
Electronic Structure Calculation of Cr3+ and Fe3+ in Phosphor Host Materials Based on Relaxed Structures by Molecular Dynamics Simulation


Abstract
:1. Introduction
- (1)
- MD reproduces the local lattice distortion induced by substitution of foreign ions (in this case, luminescent center ions) prior to the electronic structure calculations. For example, γ-LiAlO2:Fe3+ is a deep-red phosphor [9,11] in which Fe3+ replaces Al3+ in the host lattice. The ionic radii of Fe3+ and Al3+ in four-hold coordination are 0.49 Å and 0.39 Å, respectively [12], with the larger Fe3+ ion pushing the ligand oxygens away and expanding the coordination polyhedron. Such local distortion affects the electronic structure of the central Fe3+ according to the conventional crystal field theory.
- (2)
- With the development of computational techniques, ab-initio MDs such as CASTEP [13], VASP [14,15,16,17], and FMO-MD [18] have become available over the decades. In contrast to these ab-initio MDs, classical MDs describe atomic interactions between the constituent atoms by simple two-body potentials, allowing a larger number of atoms to be handled in the simulation with reasonable accuracy. It is suitable for simulating a dilute situation in which luminescent center ions are randomly dispersed in the lattice of the phosphor host crystal. Unexpected periodicity in the arrangement of luminescent center ions in the calculation cell, due to periodic boundary conditions often applied in MD simulations, can be avoided, as it can occur when the cell is constructed with a limited number of atoms.
2. Methods
2.1. Molecular Dynamics (MD) Simulation
- To investigate the effect of lattice relaxation for the isolated luminescent center ion, one Cr3+ ion was introduced to replace the Al3+ on the octahedral site in LiAl5O8 and α-Al2O3; in γ-LiAlO2, the Al3+ on the tetrahedral site was replaced by one Fe3+ ion.
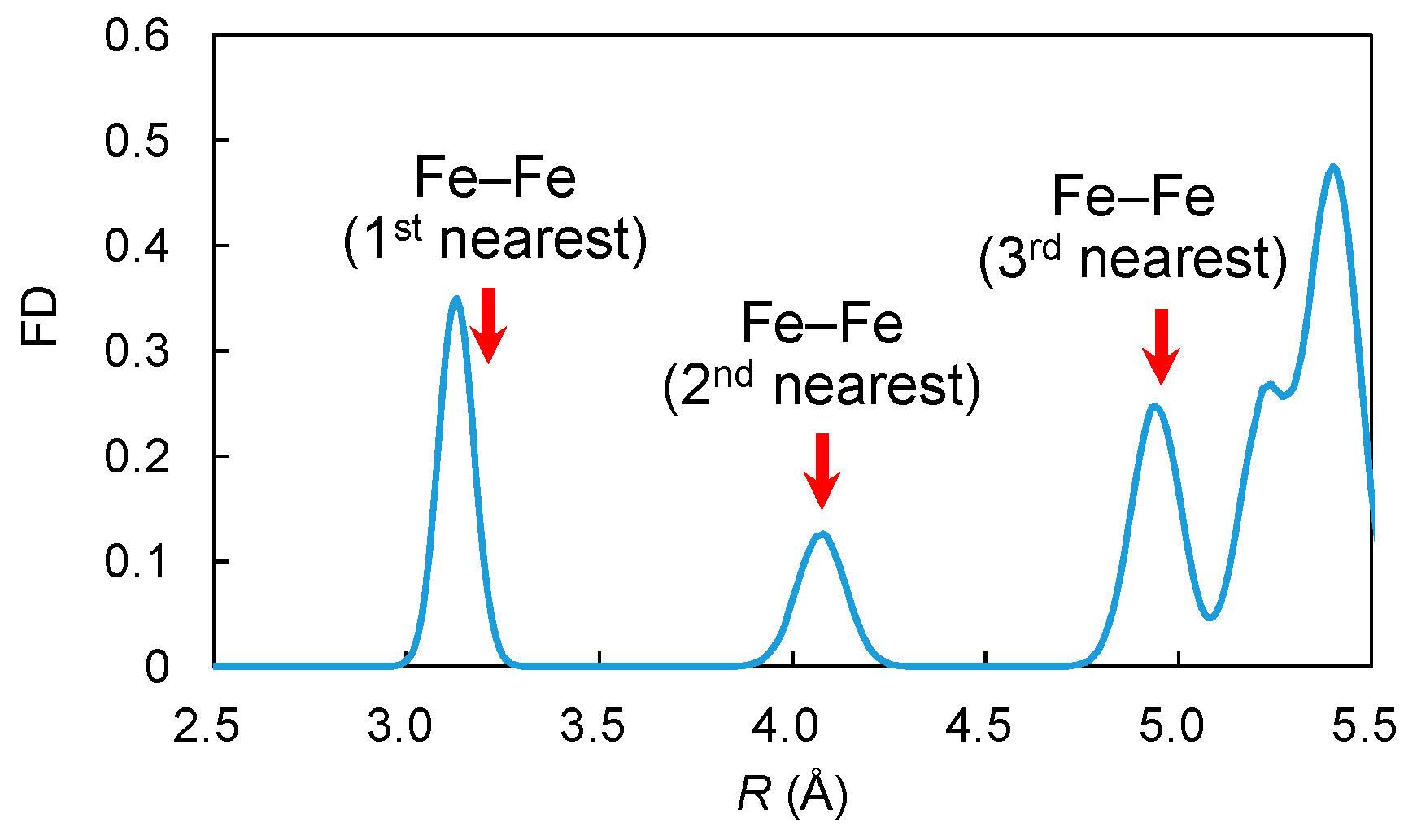
- To investigate the interaction between Fe–Fe pairs in γ-LiAlO2, two Fe3+ ions were placed at the first-, second-, and third-nearest neighboring Al3+ positions.
2.2. Molecular Orbital (MO) Calculations
3. Results and Discussion
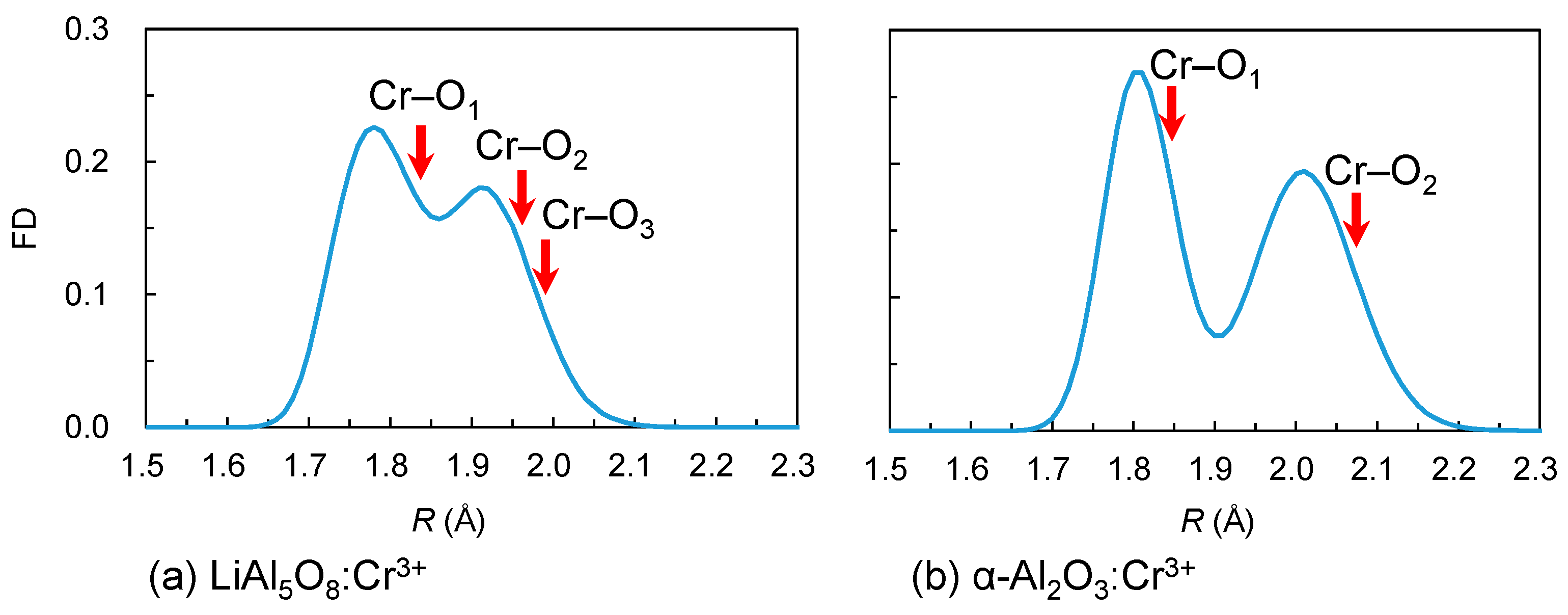
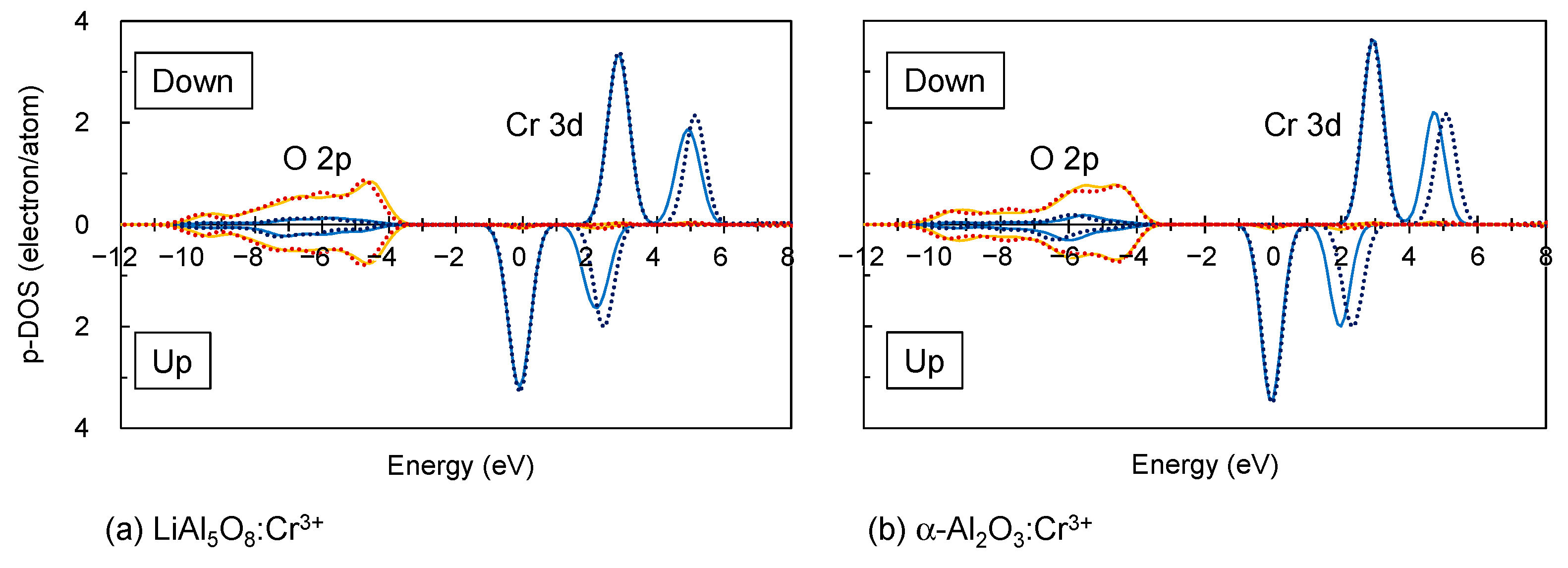
3.1. Electronic Structures of Isolated Cr3+
3.2. Electronic Structures of Fe3+
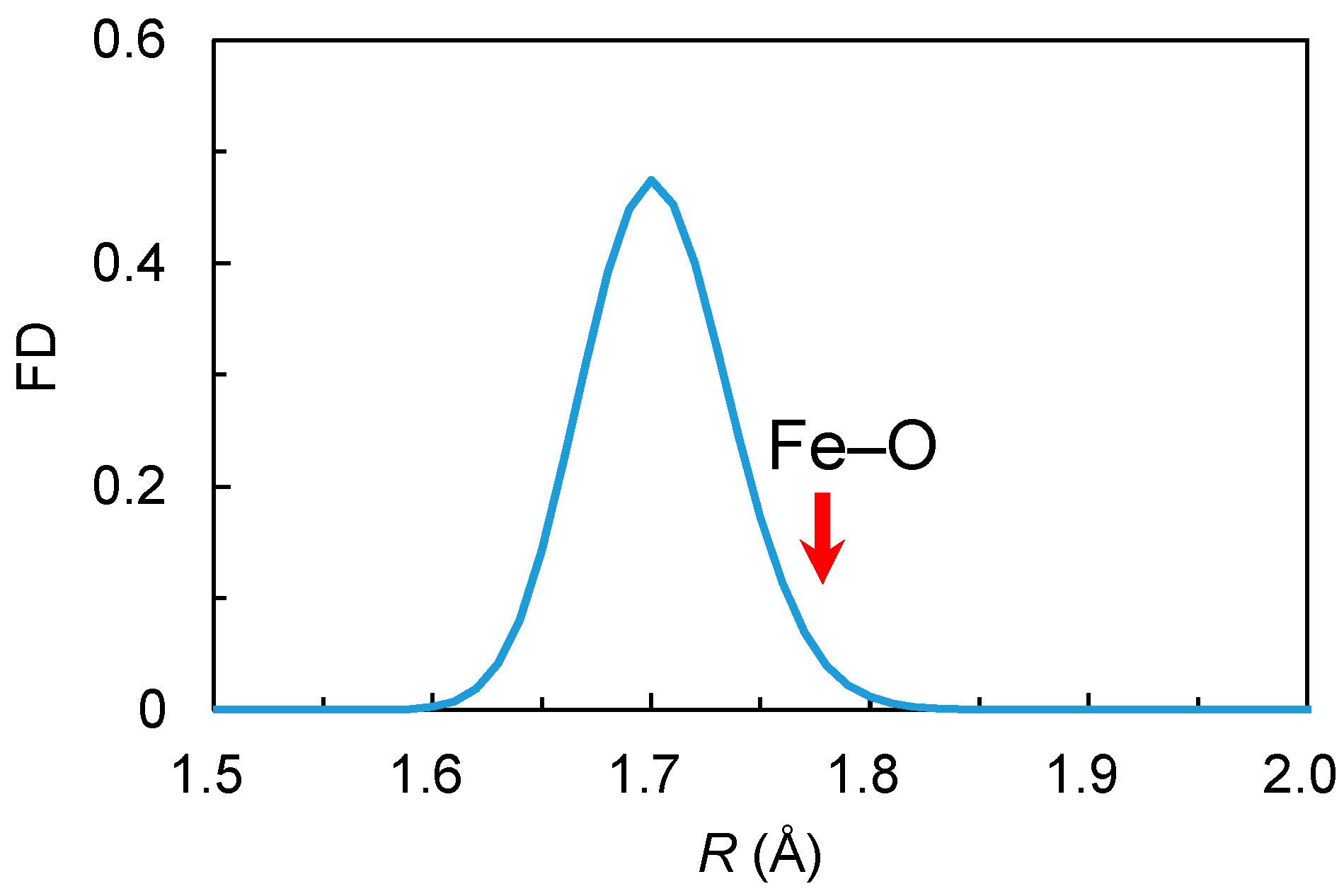
3.2.1. Isolated Fe3+
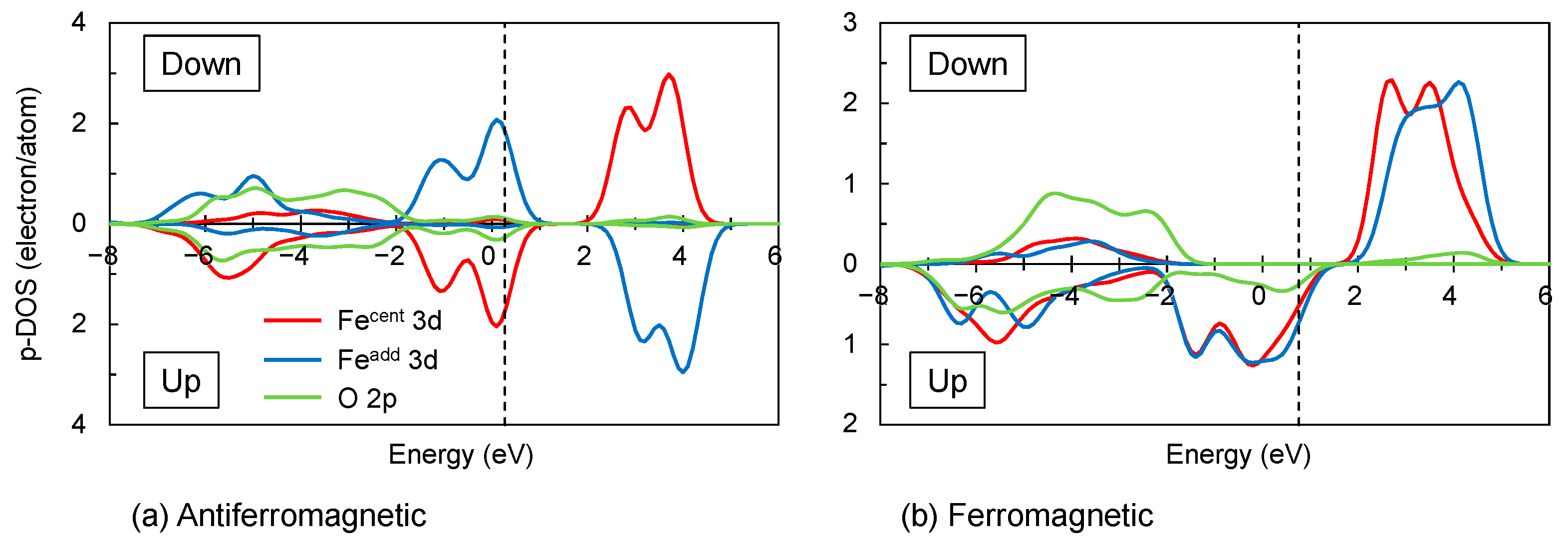
3.2.2. Fe3+–Fe3+ Interaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xie, R.-J.; Mitomo, M.; Uheda, K.; Xu, F.F.; Akimune, Y. Preparation and luminescence spectra of calcium- and rare-earth (R = Eu, Tb, and Pr)-codoped α-SiAlON ceramics. J. Am. Ceram. Soc. 2002, 85, 1229–1234. [Google Scholar] [CrossRef]
- van Krevel, J.W.H.; van Rutten, J.W.T.; Mandal, H.; Hintzen, H.T.; Metselaar, R. Luminescence properties of terbium-, cerium-, or europium-doped α-sialon materials. J. Solid State Chem. 2002, 165, 19–24. [Google Scholar] [CrossRef]
- Xie, R.-J.; Hirosaki, N.; Sakuma, K.; Yamamoto, Y.; Mitomo, M. Eu2+-doped Ca-α-SiAlON: A yellow phosphor for white light-emitting diodes. Appl. Phys. Lett. 2004, 84, 5404–5406. [Google Scholar] [CrossRef]
- Sakuma, K.; Omichi, K.; Kimura, N.; Ohashi, M.; Tanaka, D.; Hirosaki, N.; Yamamoto, Y.; Xie, R.-J.; Suehiro, T. Warm-white light-emitting diode with yellowish orange SiAlON ceramic phosphor. Opt. Lett. 2004, 29, 2001–2003. [Google Scholar] [CrossRef]
- Xie, R.-J.; Hirosaki, N.; Mitomo, M.; Yamamoto, Y.; Suehiro, T.; Sakuma, K. Optical properties of Eu2+ in α-SiAlON. J. Phys. Chem. B 2004, 108, 12027–12031. [Google Scholar] [CrossRef]
- Uheda, K.; Hirosaki, N.; Yamamoto, H. Host lattice materials in the system Ca3N2–AlN–Si3N4 for white light emitting diode. Phys. Status Solidi A 2006, 203, 2712–2717. [Google Scholar] [CrossRef]
- Uheda, K.; Hirosaki, N.; Yamamoto, Y.; Naito, A.; Nakajima, T.; Yamamoto, H. Luminescence properties of a red phosphor, CaAlSiN3: Eu2+, for white light-emitting diodes. Electrochem. Solid-State Lett. 2006, 9, H22–H25. [Google Scholar] [CrossRef]
- Watanabe, H.; Wada, H.; Seki, K.; Itou, M.; Kijima, N. Synthetic method and luminescence properties of SrxCa1−xAlSiN3:Eu2+ mixed nitride phosphors. J. Electrochem. Soc. 2008, 155, F31–F36. [Google Scholar] [CrossRef]
- Takahashi, H.; Takahashi, H.; Watanabe, K.; Kominami, H.; Hara, K.; Matsushima, Y. Fe3+ red phosphors based on lithium aluminates and an aluminum lithium oxyfluoride prepared from LiF as the Li Source. J. Lumin. 2017, 182, 53–58. [Google Scholar] [CrossRef]
- Kobayashi, R.; Tamura, H.; Kamada, Y.; Kakihana, M.; Matsushima, Y. A new host compound of aluminum lithium fluoride oxide for deep red phosphors based on Mn4+, Fe3+, and Cr3+. ECS Trans. 2018, 88, 225–236. [Google Scholar] [CrossRef]
- Aoyama, M.; Amano, Y.; Inoue, K.; Honda, S.; Hashimoto, S.; Iwamoto, Y. Synthesis and characterization of lithium aluminate red phosphors. J. Lumin. 2013, 135, 211–215. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Komeiji, Y.; Mochizuki, Y.; Nakano, T.; Mori, H. Recent advances in fragment molecular orbital-based molecular dynamics (FMO-MD) simulations. In Molecular Dynamics—Theoretical Developments and Applications in Nanotechnology and Energy; Wang, L., Ed.; InTech: London, UK, 2012; Available online: https://www.intechopen.com/chapters/34975 (accessed on 22 April 2022).
- Makino, Y.; Tanaka, I.; Kawamura, K.; Adachi, H.; Hirao, K. Electronic structure of silica glass under strong electric field. Prog. Theo. Phys. Suppl. 2000, 138, 239–240. [Google Scholar] [CrossRef]
- Famery, R.; Queyroux, F.; Gilles, J.-C.; Herpin, P. Etude structurale de la forme ordonnée de LiAl5O8. J. Solid State Chem. 1979, 30, 257–263. [Google Scholar] [CrossRef]
- Ishizawa, N.; Miyata, T.; Minato, I.; Marumo, F.; Iwai, S. A structural investigation of α-Al2O3 at 2170 K. Acta Crystallogr. B 1980, 36, 228–230. [Google Scholar] [CrossRef]
- Marezio, M. The crystal structure and anomalous dispersion of γ-LiAlO2. Acta Crystallogr. 1965, 19, 396–400. [Google Scholar] [CrossRef]
- Kawamura, K. Environmental Nano-Mechanics Molecular Simulations. Available online: https://kats-labo.jimdofree.com/mxdorto-mxdtricl/ (accessed on 5 March 2022).
- Farley, T.W.D.; Hayes, W.; Hull, S.; Hutchings, M.T.; Vrtis, M. Investigation of thermally induced Li+ ion disorder in Li2O using neutron diffraction. J. Phys. Condens. Matter 1991, 3, 4761–4781. [Google Scholar] [CrossRef]
- Marezio, M.; Remeika, J.P. High-pressure synthesis and crystal structure of α-LiAlO2. J. Chem. Phys. 1966, 44, 3143–3145. [Google Scholar] [CrossRef]
- Finger, L.W.; Hazen, R.M. Crystal structure and isothermal compression of Fe2O3, Cr2O3, and V2O3 to 50 kbars. J. Appl. Phys. 1980, 51, 5362–5367. [Google Scholar] [CrossRef]
- Lu, Z.; Dahn, J.R. Structure and electrochemistry of layered Li[CrxLi(1/3−x/3)Mn(2/3−2x/3)]O2. J. Electrochem. Soc. 2002, 149, A1454–A1459. [Google Scholar] [CrossRef]
- Maslen, E.N.; Strel’tsov, V.A.; Strel’tsova, N.R.; Ishizawa, N. Synchrotron X-ray study of the electron density in α-Fe2O3. Acta Crystallogr. B 1994, 50, 435–441. [Google Scholar] [CrossRef]
- Marin, S.J.; O’Keefe, M.; Partin, D.E. Structures and crystal chemistry of ordered spinels: LiFe5O8, LiZnNbO4, and Zn2TiO4. J. Solid State Chem. 1994, 113, 413–419. [Google Scholar] [CrossRef]
- Adachi, H.; Tsukada, M.; Satoko, C. Discrete variational Xα cluster calculations. I. Application to metal clusters. J. Phys. Soc. Jpn. 1978, 45, 875–883. [Google Scholar] [CrossRef]
- Ellis, D.E.; Painter, G.S. Discrete variational method for the energy-band problem with general crystal potentials. Phys. Rev. B 1970, 2, 2887–2898. [Google Scholar] [CrossRef]
- Evjen, H.M. On the stability of certain heteropolar crystals. Phys. Rev. 1932, 39, 675–687. [Google Scholar] [CrossRef]
- McClure, D.S. Optical spectra of transition-metal ions in corundum. J. Chem. Phys. 1962, 36, 2757–2779. [Google Scholar] [CrossRef]
- Anderson, P.W. Antiferromagnetism. Theory of superexchange interaction. Phys. Rev. 1950, 79, 350–356. [Google Scholar] [CrossRef]
- Kanamori, J. Superexchange interaction and symmetry properties of electron orbitals. J. Phys. Chem. Solids 1959, 10, 87–98. [Google Scholar] [CrossRef]
- Yamashita, J.; Kondo, J. Superexchange interaction. Phys. Rev. 1958, 109, 730–741. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom Pair | Aij (eV) | ρij (Å) |
|---|---|---|
| O–O | 1997.28 | 0.2810 |
| Al–O | 1716.41 | 0.2810 |
| Li–O | 960.0 | 0.2690 |
| Li–Li | 98.9357 | 0.2994 |
| Cr–O | 1156.68 | 0.3131 |
| Fe–O | 1200.68 | 0.3151 |
| Luminescent Center | Host Crystal | Calculation Cluster |
|---|---|---|
| Cr3+ (octahedral) | LiAl5O8 | (Li2Al10CrO38)41− |
| α-Al2O3 | (Al13CrO45)48− | |
| Fe3+ (tetrahedral) | γ-LiAlO2 | (Li9Al6FeO32)34− for isolated Fe3+ (Li9Al5Fe2O32)34− for Fe3+–Fe3+ interaction at the first- and second-nearest neighbors(Li9Al6Fe2O34)35− for Fe3+–Fe3+ interaction at the third-nearest neighbor |
| HOMO Energy (eV) | Energy Difference (eV) | ||
|---|---|---|---|
| Antiferromagnetic | Ferromagnetic | Anti.−Ferro. | |
| Nearest | 0.27 | 0.76 | −0.49 |
| Second nearest | 0.30 | 0.32 | −0.02 |
| Third nearest | 0.18 | 0.23 | −0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ichikawa, J.; Kominami, H.; Hara, K.; Kakihana, M.; Matsushima, Y. Electronic Structure Calculation of Cr3+ and Fe3+ in Phosphor Host Materials Based on Relaxed Structures by Molecular Dynamics Simulation. Technologies 2022, 10, 56. https://0-doi-org.brum.beds.ac.uk/10.3390/technologies10030056
Ichikawa J, Kominami H, Hara K, Kakihana M, Matsushima Y. Electronic Structure Calculation of Cr3+ and Fe3+ in Phosphor Host Materials Based on Relaxed Structures by Molecular Dynamics Simulation. Technologies. 2022; 10(3):56. https://0-doi-org.brum.beds.ac.uk/10.3390/technologies10030056
Chicago/Turabian StyleIchikawa, Joichiro, Hiroko Kominami, Kazuhiko Hara, Masato Kakihana, and Yuta Matsushima. 2022. "Electronic Structure Calculation of Cr3+ and Fe3+ in Phosphor Host Materials Based on Relaxed Structures by Molecular Dynamics Simulation" Technologies 10, no. 3: 56. https://0-doi-org.brum.beds.ac.uk/10.3390/technologies10030056