160 | 10 | 81 | 22,048,086 | 22,048,115 | 1.20 × 10−2 | 4.20 | 2.35 × 10−2 | 2.81 × 10−2 | 3.68 | 3.53 × 10−2 | 3.94 | ||
| 1IN | RM301 | 2 | 53.5 | 12,216,463 | 12,216,613 | 7.99 × 10−3 | 4.44 | 8.33 × 10−3 | 1.70 × 10−2 | 3.98 | 3.00 × 10−2 | 4.21 | |
| RM264 | 8 | 138.2 | 27,926,633 | 27,926,652 | 3.11 × 10−2 | 4.84 | 4.17 × 10−2 | 2.27 × 10−2 | 5.06 | 3.50 × 10−2 | 4.95 | ||
| RM257 | 9 | 48.8 | 17,570,591 | 17,570,612 | 2.20 × 10−2 | 3.83 | 3.33 × 10−2 | 5.76 × 10−4 | 5.94 | 5.00 × 10−3 | 4.88 | ||
| 2IN | RM301 | 2 | 53.5 | 12,216,463 | 12,216,613 | 1.24 × 10−3 | 5.52 | 1.36 × 10−2 | 2.13 × 10−2 | 3.85 | 3.18 × 10−2 | 4.68 | |
| RM5427 | 2 | 84.6 | 21,544,386 | 21,544,417 | 1.18 × 10−2 | 5.20 | 3.18 × 10−2 | 1.80 × 10−2 | 4.92 | 2.73 × 10−2 | 5.06 | ||
| RM348 | 4 | 160.8 | 32,650,358 | 32,650,527 | 7.39 × 10−4 | 4.57 | 4.55 × 10−3 | 1.29 × 10−3 | 4.29 | 1.82 × 10−2 | 4.43 | ||
| RM122 | 5 | 3 | 311,055 | 311,218 | 1.87 × 10−3 | 4.10 | 1.82 × 10−2 | 4.71 × 10−4 | 4.80 | 9.09 × 10−3 | 4.45 | ||
| RM305 | 5 | 96.9 | 20,944,257 | 20,944,466 | 1.89 × 10−3 | 4.89 | 2.27 × 10−2 | 1.21 × 10−3 | 5.14 | 1.36 × 10−2 | 5.02 | ||
| RM512 | 12 | 39.4 | 5,104,270 | 5,104,483 | 9.03 × 10−4 | 4.47 | 9.09 × 10−3 | 4.67 × 10−4 | 4.80 | 4.55 × 10−3 | 4.63 | ||
| 3IN | RM301 | 2 | 53.5 | 12,216,463 | 12,216,613 | 7.60 × 10−3 | 4.47 | 1.79 × 10−2 | 3.79 × 10−3 | 4.88 | 1.15 × 10−2 | 4.68 | |
| RM348 | 4 | 160.8 | 32,650,358 | 32,650,527 | 1.46 × 10−2 | 3.03 | 2.50 × 10−2 | 4.12 × 10−2 | 2.47 | 4.23 × 10−2 | 2.75 | ||
| RM122 | 5 | 3 | 311,055 | 311,218 | 7.38 × 10−4 | 4.57 | 3.57 × 10−3 | 1.25 × 10−3 | 4.30 | 3.85 × 10−3 | 4.44 | ||
| RM305 | 5 | 96.9 | 20,944,257 | 20,944,466 | 5.78 × 10−3 | 4.27 | 1.43 × 10−2 | 1.50 × 10−2 | 3.72 | 1.92 × 10−2 | 4.00 | ||
| RM3589 | 7 | 89.8 | 25,054,610 | 25,054,631 | 2.83 × 10−2 | 5.21 | 4.64 × 10−2 | 3.16 × 10−2 | 5.13 | 3.46 × 10−2 | 5.17 | ||
| RM264 | 8 | 138.2 | 27,926,633 | 27,926,652 | 2.63 × 10−2 | 4.97 | 4.29 × 10−2 | 2.08 × 10−3 | 6.67 | 7.69 × 10−3 | 5.82 | ||
| RM247 | 12 | 26.7 | 3,185,544 | 3,185,599 | 1.85 × 10−2 | 4.90 | 2.86 × 10−2 | 4.71 × 10−2 | 4.25 | 5.00 × 10−2 | 4.58 | ||
| RM512 | 12 | 39.4 | 5,104,270 | 5,104,483 | 4.51 × 10−3 | 3.65 | 1.07 × 10−2 | 5.05 × 10−3 | 3.59 | 1.54 × 10−2 | 3.62 | ||
| 4IN | RM301 | 2 | 53.5 | 12,216,463 | 12,216,613 | 5.32 × 10−4 | 5.99 | 2.94 × 10−3 | 3.49 × 10−3 | 4.93 | 6.67 × 10−3 | 5.46 | |
| RM348 | 4 | 160.8 | 32,650,358 | 32,650,527 | 2.57 × 10−2 | 2.72 | 3.82 × 10−2 | 8.00 × 10−3 | 3.35 | 1.33 × 10−2 | 3.03 | ||
| RM122 | 5 | 3 | 311,055 | 311,218 | 9.96 × 10−4 | 4.42 | 5.88 × 10−3 | 1.85 × 10−3 | 4.10 | 3.33 × 10−3 | 4.26 | ||
| RM305 | 5 | 96.9 | 20,944,257 | 20,944,466 | 4.77 × 10−3 | 4.38 | 1.76 × 10−2 | 2.52 × 10−2 | 3.42 | 3.00 × 10−2 | 3.90 | ||
| RM598 | 5 | 62.7 | 16,676,126 | 16,676,152 | 2.71 × 10−3 | 4.31 | 8.82 × 10−3 | 2.53 × 10−2 | 3.08 | 3.33 × 10−2 | 3.69 | ||
| RM225 | 6 | 26.2 | 3,416,638 | 3,416,665 | 3.19 × 10−3 | 6.39 | 1.18 × 10−2 | 3.18 × 10−2 | 4.83 | 4.33 × 10−2 | 5.61 | [38] | |
| RM234 | 7 | 93.9 | 25,420,132 | 25,420,157 | 2.47 × 10−2 | 5.61 | 3.53 × 10−2 | 4.88 × 10−2 | 5.09 | 5.00 × 10−2 | 5.35 | ||
| RM264 | 8 | 138.2 | 27,926,633 | 2,7926,652 | 3.98 × 10−3 | 6.25 | 1.47 × 10−2 | 4.34 × 10−3 | 6.19 | 1.00 × 10−2 | 6.22 | ||
| RM512 | 12 | 39.4 | 5,104,270 | 5,104,483 | 1.58 × 10−2 | 2.99 | 2.65 × 10−2 | 2.45 × 10−2 | 2.75 | 2.67 × 10−2 | 2.87 | [39] | |
| Trait (cm) | Quantitative Trait Locus | Marker Locus Linked to the QTL | Marker Location (cM) | 2018 | 2019 | ||||
|---|---|---|---|---|---|---|---|---|---|
| LOD | PVE (%) | Add (cm) | LOD | PVE (%) | Add (cm) | ||||
| PH | qPH-12 | RM19 | 20.9 | 2.96 | 17.77 | 15.10 | 2.52 | 17.19 | 14.85 |
| PL | qPL-7 | RM180 | 30.1 | 2.94 | 22.95 | 2.28 | - | - | - |
| 1IN | q1IN-7 | RM180 | 30.1 | 4.95 | 28.42 | 5.31 | - | - | - |
| q1IN-9 | RM257 | 79.7 | 3.20 | 16.92 | 5.68 | - | - | - | |
| 2IN | q2IN-7.1 | RM180 | 30.1 | 2.91 | 10.68 | 2.08 | - | - | - |
| q2IN-7.2 | RM542 | 34.7 | - | - | - | 2.60 | 10.07 | 2.80 | |
| q2IN-12 | RM19 | 20.9 | 2.87 | 10.78 | 2.87 | - | - | - | |
| 3IN | q3IN-12 | RM19 | 20.9 | - | - | - | 2.57 | 16.70 | 2.82 |
| 4IN | q4IN-8 | RM72 | 60.9 | 2.72 | 14.15 | 1.40 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sitoe, H.M.; Zhang, Y.; Chen, S.; Li, Y.; Ali, M.; Sowadan, O.; Karikari, B.; Liu, E.; Dang, X.; Qian, H.; et al. Detection of QTLs for Plant Height Architecture Traits in Rice (Oryza sativa L.) by Association Mapping and the RSTEP-LRT Method. Plants 2022, 11, 999. https://0-doi-org.brum.beds.ac.uk/10.3390/plants11070999
Sitoe HM, Zhang Y, Chen S, Li Y, Ali M, Sowadan O, Karikari B, Liu E, Dang X, Qian H, et al. Detection of QTLs for Plant Height Architecture Traits in Rice (Oryza sativa L.) by Association Mapping and the RSTEP-LRT Method. Plants. 2022; 11(7):999. https://0-doi-org.brum.beds.ac.uk/10.3390/plants11070999
Chicago/Turabian StyleSitoe, Hélder Manuel, Yuanqing Zhang, Siqi Chen, Yulong Li, Mehtab Ali, Ognigamal Sowadan, Benjamin Karikari, Erbao Liu, Xiaojing Dang, Hujun Qian, and et al. 2022. "Detection of QTLs for Plant Height Architecture Traits in Rice (Oryza sativa L.) by Association Mapping and the RSTEP-LRT Method" Plants 11, no. 7: 999. https://0-doi-org.brum.beds.ac.uk/10.3390/plants11070999