
Wheat Proteomics for Abiotic Stress Tolerance and Root System Architecture: Current Status and Future Prospects

 ,
,  ,
,  ,
,  , and
, and
Abstract
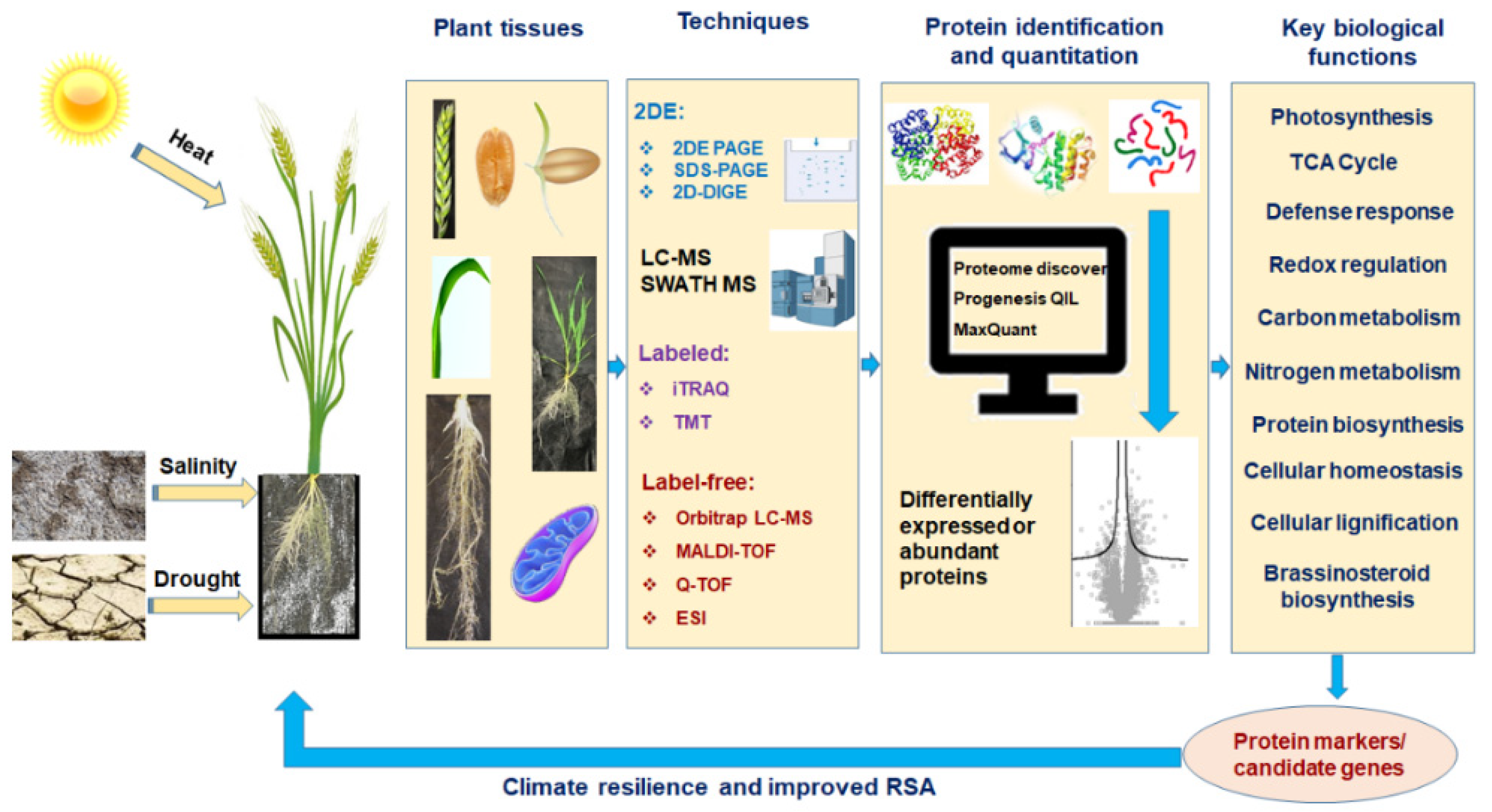
:1. Introduction
2. Proteomics: Edge over the Other Omics Techniques
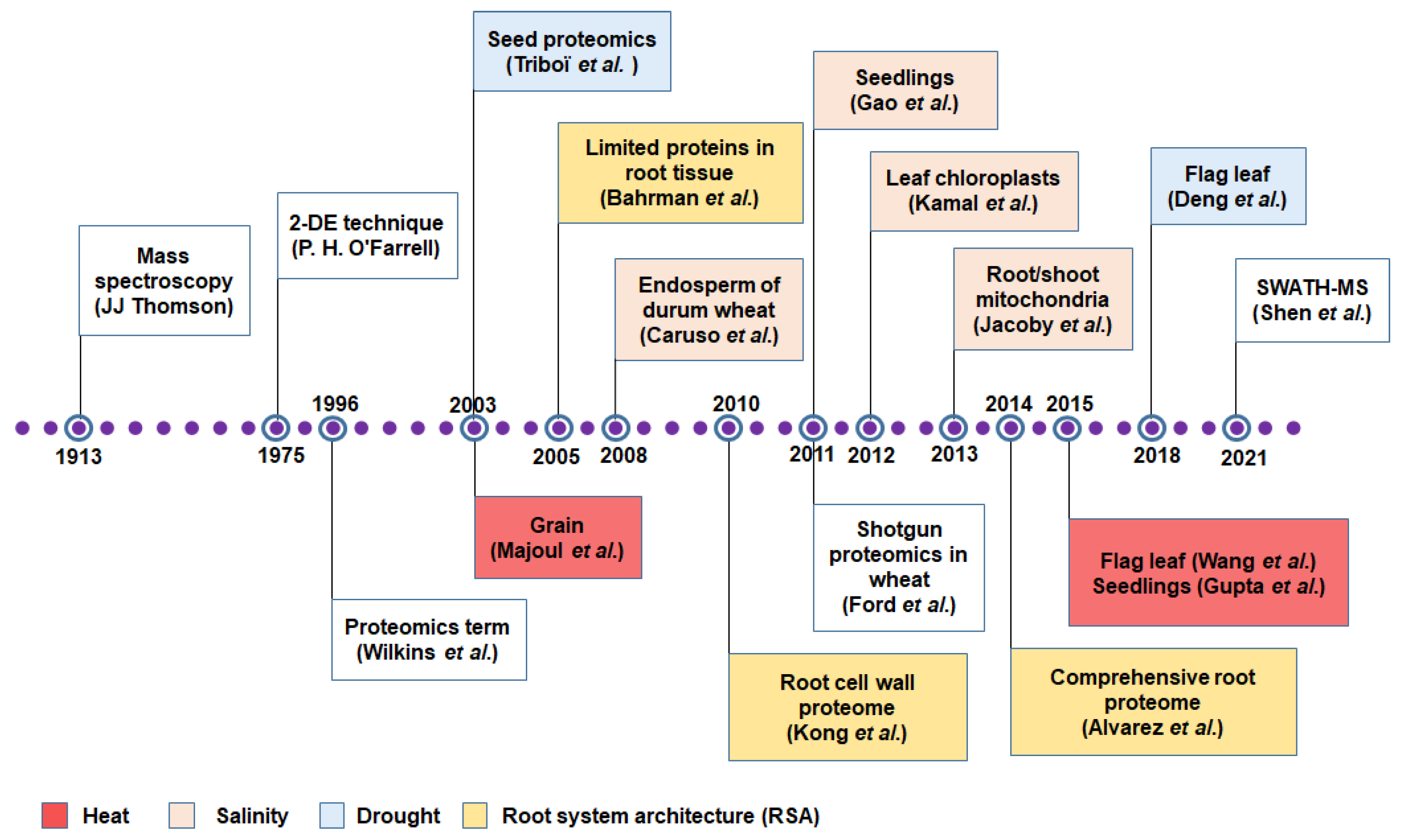
3. Protein to Proteomics: Revisiting the Journey
4. Proteomics in Abiotic Stress Tolerance in Wheat
4.1. Heat Stress

{kind=link}
{kind=link}
| Genotypes | Tissue and Developmental Stages | Treatments | Techniques | Effects | Genes/Enzymes | References |
|---|---|---|---|---|---|---|
| WH 730 (tolerant) and Raj 4014 (sensitive) with 10 extreme RILs | 10-day-old seedlings (whole seedling sampled) | 35 °C for 6 h | 2-DE, MALDI-TOF/TOF -MS/MS | RuBisCO activase A, Con A and PEP carboxylase 1 were the key DEPs. | N/A | [63] |
| 810 (tolerant) and 1039 (sensitive) | Flag leaf at 15 days post anthesis (DPA) | 35 °C/26 °C (day/night) 5 days | 2-DE, MALDI-TOF-MS | Proteins related to signal transduction, heat shock, photosynthesis, and antioxidants are upregulated, while those for nitrogen metabolism are downregulated. | N/A | [61] |
| Chinese Spring | Flag leaf (15 DPA) | 37/17 °C (day/night) for 3 days | iTRAQ, LC-MS/MS | Chlorophyll synthesis, carbon fixation, protein turnover, and redox regulation were the most remarkable heat-responsive processes. | GST and Trxs | [109] |
| Jing411 | Grains (sampled at 5, 10, 15, and 20 DPA) | 40 °C for 2 h (12:00–14:00) | iTRAQ, LC -ESI Tandem MS/MS | 256 DEPs for stimulus response, abiotic stress response, kinase activity and transferase activity. | Calcineurin B-like | [110] |
| Gaocheng 8901 | Grains (sampled at 5, 10, 15, and 20 DPA) | 40 °C for 2 h (12:00–14:00) | iTRAQ, LC-MS/MS | 207 DEPs for energy metabolism, growth and development, and stress response were identified. | N/A | [111] |
| Triso | Flag leaf (10 DPA) | 32 °C for 9 days and elevated CO2 (550 μmol/mol) | LC-MS/MS | Proteins for photosynthesis, antioxidant and protein synthesis pathways are downregulated. | GST and Trx | [112] |
| Chinese Spring | Grain in filling stage (15 DPA) | 37 °C for 4 h | SDS–PAGE, TMT | A general decrease in protein synthesis components and metabolic proteins, but a significant increase in stress-response and storage proteins was found. | N/A | [113] |
| HD2985 (tolerant) and HD2329 (sensitive) | Pooled (Spikes, Stem Flag leaf) at pollination and grain filling stages | 37 °C for 2 h | iTRAQ, LC-MS/MS | Carboxylase enzyme was the most abundant active enzyme under heat stress. | HSP17, CDPK, Cu/Zn SOD, ADP glucophosphorylase and soluble starch synthase | [114] |
| BWL4444 | Mature grains | 2 DPA to maturity; day heat stress (35/17 °C), Day–night heat stress (35/24 °C) | 2DE, MALDI-TOF- MS/MS | Proteins related to the translation, gliadins, and low-molecular-weight glutenins are upregulated. Proteins related to glycolysis, photosynthesis, defense, and high-molecular-weight glutenins are downregulated. | TaRSR1, OsbZIP58, glyceraldehyde- 3-phosphate dehydrogenase, triose phosphate translocator and sucrose transporter | [115] |
4.2. Salinity Stress
| Genotypes | Tissue and Developmental Stages | Treatments | Techniques | Effects | Genes/Enzymes | References |
|---|---|---|---|---|---|---|
| Amphiploid developed from Chinese Spring and Lophopyrum elongatum (tolerant) and Chinese Spring (sensitive) | Mitochondria of shoots and roots, seedlings | 200 mM NaCl | 2D DIGE, MALDI-TOF/TOF MS | Manganese SOD (Mn SOD), serine hydroxymethyl transferase, aconitase, malate dehydrogenase, beta (β)-cyanoalanine synthase, glutamate dehydrogenase and aspartate aminotransferase were key DEPs. | N/A | [65] |
| Waha (Triticum turgidum) (tolerant) | Seed embryo and embryo surrounding tissues (germination stage) | NaCl (250 mM) for 42 h | LC-MS/MS Orbitrap Elite hybrid ion trap-Orbitrap MS | Methionine, auxin, metabolism, ROS managing and signaling imparted in salinity stress tolerance. | S-adenosylmethionine synthetase, methionine methyltransferase, glutamate decarboxylase, 1-Cys peroxiredoxin and GST | [118] |
| Roshan (tolerant) and Ghods (sensitive) | Leaves, 4-leaf stage seedlings | Hoagland solution with 200 mM NaCl | 2DE, MALDI-TOF-TOF MS | RuBisCO activase, RuBisCO large and small subunits, chloroplastic trios-phosphate isomerase, cytosolic malate dehydrogenase upregulated. | N/A | [119] |
| T349 and T378 transgenic line with GmDREB1 gene (maize promoter) | First expanded leaves, 10 days old seedlings | Kimura B nutrient solution with 300 mM NaCl | IEF gel, MALDI-TOF MS analysis | Osmotic- and oxidative stress-associated proteins, methionine synthase, glyceraldehyde-3-phosphate dehydrogenase, glutathione transferase, NADP-dependent malic enzyme and 2-cys peroxiredoxin BAS1 upregulated. | GmDREB1 | [120] |
| Duilio (tolerant) (T. turgidum) | Leaf (5-days old seedlings) | Hydroponics-100 and 200 mM NaCl | LC-MS/MS | Plant defense, energy production and signal transduction related proteins are upregulated. | CBSX3 (cystathionine β-synthase) and dehydrin | [121] |
| T. monococcum | Leaves, seedlings | Hoagland solution with 80, 160, 240, and 320 mM NaCl | 2DE, MALDI-TOF/TOF-MS | Cu/Zn SODs, GSTs, DHNs and LEA, 64 unique DAPs upregulated. Biomarkers for salinity stress response and defense: cp31BHv, betaine-aldehyde dehydrogenase, cytosolic (GS1), Cu/Zn SOD, MAT3, leucine aminopeptidase 2, and 2-Cys peroxiredoxin BAS1 were selected. | N/A | [122] |
| Enterobacter cloacae SBP-8 bacteria inoculated wheat cv. C-309 | Whole plant, seedlings | Hoagland solution with 200 mM NaCl | LC-MS/MS | Cell wall (structure) strengthening proteins, such as tubulin, profilin, retinoblastoma, casparian strip membrane protein and xyloglucan endotransglycosylase, ion transporter (e.g., malate transporter), metabolic pathway and protein synthesis upregulated. | Clp protease, Trxs h, cysperoxiredoxin, catalase and RuBisCO | [123] |
| Han 12 (tolerant) and Jimai 19 (sensitive) | Roots, seedlings | Hoagland solution with 350 mM NaCl | iTRAQ, LC-MS, validation: RT-PCR, transgenic plant Arabidopsis | PPDK, LEA1 and LEA2 proteins imparted in salinity tolerance. | TaPPDK, TaLEA1 and TaLEA2 | [124] |
| Bobwhite | Roots and leaves, 2-week-old seedlings | Pots, 50 mM NaCl | LC-MS/MS, validation: qRT-PCR | Upregulated SODs, malate dehydrogenases, dehydrin proteins and V-ATPase protein, and Cu/Zn SODs, LEA and DHN proteins in roots and leaves, respectively. | LEA and dehydrin | [125] |
| Chinese Spring | Embryo proximal seed parts | Hoagland solution with 150 mM NaCl | Orbitrap Fusion Lumos LC-ESI MS/MS, validation: qRT-PCR | 397 DAPs (133 upregulated/264 downregulated) were identified. | N/A | [126] |
| Qingmai 6 (tolerant) | Shoots and roots, 2-week-old seedlings | Water with 150 mM NaCl, and the same combined with 100 μM ethylene precursor ACC, and 150 μM ethylene signaling inhibitor 1-MCP | iTRAQ, Shotgun (Orbitrap Q Exactive HF-X MS) | DAPs: ribosomal proteins, nucleoside diphosphate kinases, transaldolases, β-glucosidases, and phosphoenolpyruvate carboxylases were upregulated; proteins related to metabolism played role in salinity response in wheat shoots. | TaGSTU6, TaCCR, TaEXPB6, TaPOD, TaWRKY70 and TaCYP450 | [16] |
| Chinese Spring | Seeds (endosperm) | Hoagland solution with 150 mM NaCl | Orbitrap Fusion Lumos LC-ESI MS/MS, validation: qRT-PCR | 207 DEPs upregulated. | TraesCS7B02G367600.1, TraesCS4A02G246100.1, TraesCS5D02G172800.1, TraesCS6A02G357200.1, TraesCS7A02G358200.1, TraesCS3A02G150800.1, TraesCS5A02G369900.1, TraesCS6A02G059800.1, TraesCS6A02G350500.1 and TraesCS6A02G319300.1 | [127] |
| Zhongmai 175 | Leaf chloroplast, seedlings | 200 mM NaCl solution | Shotgun (Orbitrap Q Exactive HF-X MS), validation: qRT-PCR | Calvin cycle, amino acid metabolism, carbon and nitrogen metabolism, transcription and translation and antioxidation related 117 DAPs upregulated. | Allene oxide synthase 2-like, chaperone protein ClpC2, probable plastid-lipid-associated protein 2, phosphoglycerate kinase, phosphoglycolate phosphatase 1B, ribulose bisphosphate carboxylase large chain, 50S ribosomal protein L2 and sedoheptulose-1, 7-bisphos- phatase | [128] |
| Scepter | Mature roots and root tips, Emergence of the second leaf (5 days-post transplant) | 150 mM NaCl | Q-TOF, LC-MS | Translation related proteins, glycolytic enzymes, TCA cycle enzymes and ATP synthase subunits are downregulated. | S-adenosylmethionine synthase, aspartate aminotransferase, O-methyltransferase, GST and phenylalanine ammonia lyase | [26] |
4.3. Drought Stress
| Genotypes | Tissue and Developmental Stages | Treatments | Techniques | Effects | Genes/Enzymes | References |
|---|---|---|---|---|---|---|
| Triticum turgidum ssp. Dicocoides: TR39477 and TTD22, and T. turgidum ssp. Durum: Kızıltan | Leaf, seedling | No irrigation | 2-DE, nano LC-ESI–MS/MS, qRT-PCR | Eleven drought stress-specific proteins (low peptide matches) were found. TMPIT1 (integral membrane protein) upregulated in wild emmer wheat. | RuBisCO, MnSOD, GST and FNR | [138] |
| Nesser (tolerant) and Opata (sensitive) | Roots, seedling | ABA (100 μM) or EtOH with growth media | iTRAQ, LC-MS/MS, qRT-PCR | Heat shock proteins (HSPs), O-methyltransferase and 2-caffeoyl CoA-methyltransferase upregulated in tolerant genotype. | Rab24, dehydrin, fructose bisphosphate aldolase, lipoxygenase 1 and 2, calnexin, elicitor responsive protein 3-like and caffeic acid o-methyl transferase | [68] |
| Yannong 19 | Leaves, reproductive | Drought (35–60% relative water content in soil) | 2-DE, MALDI-TOF/TOF-MS | Photosynthesis and carbon metabolism associated proteins reduced yield under severe drought combined with low temperature. | Cu/Zn SOD, tAPX, MnSOD and CAT | [139] |
| Hanxuan 10 (tolerant) and Ningchun 47 (sensitive) | Leaf, seedling | 20% PEG-6000 in Hoagland solution | TiO2, label free LC-MS/MS | Sensors related to Ca2+ showed differential expression at phosphorylation. Phosphorylated proteins (H+-ATPase, MSSP2, PP2C, TaABI5, WCOR615 and WAL17) are upregulated to improve drought stress tolerance. | TaABI5-1, MYB1R1 and bHLH | [140] |
| Gaocheng 8901, Jagger and Nongda 3406 | Seed, reproductive | 7–12% soil content | SDS-PAGE, MALDI-TOF/TOF-MS | Albumin and gliadin upregulated significantly. | N/A | [135] |
| KTC86211 | Leaf, seedling | PEG- 6000 (−0.50 Mpa) spray | 2-DE, MALDI-TOF/TOF-MS | ROS scavenging proteins (ascorbate peroxidase, GST, thiol-specific antioxidant protein) primarily upregulated. | N/A | [141] |
| SERI M 82 (tolerant) and SW89.5193/kAu2 (sensitive) | Leaf and root, seedling | 20% field capacity | 2-DE, nanoLC-MS/MS, qRT-PCR | Cell biogenesis and degradation-related proteins significantly upregulated in leaf and root of tolerant genotype. | Ascorbate peroxidase, ATP synthase subunit β, GST and 16.9 kDA HSPs | [142] |
| Hanxuan 10 (tolerant) and Chinese Spring (sensitive) | Roots, leaf, and intermediate sections (IS) between roots and leaf (IS), seedling | 20% PEG-6000 in ½ Hoagland solution, drought recovery | 2-DE, MALDI-TOF/TOF-MS | A higher percentage of proteins upregulated in roots than in leaves and IS during drought stress but downregulated during recovery. HSPs significantly upregulated in all organs. | N/A | [130] |
| Erebuni (T. boeoticum) | Leaf and root | 20% PEG-6000 in 1/2 Hoagland solution | 2-DE, MALDI-TOF/TOF-MS | Abscisic acid increased higher in leaves than roots to improve drought stress tolerance. Signal transduction proteins, and UDP-glucose/GDP mannose dehydrogenase, ribulose-phosphate 3-epimerase, transketolase and transaldolase-like protein are upregulated, but proteins related to protein metabolism and glycolysis are downregulated in roots. | N/A | [143] |
| Transgenic wheat lines (08 T(1)-27 and 08 T(1)-47) containing maize phosphoenolpyruvate carboxylase (PEPC) gene developed from Zhoumai19 | Leaf and root, reproductive | 30–35% soil moisture | 2-DE, MALDI-TOF-MS | ATP synthesis subunits, ferredoxin-NADP reductase and S-adenosylmethionine, chloroplast glyceraldehyde-3-phosphate dehydrogenase, chlorophyll A-B binding protein and phosphate diakinase upregulated in transgenic wheat. | PEPC | [144] |
| Xihan No. 2 (tolerant) and Longchun 23 (sensitive) | Leaf and root, seedling | 30% moisture content | 2-DE, MALDI-TOF/TOF-MS | Proteins associated with photosynthesis, stress defense and detoxification played the most important role in yield improvement during drought stress. | N/A | [145] |
| Kundan (tolerant) and Lok1 (sensitive) | Leaf, seedling and reproductive | 50% and 75% relative water content in leaf and rehydration for recovery | 2-DE, MALDI-TOF-MS, western blotting | Proteins related to carbon metabolism, amino acid, defense and antioxidation took part in drought stress tolerance. | N/A | [146] |
| Yumai34 | Leaf, seedling | 0.05 mM NaHS and PEG 6000 in Hogland solution | SDS-PAGE, iTRAQ, nano-LC-MS/MS, RT-PCR | Carbon metabolism and protein synthesis associated proteins increased, and photosynthesis and signal transduction proteins downregulated in PEG with NaHS treated genotypes. | Genes associated with W5A5Z6, W5A2Y8, W5BBW7, W5IAG4, W5F3S8, W5EDB0, W5H6J0, W5BQ07, and C1K737 proteins | [147] |
| Shaanhe 6 (tolerant) and Zhengyin 1 (sensitive) | Leaf, seedling | 70%, 50%, 40%, 30%, and 20% field capacity | SDS-PAGE, LC-MS/MS, western blotting | LEA protein helped in drought stress tolerance. | lea genes | [148] |
| Kavir (tolerant) and Bahar (sensitive) | Leaf, seedling | No irrigation for a week | 2D-PAGE, LC-MS/MS | ADP-glucose pyrophosphatase, GST, glyoxalase enzymes and phosphoribulokinase downregulated in the sensitive genotype, and soluble inorganic pyrophosphatase is downregulated in both genotypes. | N/A | [149] |
| Zhongmai 175 | Flag leaf and grain, reproductive | No irrigation | 2D-DIGE, MALDI-TOF/TOF-MS, western blotting, qRT-PCR | Proteins associated with photosynthesis and energy metabolism, and carbon metabolism and stress found in flag leaves and grain, respectively, responded during drought stress. | N/A | [69] |
| Jinmai 47 | Leaf, seedling | 20% PEG-6000 in 1/2 Hoagland solution | iTRAQ, LC/MS, qRT-PCR | Citrate synthase, pyruvate dehydrogenase E1 component subunit alpha and aconitate hydratase upregulated during drought stress. Redox regulating proteins, chaperone proteins and enzymes proline biosynthesis are also upregulated, but RuBisCO activase small subunit downregulated. | N/A | [150] |
| Yan995 | Leaf, seedling | 25% PEG-6000 in 1/2 Hoagland solution and 40% field capacity | iTRAQ, MS/MS, qRT-PCR | Formate tetrahydrofolate ligase, glyceraldehyde-3-phosphate dehydrogenase, malate dehydrogenase 2, phosphoglycerate kinase, RuBisCO, and serine hydroxymethyl-transferase significantly downregulated in both type stresses. Amino acid synthesis associated proteins hampered plant growth during stress. | Genes associated with W5E659, W5EN32, W5ATV6, W5BAB9, W5ETI9, G8D5C5, W5DTC2 and W5FL86 proteins | [151] |
| Arg (tolerant) and Arta (sensitive) and F6 lines of their cross | Leaf, seedling | No irrigation | Linear ion trap mass spectrometer | Photosynthesis and stress-associated proteins downregulated and upregulated, respectively. Proline and malondialdehyde played a significant role in drought stress improvement. | N/A | [152] |
| Chinese Spring | Seed, reproductive | 20% PEG 6000 in modified 1/4 Hoagland solution | Label-free nano LC-MS/MS | 4-coumarate-CoA ligase, shikimate O-hydroxycinnamoyl transferase, caffeic acid O-methyltransferase, caffeoyl CoA O-methyltransferase, cinnamyl-alcohol dehydrogenase, and peroxidases downregulated. | N/A | [126] |
| Arg (tolerant) and Moghan3 (sensitive) | Leaf, reproductive | No irrigation after pollination to harvest | 2-DE, MALDI TOF/TOF-MS | Proteins associated with photosynthesis, stress defense and detoxification played the most important role in higher yield during stress. | N/A | [153] |
| PAN3478 | Seed, reproductive | No irrigation | 2-DE, LC–MS/MS | α-gliadin upregulated. High molecular weight glutenin proteins expressed differentially for wheat quality. | N/A | [136] |
| Yangmai 16 | Root apex, seedling | Drought priming by 5% (−0.37 MPa) and 15% (−0.78 MPa) PEG in Hoagland solution | iTRAQ, MS/MS | Phytohormones (auxin, cytokinin, brassinosteroids, ethylene, abscisic acid, jasmonic acid and salicylic acid) downregulated during drought stress. | N/A | [51] |
| TRI 5630 (tolerant) and White Fife (sensitive) | roots, leaves and seeds, reproductive | 71.11% field capacity | SDS-PAGE, LC−MS/MS | 3-ketoacyl-CoA synthase and ATP-binding cassette transporter regulated cuticular wax biosynthesis in wheat leaf and improved drought stress tolerance. | N/A | [154] |
5. Proteomic Approaches to Study RSA
| Root Traits | Genotypes | Tissues | Techniques | Important Findings | Genes/Enzymes | Validation | Treatments | References |
|---|---|---|---|---|---|---|---|---|
| Dry mass | Yumai 34 | Total root | 2-DE, tandem MS | Higher lipid peroxidation by malondialdehyde (MDA) at roots caused more sensitivity of roots than leaves under copper (Cu) toxicity. Upregulated glutathione S-transferase (GST) and downregulated MDA led to improved Cu stress tolerance. | GST | Quantitative real-time PCR (qPCR) | Cu stress (100 µM CuSO4·5H2O) | [163] |
| N/A | Opata and Nesser | Total root | iTRAQ, LC-MS/MS | Heat shock proteins, signal transduction pathway, secondary metabolism, and lignin metabolism associated proteins helped to drought stress tolerance through improved root growth. | Rab24 | qPCR | Drought (ABA (100 μM) or EtOH with growth media) | [68] |
| N/A | Keumgang | Mitochondria from root | Tricine SDS-PAGE, LTQ–FTICR MS | Proteins associated with translation, energy metabolism and amino acid synthesis were important to supply energy for root growth. | N/A | N/A | Controlled (soil in a greenhouse) | [164,165] |
| Depth | F2 generation from QTL isolines 178A and 178B, and 10 commercial varieties | Total root | 2D-DIGE | Primary rooting depth was reduced due to accumulation of oxygen in root tip and size of meristem and inhibition of peroxidases (PODs) activity, brassinosteroid (BR) by TaTRIP1. 24-epibrassinolide increased root meristem size. | TaTRIP1 and POD | qPCR and western blot | Controlled (hydroponic) | [166] |
| Depth, fresh mass | Keumkang | Total root | 2-DE gel, nano-LC/MS | Root elongation is reduced with high Aluminum (Al) concentration due to upregulation and downregulation of 19 and 28 proteins, respectively. | N/A | N/A | Al stress (0, 100 and 150 µM AlCl3) | [23] |
| Depth | Yumai 34 | Total root and leaf | iTRAQ, LC-MS/MS | 117 differential expressed proteins (DAPs) were found in wheat root under mercury (Hg) stress. Upregulated ADP-ribosylation factor GTPase- activating protein and antioxidant enzymes regulated root growth under Hg stress. | N/A | N/A | Heavy metal (different concentrations of HgCl2 with Hoagland solution) | [167] |
| Dry mass | Transgenic wheat containing Phosphoenolpyruvate carboxylase (PEPC) gene of maize developed from wheat variety Zhoumai19 | Leaf | 2-DE gel, MALDI-TOF-MS | Prostatic acid phosphatase fibrillin and protein related to methionine synthesis increased root growth and root mass due to the influence of PEPC, and so, improved drought stress tolerance. | PEPC | N/A | Drought (30–35% relative soil water) | [144] |
| Depth, fresh mass | Jiami 19 (sensitive) and Han 12 (tolerant) | Total root | iTRAQ, nano LC-MS/MS | Pyruvate, phosphate dikinase, late embryogenesis abundant (LEA) protein1 and LEA2 proteins increased rooting depth and fresh root mass and thereby improved salinity stress tolerance. | TaPPDK1, TaLEA1 and TaLEA2 | RT PCR, transgenic Arabidopsis and soybean | Salinity (0.4% soil salinity, 150 and 200 mM NaCl) | [133] |
| Depth and volume | Seri M82 (sensitive) and CIGM90.863 (tolerant) | Total root | TMT, LC-MS/MS | Upregulated proteins related to anaerobic adaptation and fermentation, such as alcohol dehydrogenases might increase root volume to improve waterlogging tolerance. | TaBWPR-1.2#2 and TaBWPR-1.2#13, Mn-SOD and NADK3 | qPCR | Waterlogging (hypoxic by N2 gas bubbling and 2.0 mg/L O2 in water) | [50] |
| Depth | XY54 and J411 | Total root | iTRAQ, LC-ESI-MS/MS | Eighty differentially expressed proteins (DEPs) associated with the steroid biosynthesis pathway, and peroxidases controlled rooting depth (primary rooting depth, and total rooting depth). Brassinosteroid biosynthesis pathway mediated ROS distribution contributed to long primary root growth through determining root meristem size. | Peroxidases related genes | qPCR | Controlled(greenhouse) | [168] |
| Depth and dry mass | Yumai 34 | Total root and leaf | iTRAQ, LC-MS/MS | Eight-hundred and sixty-six nitrogen (N2) deficiency associated proteins were found in the root. Wheat seedlings with silenced zeaxanthin epoxidase had reduced dry mass and high sensitivity to stress. | N/A | N/A | N2 stress (N2 free Hoagland solution) | [169] |
| Depth | RIL from XY54 × J411 | Total root and leaf | iTRAQ, LC-ESI-MS/MS | Lower N2 promotes longer root growth; 84 DAPs increased root growth. Four and one of glutathione metabolism related DAPs were upregulated and downregulated, respectively, and associated with longer root growth under lower N2. | N/A | qPCR | N2 stress | [170] |
| N/A | M1019 (tolerant) and Xinong20 (sensitive) | Total root | TMT, LC-MS/MS | Tolerant genotype had higher cadmium (Cd) in root cell walls than cell fluid and cytoplasm. Upregulation of DEPs associated with transferase activity, transferring glycosyl groups and metal iron binding helped in Cd stress tolerance. | N/A | N/A | Cd (CdCl2 stress) | [171] |
| N/A | HD2985 (tolerant) and HD2329 (sensitive) | Leaf, stem, and spike | iTRAQ, LC-MS/MS | HSP17 and HSP70, calcium-dependent protein kinase (CDPK) and Cu/Zn SOD, and defense associated proteins were upregulated in roots which might improve heat stress tolerance. | β-actin, HSP70, HSP17, CDPK, Cu/Zn SOD, f, Rca, OEEP, SucSyn, AGPase, SSS, SBE, and α-amylase | qPCR and immunoblotting | Heat stress | [114] |
| N/A | Qingmai 6 | Total root and leaf | iTRAQ, LC-MS/MS | Sixteen and three DAPs were found in roots at ethylene precursor ACC and ethylene inhibitor treatment, respectively. Ethylene dependent salinity response in root changed significantly due to the accumulation of 48 ribosomal proteins. | LOXs, UDPGs, GLUDs, PALs, 6-PGDHs, GSTs, BGLUs, PODs, and OXOs | qPCR | Salinity stress (ethylene dependent salinity stress) | [16] |
| Depth | TRI 5630 (tolerant) and White Fife (sensitive) | Total root | SDS-PAGE, LC-MS/MS | The rooting depth of both genotypes increased under drought stress might be due to the upregulation of β-glucosidase. | Drought (71.11% field capacity) | [154] | ||
| Total length, number, average diameter, dry mass and specific length | Jimai 22 | Total root | SDS-PAGE, LC-MS/MS | Total root length and specific root length decreased significantly due to upregulated peroxidase enzyme and phenylalanine ammonia-lyase. Proteins related to GST and phenylpropanoid biosynthesis upregulated and played an important role in root development and oxidative stress tolerance. | A0A3B6K9P2, Q8RW0, A0A3B6JL78, TraesCS3D02G344800, and TraesCS3A02G350800 | N/A | NH4+/NO3− ratios | [171] |
| N/A | Wyalkatchem | Root tip and root mucilage | Q-TOF/LC-MS | Root mucilage proteins, such as endopeptidase and oxidoreductase or carbohydrate binding played role in root development. Cell wall modified and defense mechanism influenced by P-starvation induced proteins, peroxidase, protease and chitinase localized at the root tip apoplast. | N/A | Multiple rection monitoring | Phosphorus starvation (250 μM KH2PO4 for 10 days) | [25] |
| Total length and dry mass | Scepter | Total root | Q-TOF/LC-MS | Root tip growth reduced more than mature root under salinity stress due to decreased abundance of TCA cycle enzymes, such as aconitate hydratase, and ATP synthase subunits, such as subunit β. | TraesCS5A01G505000.2 and TaesCS1A01G379000.1 | N/A | Salinity (150 mM NaCl) | [26] |
6. Limitations and Potential of Proteomics for Abiotic Stress Tolerance and RSA in Wheat
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. Cereal Production, Utilization, and Trade Reaching Record Levels in 2021/22; Food and Agriculture Organization: Rome, Italy, 2022. [Google Scholar]
- Bruinsma, J. Prospects for aggregate agriculture and major commodity groups. In World Agriculture: Towards 2015/2030: An FAO Study; Bruinsma, J., Ed.; Routledge: London, UK, 2017; p. 444. [Google Scholar]
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CIMMYT. Wheat Research. Available online: https://www.cimmyt.org/work/wheat-research/ (accessed on 5 May 2020).
- Singh, R. Wheat Strategy Delivers Stronger Grains; Consultative Group for International Agricultural Research (CGIAR): El Batán, Mexico, 2019. [Google Scholar]
- Brown, A. Wheat: March Quarter 2020; Australian Bureau of Agricultural and Resource Economics: Canberra, Australia, 2020.
- Zhang, J.; Zhang, S.; Cheng, M.; Jiang, H.; Zhang, X.; Peng, C.; Lu, X.; Zhang, M.; Jin, J. Effect of drought on agronomic traits of rice and wheat: A meta-analysis. Int. J. Environ. Res. Public Health 2018, 15, 839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, H.; Xiao, D.; Wang, B.; Liu, D.L.; Feng, P.; Tang, J. Multi-model ensemble of CMIP6 projections for future extreme climate stress on wheat in the North China Plain. Int. J. Climatol. 2021, 41, E171–E186. [Google Scholar] [CrossRef]
- Sabagh, A.E.; Islam, M.S.; Skalicky, M.; Raza, M.A.; Singh, K.; Hossain, M.A.; Hossain, A.; Mahboob, W.; Iqbal, M.; Ratnasekera, D. Salinity stress in wheat (Triticum aestivum L.) in the changing climate: Adaptation and management strategies. Front. Agron. 2021, 3, 661932. [Google Scholar] [CrossRef]
- Qadir, M.; Quillérou, E.; Nangia, V.; Murtaza, G.; Singh, M.; Thomas, R.J.; Drechsel, P.; Noble, A.D. Economics of salt-induced land degradation and restoration. Nat. Resour. Forum. 2014, 38, 282–295. [Google Scholar] [CrossRef]
- Chen, Y.L.; Djalovic, I.; Rengel, Z. Phenotyping for root traits. In Phenomics in Crop Plants: Trends, Options and Limitations; Springer: New Delhi, India, 2015; pp. 101–128. [Google Scholar]
- Lynch, J.P. Roots of the second green revolution. Aust. J. Bot. 2007, 55, 493–512. [Google Scholar] [CrossRef]
- Halder, T.; Liu, H.; Chen, Y.; Yan, G.; Siddique, K.H.M. Identification of Candidate Genes for Root Traits Using Genotype–Phenotype Association Analysis of Near-Isogenic Lines in Hexaploid Wheat (Triticum aestivum L.). Int. J. Mol. Sci. 2021, 22, 3579. [Google Scholar] [CrossRef]
- Lynch, J.P. Harnessing root architecture to address global challenges. Plant J. 2022, 109, 415–431. [Google Scholar] [CrossRef]
- Al-Othman, Z.A.; Ali, R.; Al-Othman, A.M.; Ali, J.; Habila, M.A. Assessment of toxic metals in wheat crops grown on selected soils, irrigated by different water sources. Arab. J. Chem. 2016, 9, S1555–S1562. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Shi, C.; Su, C.; Liu, Y. Complementary analyses of the transcriptome and iTRAQ proteome revealed mechanism of ethylene dependent salt response in bread wheat (Triticum aestivum L.). Food Chem. 2020, 325, 126866. [Google Scholar] [CrossRef]
- Neumann, G.; George, T.S.; Plassard, C. Strategies and methods for studying the rhizosphere—The plant science toolbox. Plant Soil 2009, 321, 431–456. [Google Scholar] [CrossRef]
- Atkinson, J.A.; Wingen, L.U.; Griffiths, M.; Pound, M.P.; Gaju, O.; Foulkes, M.J.; Le Gouis, J.; Griffiths, S.; Bennett, M.J.; King, J.; et al. Phenotyping pipeline reveals major seedling root growth QTL in hexaploid wheat. J. Exp. Bot. 2015, 66, 2283–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Gao, K.; Shan, S.; Gu, R.; Wang, Z.; Craft, E.J.; Mi, G.; Yuan, L.; Chen, F. Comparative analysis of root traits and the associated QTLs for maize seedlings grown in paper roll, hydroponics and vermiculite culture system. Front. Plant Sci. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Djalovic, I.; Siddique, K.H.M. Advances in understanding grain legume physiology: Understanding root architecture, nutrient uptake and response to abiotic stress. In Achieving Sustainable Cultivation of Grain Legumes; Burleigh Dodds Series in Agricultural Science; Burleigh Dodds Science Publishing Limited: Cambridgeshire, UK, 2018; Volume 1, pp. 11–28. [Google Scholar]
- Chen, Y.; Palta, J.; Prasad, P.V.V.; Siddique, K.H.M. Phenotypic variability in bread wheat root systems at the early vegetative stage. BMC Plant Biol. 2020, 20, 185. [Google Scholar] [CrossRef] [PubMed]
- Amudha, J.; Balasubramani, G. Recent molecular advances to combat abiotic stress tolerance in crop plants. Biotechnol. Mol. Biol. Rev. 2011, 6, 31–58. [Google Scholar]
- Oh, M.W.; Roy, S.K.; Kamal, A.H.; Cho, K.; Cho, S.W.; Park, C.S.; Choi, J.S.; Komatsu, S.; Woo, S.H. Proteome analysis of roots of wheat seedlings under aluminum stress. Mol. Biol. Rep. 2014, 41, 671–681. [Google Scholar] [CrossRef]
- Komatsu, S.; Kamal, A.H.; Hossain, Z. Wheat proteomics: Proteome modulation and abiotic stress acclimation. Front. Plant Sci. 2014, 5, 684. [Google Scholar] [CrossRef] [Green Version]
- Staudinger, C.; Dissanayake, B.M.; Duncan, O.; Millar, A.H. The wheat secreted root proteome: Implications for phosphorus mobilisation and biotic interactions. J. Proteom. 2022, 252, 104450. [Google Scholar] [CrossRef]
- Dissanayake, B.M.; Staudinger, C.; Munns, R.; Taylor, N.L.; Millar, A.H. Distinct salinity-induced changes in wheat metabolic machinery in different root tissue types. J. Proteom. 2022, 256, 104502. [Google Scholar] [CrossRef]
- Mustafa, G.; Komatsu, S. Plant proteomic research for improvement of food crop under stresses: A review. Mol. Omics 2021, 17, 860–880. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, M.; Li, F.; Lv, H.; Li, C.; Xia, G. A proteomic study of the response to salinity and drought stress in an introgression strain of bread wheat. Mol. Cell. Proteom. 2009, 8, 2676–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, P.; Abdel Latef, A.A.H.; Rasool, S.; Akram, N.A.; Ashraf, M.; Gucel, S. Role of proteomics in crop stress tolerance. Front. Plant Sci. 2016, 7, 1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holman, J.D.; Dasari, S.; Tabb, D.L. Informatics of protein and posttranslational modification detection via shotgun proteomics. Methods Mol. Biol. 2013, 1002, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Rampitsch, C.; Bykova, N.V. Advances in plant proteomics toward improvement of crop productivity and stress resistance. Front. Plant Sci. 2015, 6, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrill, P.; Harrington, S.A.; Uauy, C. Applying the latest advances in genomics and phenomics for trait discovery in polyploid wheat. Plant J. 2019, 97, 56–72. [Google Scholar] [CrossRef]
- Afzal, M.; Sielaff, M.; Curella, V.; Neerukonda, M.; El Hassouni, K.; Schuppan, D.; Tenzer, S.; Longin, C.F.H. Characterization of 150 wheat cultivars by LC-MS-based label-free quantitative proteomics unravels possibilities to design wheat better for baking quality and human health. Plants 2021, 10, 424. [Google Scholar] [CrossRef]
- Hochholdinger, F.; Marcon, C.; Baldauf, J.A.; Yu, P.; Frey, F.P. Proteomics of maize root development. Front. Plant Sci. 2018, 9, 143. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, R.P.; Millar, A.H.; Taylor, N.L. Opportunities for wheat proteomics to discover the biomarkers for respiration-dependent biomass production, stress tolerance and cytoplasmic male sterility. J. Proteom. 2016, 143, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Vanderschuren, H.; Lentz, E.; Zainuddin, I.; Gruissem, W. Proteomics of model and crop plant species: Status, current limitations and strategic advances for crop improvement. J. Proteom. 2013, 93, 5–19. [Google Scholar] [CrossRef]
- Parvaiz, A.; Latef, A.; Saiema, R.; Akram, N.A.; Muhammad, A.; Salih, G. Role of proteomics in crop stress tolerance. Front. Plant Sci. 2016, 7, 1336. [Google Scholar]
- Kosová, K.; Vítámvás, P.; Prášil, I.T. Proteomics of stress responses in wheat and barley—Search for potential protein markers of stress tolerance. Front. Plant Sci. 2014, 5, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canovas, F.M.; Dumas-Gaudot, E.; Recorbet, G.; Jorrin, J.; Mock, H.P.; Rossignol, M. Plant proteome analysis. Proteomics 2004, 4, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Jorrín-Novo, J.V.; Pascual, J.; Sánchez-Lucas, R.; Romero-Rodríguez, M.C.; Rodríguez-Ortega, M.J.; Lenz, C.; Valledor, L. Fourteen years of plant proteomics reflected in Proteomics: Moving from model species and 2DE-based approaches to orphan species and gel-free platforms. Proteomics 2015, 15, 1089–1112. [Google Scholar] [CrossRef] [PubMed]
- University of Oxford. Proteomics. Available online: https://massspec.chem.ox.ac.uk/proteomics (accessed on 29 March 2022).
- Ilyakalinin. Agricultural Crops. Rye, Rice Maize Wheat and Soybean Plant. Vector Illustration. Secale cereale. Agriculture Cultivated Plant. Available online: https://www.dreamstime.com/agricultural-crops-rye-rice-maize-wheat-soybean-plant-vector-illustration-secale-cereale-agriculture-cultivated-green-leaves-image144102338 (accessed on 29 March 2022).
- Kwon, Y.W.; Jo, H.S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J.H. Application of proteomics in cancer: Recent trends and approaches for biomarkers discovery. Front. Med. 2021, 8, 747333. [Google Scholar] [CrossRef]
- SkieTheAce. Available online: https://pixabay.com/illustrations/cell-mitochondria-biology-organelle-6474673/ (accessed on 2 April 2022).
- Yang, Y.; Saand, M.A.; Huang, L.; Abdelaal, W.B.; Zhang, J.; Wu, Y.; Li, J.; Sirohi, M.H.; Wang, F. Applications of multi-omics technologies for crop improvement. Front. Plant Sci. 2021, 12, 563953. [Google Scholar] [CrossRef]
- Pérez-Clemente, R.M.; Vives, V.; Zandalinas, S.I.; López-Climent, M.F.; Muñoz, V.; Gómez-Cadenas, A. Biotechnological approaches to study plant responses to stress. BioMed Res. Int. 2013, 2013, 654120. [Google Scholar] [CrossRef]
- He, M.; He, C.-Q.; Ding, N.-Z. Abiotic stresses: General defenses of land plants and chances for engineering multistress tolerance. Front. Plant Sci. 2018, 9, 1771. [Google Scholar] [CrossRef] [Green Version]
- Ingolia, N.T. Ribosome profiling: New views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014, 15, 205–213. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, S.; Liu, K.; Wang, S.; Huang, L.; Guo, L. Proteomics: A powerful tool to study plant responses to biotic stress. Plant Methods 2019, 15, 135. [Google Scholar] [CrossRef]
- Pan, R.; He, D.; Xu, L.; Zhou, M.; Li, C.; Wu, C.; Xu, Y.; Zhang, W. Proteomic analysis reveals response of differential wheat (Triticum aestivum L.) genotypes to oxygen deficiency stress. BMC Genom. 2019, 20, 60. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, J.; Ge, J.; Huang, M.; Cai, J.; Zhou, Q.; Dai, T.; Mur, L.A.J.; Jiang, D. The different root apex zones contribute to drought priming induced tolerance to a reoccurring drought stress in wheat. Crop J. 2021, 9, 1088–1097. [Google Scholar] [CrossRef]
- Kaur, B.; Sandhu, K.S.; Kamal, R.; Kaur, K.; Singh, J.; Röder, M.S.; Muqaddasi, Q.H. Omics for the improvement of abiotic, biotic, and agronomic traits in major cereal crops: Applications, challenges, and prospects. Plants 2021, 10, 1989. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Xu, J. Abiotic stress responses in plant roots: A proteomics perspective. Front. Plant Sci. 2014, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and their applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Cristea, I.M.; Gaskell, S.J.; Whetton, A.D. Proteomics techniques and their application to hematology. Blood 2004, 103, 3624–3634. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, M.R.; Sanchez, J.-C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with proteome projects: Why all proteins expressed by a genome should be identified and how to do it. Biotechnol. Genet. Eng. Rev. 1996, 13, 19–50. [Google Scholar] [CrossRef] [Green Version]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef]
- Thomson, J.J. Bakerian Lecture:—Rays of positive electricity. In Proceedings of the Royal Society of London, London, UK, 1 August 1913; pp. 1–20. [Google Scholar]
- Ford, K.L.; Cassin, A.; Bacic, A.F.A.A. Quantitative proteomic analysis of wheat cultivars with differing drought stress tolerance. Front. Plant Sci. 2011, 2, 44. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.-C.; Chen, M.-X.; Xiao, T.; Zhang, C.; Shang, J.; Zhang, K.-L.; Zhu, F.-Y. Global proteome response to Pb (II) toxicity in poplar using SWATH-MS-based quantitative proteomics investigation. Ecotoxicol. Environ. Saf. 2021, 220, 112410. [Google Scholar] [CrossRef]
- Wang, X.; Dinler, B.S.; Vignjevic, M.; Jacobsen, S.; Wollenweber, B. Physiological and proteome studies of responses to heat stress during grain filling in contrasting wheat cultivars. Plant Sci. 2015, 230, 33–50. [Google Scholar] [CrossRef]
- Majoul, T.; Bancel, E.; Triboï, E.; Ben Hamida, J.; Branlard, G. Proteomic analysis of the effect of heat stress on hexaploid wheat grain: Characterization of heat-responsive proteins from total endosperm. Proteomics 2003, 3, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Gupta, O.P.; Mishra, V.; Singh, N.; Tiwari, R.; Sharma, P.; Gupta, R.; Sharma, I. Deciphering the dynamics of changing proteins of tolerant and intolerant wheat seedlings subjected to heat stress. Mol. Biol. Rep. 2015, 42, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yan, X.; Li, X.; Guo, G.; Hu, Y.; Ma, W.; Yan, Y. Proteome analysis of wheat leaf under salt stress by two-dimensional difference gel electrophoresis (2D-DIGE). Phytochemistry 2011, 72, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, R.P.; Millar, A.H.; Taylor, N.L. Investigating the role of respiration in plant salinity tolerance by analyzing mitochondrial proteomes from wheat and a salinity-tolerant Amphiploid (wheat× Lophopyrum elongatum). J. Proteome Res. 2013, 12, 4807–4829. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Cavaliere, C.; Guarino, C.; Gubbiotti, R.; Foglia, P.; Laganà, A. Identification of changes in Triticum durum L. leaf proteome in response to salt stress by two-dimensional electrophoresis and MALDI-TOF mass spectrometry. Anal. Bioanal. Chem. 2008, 391, 381–390. [Google Scholar] [CrossRef]
- Triboï, E.; Martre, P.; Triboï-Blondel, A.M. Environmentally-induced changes in protein composition in developing grains of wheat are related to changes in total protein content. J. Exp. Bot. 2003, 54, 1731–1742. [Google Scholar] [CrossRef]
- Alvarez, S.; Roy Choudhury, S.; Pandey, S. Comparative quantitative proteomics analysis of the ABA response of roots of drought-sensitive and drought-tolerant wheat varieties identifies proteomic signatures of drought adaptability. J. Proteome Res. 2014, 13, 1688–1701. [Google Scholar] [CrossRef]
- Deng, X.; Liu, Y.; Xu, X.; Liu, D.; Zhu, G.; Yan, X.; Wang, Z.; Yan, Y. Comparative proteome analysis of wheat flag leaves and developing grains under water deficit. Front. Plant Sci. 2018, 9, 425. [Google Scholar] [CrossRef] [Green Version]
- Bahrman, N.; Gouy, A.; Devienne-Barret, F.; Hirel, B.; Vedele, F.; Le Gouis, J. Differential change in root protein patterns of two wheat varieties under high and low nitrogen nutrition levels. Plant Sci. 2005, 168, 81–87. [Google Scholar] [CrossRef]
- Kong, F.-J.; Oyanagi, A.; Komatsu, S. Cell wall proteome of wheat roots under flooding stress using gel-based and LC MS/MS-based proteomics approaches. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2010, 1804, 124–136. [Google Scholar] [CrossRef]
- Kamal, A.H.M.; Cho, K.; Choi, J.-S.; Bae, K.-H.; Komatsu, S.; Uozumi, N.; Woo, S.H. The wheat chloroplastic proteome. J. Proteom. 2013, 93, 326–342. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. Is proteomics the new genomics? Cell 2007, 130, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurien, B.T.; Scofield, R.H. Western blotting. Methods 2006, 38, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Lequin, R.M. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 2005, 51, 2415–2418. [Google Scholar] [CrossRef] [Green Version]
- Waltraud, X.S.; Björn, U. Quantitation in mass-spectrometry-based proteomics. Annu. Rev. Plant Biol. 2010, 61, 491–516. [Google Scholar] [CrossRef] [Green Version]
- Magdeldin, S.; Enany, S.; Yoshida, Y.; Xu, B.; Zhang, Y.; Zureena, Z.; Lokamani, I.; Yaoita, E.; Yamamoto, T. Basics and recent advances of two dimensional- polyacrylamide gel electrophoresis. Clin. Proteom. 2014, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- Millikan, R. Rays of Positive Electricity and their Application to Chemical Analysis. By SIR JJ THOMSON. Longmans, Green & Co. 1913.Pp. vi+ 132. Science 1914, 40, 174. [Google Scholar]
- Lee, P.Y.; Saraygord-Afshari, N.; Low, T.Y. The evolution of two-dimensional gel electrophoresis-from proteomics to emerging alternative applications. J. Chromatogr. A 2020, 1615, 460763. [Google Scholar] [CrossRef]
- Meleady, P. Two-dimensional gel electrophoresis and 2D-DIGE. In Difference Gel Electrophoresis. Methods in Molecular Biology; Ohlendieck, K., Ed.; Humana Press: New York, NY, USA, 2018; Volume 1664, pp. 3–14. [Google Scholar]
- Abdallah, C.; Dumas-Gaudot, E.; Renaut, J.; Sergeant, K. Gel-based and gel-free quantitative proteomics approaches at a glance. Int. J. Plant Genom. 2012, 2012, 494572. [Google Scholar] [CrossRef] [Green Version]
- Cunsolo, V.; Muccilli, V.; Saletti, R.; Foti, S. Mass spectrometry in food proteomics: A tutorial. J. Mass Spectrom. 2014, 49, 768–784. [Google Scholar] [CrossRef]
- Agregán, R.; Echegaray, N.; López-Pedrouso, M.; Aadil, R.M.; Hano, C.; Franco, D.; Lorenzo, J.M. Proteomic advances in cereal and vegetable crops. Molecules 2021, 26, 4924. [Google Scholar] [CrossRef] [PubMed]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef]
- Glinski, M.; Weckwerth, W. The role of mass spectrometry in plant systems biology. Mass Spectrom. Rev. 2006, 25, 173–214. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Silva, L.P. MALDI-TOF MS profiling approach: How much can we get from it? Front. Plant Sci. 2015, 6, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazari, M.; Moosavi, S.S.; Maleki, M.; Jamshidi Goharrizi, K. Chloroplastic acyl carrier protein synthase I and chloroplastic 20 kDa chaperonin proteins are involved in wheat (Triticum aestivum) in response to moisture stress. J. Plant Interact. 2020, 15, 180–187. [Google Scholar] [CrossRef]
- Ghatak, A.; Chaturvedi, P.; Weckwerth, W. Cereal crop proteomics: Systemic analysis of crop drought stress responses towards marker-assisted selection breeding. Front. Plant Sci. 2017, 8, 757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, N.; Shimada, T. Recent advances in mass spectrometry-based approaches for proteomics and biologics: Great contribution for developing therapeutic antibodies. Pharmacol. Ther. 2018, 185, 147–154. [Google Scholar] [CrossRef]
- Wu, C.C.; MacCoss, M.J. Shotgun proteomics: Tools for the analysis of complex biological systems. Curr. Opin. Mol. Ther. 2002, 4, 242–250. [Google Scholar]
- Komatsu, S. Plant Proteomic Research 2.0: Trends and perspectives. Int. J. Mol. Sci. 2019, 20, 2495. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.C.; Lim, Y.S.; Lau, S.-E. Proteomics in commercial crops: An overview. J. Proteom. 2017, 169, 176–188. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, L.; Shang, H.; Liu, S.; Peng, J.; Gong, W.; Shi, Y.; Zhang, S.; Li, J.; Gong, J. iTRAQ-based quantitative proteomic analysis of cotton roots and leaves reveals pathways associated with salt stress. PLoS ONE 2016, 11, e0148487. [Google Scholar] [CrossRef] [PubMed]
- Ow, S.Y.; Salim, M.; Noirel, J.; Evans, C.; Rehman, I.; Wright, P.C. iTRAQ Underestimation in Simple and Complex Mixtures: “The Good, the Bad and the Ugly”. J. Proteome Res. 2009, 8, 5347–5355. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Liu, L.; Zhang, Y.; Sun, H.; Zhang, K.; Bai, Z.; Dong, H.; Liu, Y.; Li, C. Tandem mass tag-based (TMT) quantitative proteomics analysis reveals the response of fine roots to drought stress in cotton (Gossypium hirsutum L.). BMC Plant Biol. 2020, 20, 328. [Google Scholar] [CrossRef] [PubMed]
- Pichler, P.; Köcher, T.; Holzmann, J.; Mazanek, M.; Taus, T.; Ammerer, G.; Mechtler, K. Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ orbitrap. Anal. Chem. 2010, 82, 6549–6558. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Timp, G. Beyond mass spectrometry, the next step in proteomics. Sci. Adv. 2020, 6, eaax8978. [Google Scholar] [CrossRef] [Green Version]
- Lundgren, D.H.; Hwang, S.-I.; Wu, L.; Han, D.K. Role of spectral counting in quantitative proteomics. Expert Rev. Proteom. 2010, 7, 39–53. [Google Scholar] [CrossRef]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A critical review of bottom-up proteomics: The good, the bad, and the future of this field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Al Shweiki, M.H.D.R.; Mönchgesang, S.; Majovsky, P.; Thieme, D.; Trutschel, D.; Hoehenwarter, W. Assessment of label-free quantification in discovery proteomics and impact of technological factors and natural variability of protein abundance. J. Proteome Res. 2017, 16, 1410–1424. [Google Scholar] [CrossRef]
- O’Connell, J.D.; Paulo, J.A.; O’Brien, J.J.; Gygi, S.P. Proteome-wide evaluation of two common protein quantification methods. J. Proteome Res. 2018, 17, 1934–1942. [Google Scholar] [CrossRef]
- Palmblad, M.; Mills, D.J.; Bindschedler, L.V.; Cramer, R. Chromatographic alignment of LC-MS and LC-MS/MS datasets by genetic algorithm feature extraction. J. Am. Soc. Mass Spectrom. 2007, 18, 1835–1843. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-X.; Zhang, Y.; Fernie, A.R.; Liu, Y.-G.; Zhu, F.-Y. SWATH-MS-based proteomics: Strategies and applications in plants. Trends Biotechnol. 2021, 39, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Nahar, K.; Alam, M.; Roychowdhury, R.; Fujita, M. Physiological, biochemical, and molecular mechanisms of heat stress tolerance in plants. Int. J. Mol. Sci. 2013, 14, 9643–9684. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Bramley, H.; Palta, J.A.; Siddique, K.H. Heat stress in wheat during reproductive and grain-filling phases. Crit. Rev. Plant Sci. 2011, 30, 491–507. [Google Scholar] [CrossRef]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Wang, X.; Cai, J.; Jiang, D.; Liu, F.; Dai, T.; Cao, W. Pre-anthesis high-temperature acclimation alleviates damage to the flag leaf caused by post-anthesis heat stress in wheat. J. Plant Physiol. 2011, 168, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Prasad, P.V.V.; Fritz, A.K.; Boyle, D.L.; Gill, B.S. Impact of high night-time and high daytime temperature stress on winter wheat. J. Agron. Crop Sci. 2015, 201, 206–218. [Google Scholar] [CrossRef]
- Lu, Y.; Li, R.; Wang, R.; Wang, X.; Zheng, W.; Sun, Q.; Tong, S.; Dai, S.; Xu, S. Comparative proteomic analysis of flag leaves reveals new insight into wheat heat adaptation. Front. Plant Sci. 2017, 8, 1086. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pan, J.; Huang, X.; Guo, D.; Lou, H.; Hou, Z.; Su, M.; Liang, R.; Xie, C.; You, M. Differential effects of a post-anthesis heat stress on wheat (Triticum aestivum L.) grain proteome determined by iTRAQ. Sci. Rep. 2017, 7, 3468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lou, H.; Guo, D.; Zhang, R.; Su, M.; Hou, Z.; Zhou, H.; Liang, R.; Xie, C.; You, M. Identifying changes in the wheat kernel proteome under heat stress using iTRAQ. Crop J. 2018, 6, 600–610. [Google Scholar] [CrossRef]
- Zhang, X.; Högy, P.; Wu, X.; Schmid, I.; Wang, X.; Schulze, W.X.; Jiang, D.; Fangmeier, A. Physiological and proteomic evidence for the interactive effects of post-anthesis heat stress and elevated CO2 on wheat. Proteomics 2018, 18, 1800262. [Google Scholar] [CrossRef]
- Wang, X.; Hou, L.; Lu, Y.; Wu, B.; Gong, X.; Liu, M.; Wang, J.; Sun, Q.; Vierling, E.; Xu, S. Metabolic adaptation of wheat grain contributes to a stable filling rate under heat stress. J. Exp. Bot. 2018, 69, 5531–5545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.R.; Singh, K.; Ahuja, S.; Tasleem, M.; Singh, I.; Kumar, S.; Grover, M.; Mishra, D.; Rai, G.K.; Goswami, S. Quantitative proteomic analysis reveals novel stress-associated active proteins (SAAPs) and pathways involved in modulating tolerance of wheat under terminal heat. Funct. Integr. Genom. 2019, 19, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Chunduri, V.; Kaur, A.; Kaur, S.; Kumar, A.; Sharma, S.; Sharma, N.; Singh, P.; Kapoor, P.; Kaur, S.; Kumari, A. Gene expression and proteomics studies suggest an involvement of multiple pathways under day and day–night combined heat stresses during grain filling in wheat. Front. Plant Sci. 2021, 12, 660446. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Choudhary, M.; Halder, T.; Prakash, N.R.; Singh, V.; Sheoran, S.; Longmei, N.; Rakshit, S.; Siddique, K.H.M. Salinity stress tolerance and omics approaches: Revisiting the progress and achievements in major cereal crops. Heredity 2022, 1–22. [Google Scholar] [CrossRef]
- Kamal, A.H.M.; Cho, K.; Kim, D.-E.; Uozumi, N.; Chung, K.-Y.; Lee, S.Y.; Choi, J.-S.; Cho, S.-W.; Shin, C.-S.; Woo, S.H. Changes in physiology and protein abundance in salt-stressed wheat chloroplasts. Mol. Biol. Rep. 2012, 39, 9059–9074. [Google Scholar] [CrossRef]
- Fercha, A.; Capriotti, A.L.; Caruso, G.; Cavaliere, C.; Samperi, R.; Stampachiacchiere, S.; Laganà, A. Comparative analysis of metabolic proteome variation in ascorbate-primed and unprimed wheat seeds during germination under salt stress. J. Proteom. 2014, 108, 238–257. [Google Scholar] [CrossRef]
- Maleki, M.; Naghavi, M.R.; Alizadeh, H.; Poostini, K.; Abd Mishani, C. Comparison of protein changes in the leaves of two bread wheat cultivars with different sensitivity under salt stress. Annu. Res. Rev. Biol. 2014, 11, 1784–1797. [Google Scholar] [CrossRef]
- Jiang, Q.; Hu, Z.; Zhang, H.; Ma, Y. Overexpression of GmDREB1 improves salt tolerance in transgenic wheat and leaf protein response to high salinity. Crop J. 2014, 2, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, A.L.; Borrelli, G.M.; Colapicchioni, V.; Papa, R.; Piovesana, S.; Samperi, R.; Stampachiacchiere, S.; Laganà, A. Proteomic study of a tolerant genotype of durum wheat under salt-stress conditions. Anal. Bioanal. Chem. 2014, 406, 1423–1435. [Google Scholar] [CrossRef]
- Lv, D.-W.; Zhu, G.-R.; Zhu, D.; Bian, Y.-W.; Liang, X.-N.; Cheng, Z.-W.; Deng, X.; Yan, Y.-M. Proteomic and phosphoproteomic analysis reveals the response and defense mechanism in leaves of diploid wheat T. monococcum under salt stress and recovery. J. Proteom. 2016, 143, 93–105. [Google Scholar] [CrossRef]
- Singh, R.P.; Runthala, A.; Khan, S.; Jha, P.N. Quantitative proteomics analysis reveals the tolerance of wheat to salt stress in response to Enterobacter cloacae SBP-8. PLoS ONE 2017, 12, e0183513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Li, X.; Niu, F.; Sun, X.; Hu, Z.; Zhang, H. iTRAQ-based quantitative proteomic analysis of wheat roots in response to salt stress. Proteomics 2017, 17, 1600265. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Xiao, C.; Xiao, B.; Wang, M.; Liu, J.; Bhanbhro, N.; Khan, A.; Wang, H.; Wang, H.; Yang, C. Proteomic profiling sheds light on alkali tolerance of common wheat (Triticum aestivum L.). Plant Physiol. Biochem. 2019, 138, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Xue, C.; Xiong, Y.; Meng, X.; Li, B.; Shen, R.; Lan, P. Proteomic dissection of the similar and different responses of wheat to drought, salinity and submergence during seed germination. J. Proteom. 2020, 220, 103756. [Google Scholar] [CrossRef]
- Yan, M.; Zheng, L.; Li, B.; Shen, R.; Lan, P. Comparative proteomics reveals new insights into the endosperm responses to drought, salinity and submergence in germinating wheat seeds. Plant Mol. Biol. 2021, 105, 287–302. [Google Scholar] [CrossRef]
- Zhu, D.; Luo, F.; Zou, R.; Liu, J.; Yan, Y. Integrated physiological and chloroplast proteome analysis of wheat seedling leaves under salt and osmotic stresses. J. Proteom. 2021, 234, 104097. [Google Scholar] [CrossRef]
- Oyiga, B.C.; Ogbonnaya, F.C.; Sharma, R.C.; Baum, M.; Léon, J.; Ballvora, A. Genetic and transcriptional variations in NRAMP-2 and OPAQUE1 genes are associated with salt stress response in wheat. Theor. Appl. Genet. 2019, 132, 323–346. [Google Scholar] [CrossRef] [Green Version]
- Hao, P.; Zhu, J.; Gu, A.; Lv, D.; Ge, P.; Chen, G.; Li, X.; Yan, Y. An integrative proteome analysis of different seedling organs in tolerant and sensitive wheat cultivars under drought stress and recovery. Proteomics 2015, 15, 1544–1563. [Google Scholar] [CrossRef]
- Ashraf, M.; Harris, P.J.C. Photosynthesis under stressful environments: An overview. Photosynthetica 2013, 51, 163–190. [Google Scholar] [CrossRef]
- Caruso, G.; Cavaliere, C.; Foglia, P.; Gubbiotti, R.; Samperi, R.; Laganà, A. Analysis of drought responsive proteins in wheat (Triticum durum) by 2D-PAGE and MALDI-TOF mass spectrometry. Plant Sci. 2009, 177, 570–576. [Google Scholar] [CrossRef]
- Nezhadahmadi, A.; Prodhan, Z.H.; Faruq, G. Drought tolerance in wheat. Sci. World J. 2013, 2013, 610721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajheidari, M.; Eivazi, A.; Buchanan, B.B.; Wong, J.H.; Majidi, I.; Salekdeh, G.H. Proteomics uncovers a role for redox in drought tolerance in wheat. J. Proteome Res. 2007, 6, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Huang, X.-W.; Wang, L.-L.; Wei, L.; Wu, Z.-H.; You, M.-S.; Li, B.-Y. Proteomic analysis of wheat seed in response to drought stress. J. Integr. Agric. 2014, 13, 919–925. [Google Scholar] [CrossRef]
- Labuschagne, M.; Masci, S.; Tundo, S.; Muccilli, V.; Saletti, R.; van Biljon, A. Proteomic analysis of proteins responsive to drought and low temperature stress in a hard red spring wheat cultivar. Molecules 2020, 25, 1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.-S.; Liang, X.-N.; Li, X.; Wang, S.-L.; Lv, D.-W.; Ma, C.-Y.; Li, X.-H.; Ma, W.-J.; Yan, Y.-M. Wheat drought-responsive grain proteome analysis by linear and nonlinear 2-DE and MALDI-TOF mass spectrometry. Int. J. Mol. Sci. 2012, 13, 16065–16083. [Google Scholar] [CrossRef] [Green Version]
- Budak, H.; Akpinar, B.A.; Unver, T.; Turktas, M. Proteome changes in wild and modern wheat leaves upon drought stress by two-dimensional electrophoresis and nanoLC-ESI–MS/MS. Plant Mol. Biol. 2013, 83, 89–103. [Google Scholar] [CrossRef]
- Li, X.; Cai, J.; Liu, F.; Dai, T.; Cao, W.; Jiang, D. Physiological, proteomic and transcriptional responses of wheat to combination of drought or waterlogging with late spring low temperature. Funct. Plant Biol. 2014, 41, 690–703. [Google Scholar] [CrossRef]
- Zhang, M.; Lv, D.; Ge, P.; Bian, Y.; Chen, G.; Zhu, G.; Li, X.; Yan, Y. Phosphoproteome analysis reveals new drought response and defense mechanisms of seedling leaves in bread wheat (Triticum aestivum L.). J. Proteom. 2014, 109, 290–308. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, L.; Lv, H.; Yu, Z.; Zhang, D.; Zhu, W. Identification of changes in Triticum aestivum L. leaf proteome in response to drought stress by 2D-PAGE and MALDI-TOF/TOF mass spectrometry. Acta Physiol. Plant. 2014, 36, 1385–1398. [Google Scholar] [CrossRef]
- Faghani, E.; Gharechahi, J.; Komatsu, S.; Mirzaei, M.; Khavarinejad, R.A.; Najafi, F.; Farsad, L.K.; Salekdeh, G.H. Comparative physiology and proteomic analysis of two wheat genotypes contrasting in drought tolerance. J. Proteom. 2015, 114, 1–15. [Google Scholar] [CrossRef]
- Liu, H.; Sultan, M.A.R.F.; Liu, X.L.; Zhang, J.; Yu, F.; Zhao, H.X. Physiological and comparative proteomic analysis reveals different drought responses in roots and leaves of drought-tolerant wild wheat (Triticum boeoticum). PLoS ONE 2015, 10, e0121852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, N.; Xu, W.; Hu, L.; Li, Y.; Wang, H.; Qi, X.; Fang, Y.; Hua, X. Drought tolerance and proteomics studies of transgenic wheat containing the maize C4 phosphoenolpyruvate carboxylase (PEPC) gene. Protoplasma 2016, 253, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, Y.; He, Q.; Li, H.; Zhang, X.; Zhang, F. Comparative proteomics illustrates the complexity of drought resistance mechanisms in two wheat (Triticum aestivum L.) cultivars under dehydration and rehydration. BMC Plant Biol. 2016, 16, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Gupta, S.K.; Majumder, B.; Maurya, V.K.; Deeba, F.; Alam, A.; Pandey, V. Salicylic acid mediated growth, physiological and proteomic responses in two wheat varieties under drought stress. J. Proteom. 2017, 163, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Han, Q.; Ma, D.; Hou, J.; Huang, X.; Wang, C.; Xie, Y.; Kang, G.; Guo, T. Characterizing physiological and proteomic analysis of the action of H2S to mitigate drought stress in young seedling of wheat. Plant Mol. Biol. Rep. 2018, 36, 45–57. [Google Scholar] [CrossRef]
- Li, N.; Zhang, S.; Liang, Y.; Qi, Y.; Chen, J.; Zhu, W.; Zhang, L. Label-free quantitative proteomic analysis of drought stress-responsive late embryogenesis abundant proteins in the seedling leaves of two wheat (Triticum aestivum L.) genotypes. J. Proteom. 2018, 172, 122–142. [Google Scholar] [CrossRef] [PubMed]
- Michaletti, A.; Naghavi, M.R.; Toorchi, M.; Zolla, L.; Rinalducci, S. Metabolomics and proteomics reveal drought-stress responses of leaf tissues from spring-wheat. Sci. Rep. 2018, 8, 5710. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, X.; Huang, G.; Feng, F.; Liu, X.; Guo, R.; Gu, F.; Zhong, X.; Mei, X. iTRAQ-based quantitative analysis of responsive proteins under PEG-induced drought stress in wheat leaves. Int. J. Mol. Sci. 2019, 20, 2621. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Zhao, Y.; Zhang, J.; Chao, M.; Xie, K.; Zhang, C.; Sun, F.; Liu, S.; Xi, Y. Proteomic analysis of the similarities and differences of soil drought and polyethylene glycol stress responses in wheat (Triticum aestivum L.). Plant Mol. Biol. 2019, 100, 391–410. [Google Scholar] [CrossRef]
- Nemati, M.; Piro, A.; Norouzi, M.; Vahed, M.M.; Nisticò, D.M.; Mazzuca, S. Comparative physiological and leaf proteomic analyses revealed the tolerant and sensitive traits to drought stress in two wheat parental lines and their F6 progenies. Environ. Exp. Bot. 2019, 158, 223–237. [Google Scholar] [CrossRef]
- Shayan, S.; Norouzi, M.; Vahed, M.M.; Mohammadi, S.A.; Toorchi, M. Leaf proteome pattern of two bread wheat varieties under water deficit stress conditions. Curr. Plant Biol. 2020, 23, 100146. [Google Scholar] [CrossRef]
- Ghatak, A.; Chaturvedi, P.; Bachmann, G.; Valledor, L.; Ramšak, Ž.; Bazargani, M.M.; Bajaj, P.; Jegadeesan, S.; Li, W.; Sun, X. Physiological and proteomic signatures reveal mechanisms of superior drought resilience in pearl millet compared to wheat. Front. Plant Sci. 2021, 11, 1965. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Li, G.; Xu, W.; Peng, X.; Han, Q.; Zhu, Y.; Guo, T. Proteomics reveals the effects of salicylic acid on growth and tolerance to subsequent drought stress in wheat. J. Proteome Res. 2012, 11, 6066–6079. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Noctor, G. Redox signaling in plants. Antioxid. Redox Signal. 2013, 18, 2087–2090. [Google Scholar] [CrossRef]
- Osipova, S.V.; Permyakov, A.V.; Permyakova, M.D.; Pshenichnikova, T.A.; Börner, A. Leaf dehydroascorbate reductase and catalase activity is associated with soil drought tolerance in bread wheat. Acta Physiol. Plant. 2011, 33, 2169–2177. [Google Scholar] [CrossRef]
- Dresselhaus, T.; Hückelhoven, R. Biotic and abiotic stress responses in crop plants. Agronomy 2018, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Ayalew, H.; Liu, H.; Yan, G. Identification and validation of root length QTLs for water stress resistance in hexaploid wheat (Triticum aestivum L.). Euphytica 2017, 213, 126. [Google Scholar] [CrossRef]
- Ferguson, D.L.; Guikema, J.A.; Paulsen, G.M. Ubiquitin pool modulation and protein degradation in wheat roots during high temperature stress. Plant Physiol. 1990, 92, 740–746. [Google Scholar] [CrossRef] [Green Version]
- Picton, S.J.; Richards, K.D.; Gardner, R.C. Protein profiles in root-tips of two wheat (Triticum aestivum L.) cultivars with differential tolerance to aluminium. In Plant-Soil Interactions at Low pH, 1st ed.; Wright, R.J., Baligar, V.C., Murrmann, R.P., Eds.; Springer: Beckley West Virginia, VA, USA, 1991; Volume 45, pp. 1063–1070. [Google Scholar]
- Basu, A.; Basu, U.; Taylor, G.J. Induction of microsomal membrane proteins in roots of an aluminum-resistant cultivar of Triticum aestivum L. under conditions of aluminum stress. Plant Physiol. 1994, 104, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Peng, X.; Xuan, H.; Wei, L.; Yang, Y.; Guo, T.; Kang, G. Proteomic analysis of leaves and roots of common wheat (Triticum aestivum L.) under copper-stress conditions. J. Proteome Res. 2013, 12, 4846–4861. [Google Scholar] [CrossRef]
- Kim, D.-E.; Roy, S.K.; Kamal, A.H.M.; Cho, K.; Kwon, S.J.; Cho, S.-W.; Park, C.-S.; Choi, J.-S.; Komatsu, S.; Lee, M.-S. Profiling of mitochondrial proteome in wheat roots. Mol. Biol. Rep. 2014, 41, 5359–5366. [Google Scholar] [CrossRef] [PubMed]
- Van Dingenen, J.; Blomme, J.; Gonzalez, N.; Inzé, D. Plants grow with a little help from their organelle friends. J. Exp. Bot. 2016, 67, erw399. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Fang, J.; Li, J.; Qu, B.; Ren, Y.; Ma, W.; Zhao, X.; Li, B.; Wang, D.; Li, Z. A genotypic difference in primary root length is associated with the inhibitory role of transforming growth factor-beta receptor-interacting protein-1 on root meristem size in wheat. Plant J. 2014, 77, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Li, G.; Wang, L.; Wei, L.; Yang, Y.; Wang, P.; Yang, Y.; Wang, Y.; Feng, W.; Wang, C. Hg-responsive proteins identified in wheat seedlings using iTRAQ analysis and the role of ABA in Hg stress. J. Proteome Res. 2015, 14, 249–267. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, Y.; Ren, Y.; Guo, Z.; Li, J.; Tong, Y.; Lin, T.; Cui, D. Comparative proteomic analysis provides insights into the regulatory mechanisms of wheat primary root growth. Sci. Rep. 2019, 9, 11741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, G.; Wu, Y.; Li, G.; Wang, P.; Han, Q.; Wang, Y.; Xie, Y.; Feng, W.; Ma, D.; Wang, C. Proteomics combined with BSMV-VIGS methods identified some N deficiency-responsive protein species and ABA role in wheat seedling. Plant Soil 2019, 444, 177–191. [Google Scholar] [CrossRef]
- Xu, Y.; Ren, Y.; Li, J.; Li, L.; Chen, S.; Wang, Z.; Xin, Z.; Chen, F.; Lin, T.; Cui, D. Comparative proteomic analysis provides new insights into low nitrogen-promoted primary root growth in hexaploid wheat. Front. Plant Sci. 2019, 10, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Zhao, J.; Bi, C.; Li, L.; Wang, Z. Transcriptome and proteomics analysis of wheat seedling roots reveals that increasing NH4+/NO3– ratio induced root lignification and reduced nitrogen utilization. Front. Plant Sci. 2022, 12, 797260. [Google Scholar] [CrossRef]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.A.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Liu, H.; Wu, Y.; Yan, G. Wheat genotypes tolerant to heat at seedling stage tend to be also tolerant at adult stage: The possibility of early selection for heat tolerance breeding. Crop J. 2022; in press. [Google Scholar] [CrossRef]
- Jian, M.; Zhang, D.; Wang, X.; Wei, S.; Zhao, Y.; Ding, Q.; Han, Y.; Ma, L. Differential expression pattern of the proteome in response to cadmium stress based on proteomics analysis of wheat roots. BMC Genom. 2020, 21, 343. [Google Scholar] [CrossRef]
- Beyer, S.; Daba, S.; Tyagi, P.; Bockelman, H.; Brown-Guedira, G.; IWGSC; Mohammadi, M. Loci and candidate genes controlling root traits in wheat seedlings—A wheat root GWAS. Funct. Integr. Genom. 2019, 19, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, R.; Mao, X.; Li, L.; Chang, X.; Zhang, X.; Jing, R. TaARF4 genes are linked to root growth and plant height in wheat. Ann. Bot. 2019, 124, 903–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmy, M.; Sugiyama, N.; Tomita, M.; Ishihama, Y. The rice proteogenomics database OryzaPG-DB: Development, expansion, and new features. Front. Plant Sci. 2012, 3, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Zybailov, B.; Majeran, W.; Friso, G.; Olinares, P.D.; van Wijk, K.J. PPDB, the Plant Proteomics Database at Cornell. Nucleic Acids Res. 2009, 37, D969–D974. [Google Scholar] [CrossRef]
- Duncan, O.; Trösch, J.; Fenske, R.; Taylor, N.L.; Millar, A.H. Resource: Mapping the Triticum aestivum proteome. Plant J. 2017, 89, 601–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemori, N.; Takemori, A.; Matsuoka, K.; Morishita, R.; Matsushita, N.; Aoshima, M.; Takeda, H.; Sawasaki, T.; Endo, Y.; Higashiyama, S. High-throughput synthesis of stable isotope-labeled transmembrane proteins for targeted transmembrane proteomics using a wheat germ cell-free protein synthesis system. Mol. BioSyst. 2015, 11, 361–365. [Google Scholar] [CrossRef]
- Niu, L.; Yuan, H.; Gong, F.; Wu, X.; Wang, W. Protein extraction methods shape much of the extracted proteomes. Front. Plant Sci. 2018, 9, 802. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Cong, X.; Zhai, L.; Hu, H.; Xu, J.-Y.; Zhao, W.; Zhu, M.; Tan, M.; Ye, B.-C. Comparative evaluation of label-free quantification strategies. J. Proteom. 2020, 215, 103669. [Google Scholar] [CrossRef]
- Šafář, J.; Šimková, H.; Kubaláková, M.; Číhalíková, J.; Suchánková, P.; Bartoš, J.; Doležel, J. Development of chromosome-specific BAC resources for genomics of bread wheat. Cytogenet. Genome Res. 2010, 129, 211–223. [Google Scholar] [CrossRef]
- Ribeiro, M.; Nunes-Miranda, J.D.; Branlard, G.; Carrillo, J.M.; Rodriguez-Quijano, M.; Igrejas, G. One hundred years of grain omics: Identifying the glutens that feed the world. J. Proteome Res. 2013, 12, 4702–4716. [Google Scholar] [CrossRef]
- Cozzolino, F.; Landolfi, A.; Iacobucci, I.; Monaco, V.; Caterino, M.; Celentano, S.; Zuccato, C.; Cattaneo, E.; Monti, M. New label-free methods for protein relative quantification applied to the investigation of an animal model of Huntington Disease. PLoS ONE 2020, 15, e0238037. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Zhu, D.; Han, Z.; Zhang, J.; Zhang, M.; Yan, Y. Label-free quantitative proteome analysis reveals the underlying mechanisms of grain nuclearproteins involved in wheat water-deficit response. Front. Plant Sci. 2021, 12, 748487. [Google Scholar] [CrossRef]
- Vasil, V.; Ana, M.C.; Michael, E.F.; Vasi, I.K. Herbicide resistant fertile transgenic wheat plants obtained by microprojectile bombardment of regenerable embryogenic callus. Nat. Biotechnol. 1992, 10, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Shan, Q.; Wang, Y.; Li, J.; Zhang, Y.; Chen, K.; Liang, Z.; Zhang, K.; Liu, J.; Xi, J.J.; Qiu, J.-L. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 686–688. [Google Scholar] [CrossRef]
- Shan, Q.; Wang, Y.; Li, J.; Gao, C. Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 2014, 9, 2395–2410. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Singh, K.; Grewal, S.; Nath, M. Impact of “omics” in improving drought tolerance in wheat. Crit. Rev. Plant Sci. 2020, 39, 222–235. [Google Scholar] [CrossRef]
- Nazir, R.; Mandal, S.; Mitra, S.; Ghorai, M.; Das, N.; Jha, N.K.; Majumder, M.; Pandey, D.K.; Dey, A. CRISPR/Cas genome-editing toolkit to enhance salt stress tolerance in rice and wheat. Physiol. Plant. 2022, 174, e13642. [Google Scholar] [CrossRef]
- Kumar, K.; Gambhir, G.; Dass, A.; Tripathi, A.K.; Singh, A.; Jha, A.K.; Yadava, P.; Choudhary, M.; Rakshit, S. Genetically modified crops: Current status and future prospects. Planta 2020, 251, 91. [Google Scholar] [CrossRef]
- Jouanin, A.; Gilissen, L.J.W.J.; Schaart, J.G.; Leigh, F.J.; Cockram, J.; Wallington, E.J.; Boyd, L.A.; van den Broeck, H.C.; van der Meer, I.M.; America, A.H.P. CRISPR/Cas9 gene editing of gluten in wheat to reduce gluten content and exposure—Reviewing methods to screen for coeliac safety. Front. Nutr. 2020, 7, 51. [Google Scholar] [CrossRef]
- Dolgalev, G.; Poverennaya, E. Applications of CRISPR-Cas technologies to proteomics. Genes 2021, 12, 1790. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Sacco, R.; Stukalov, A.; Van Drogen, A.; Planyavsky, M.; Hauri, S.; Aebersold, R.; Bennett, K.L.; Colinge, J.; Gstaiger, M. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat. Methods 2013, 10, 307–314. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halder, T.; Choudhary, M.; Liu, H.; Chen, Y.; Yan, G.; Siddique, K.H.M. Wheat Proteomics for Abiotic Stress Tolerance and Root System Architecture: Current Status and Future Prospects. Proteomes 2022, 10, 17. https://0-doi-org.brum.beds.ac.uk/10.3390/proteomes10020017
Halder T, Choudhary M, Liu H, Chen Y, Yan G, Siddique KHM. Wheat Proteomics for Abiotic Stress Tolerance and Root System Architecture: Current Status and Future Prospects. Proteomes. 2022; 10(2):17. https://0-doi-org.brum.beds.ac.uk/10.3390/proteomes10020017
Chicago/Turabian StyleHalder, Tanushree, Mukesh Choudhary, Hui Liu, Yinglong Chen, Guijun Yan, and Kadambot H. M. Siddique. 2022. "Wheat Proteomics for Abiotic Stress Tolerance and Root System Architecture: Current Status and Future Prospects" Proteomes 10, no. 2: 17. https://0-doi-org.brum.beds.ac.uk/10.3390/proteomes10020017