
The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle

 , , and
, , and 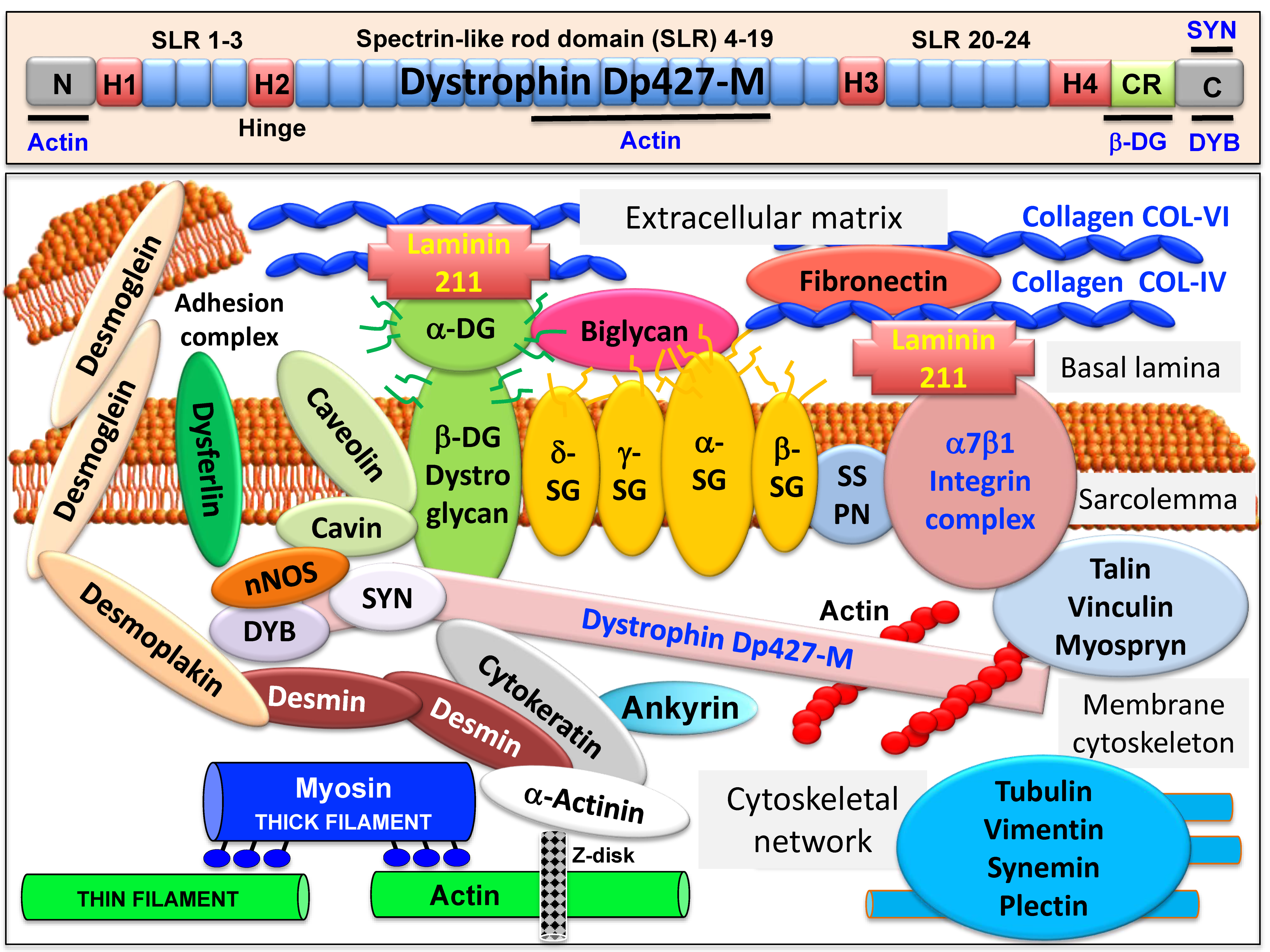{kind=link}
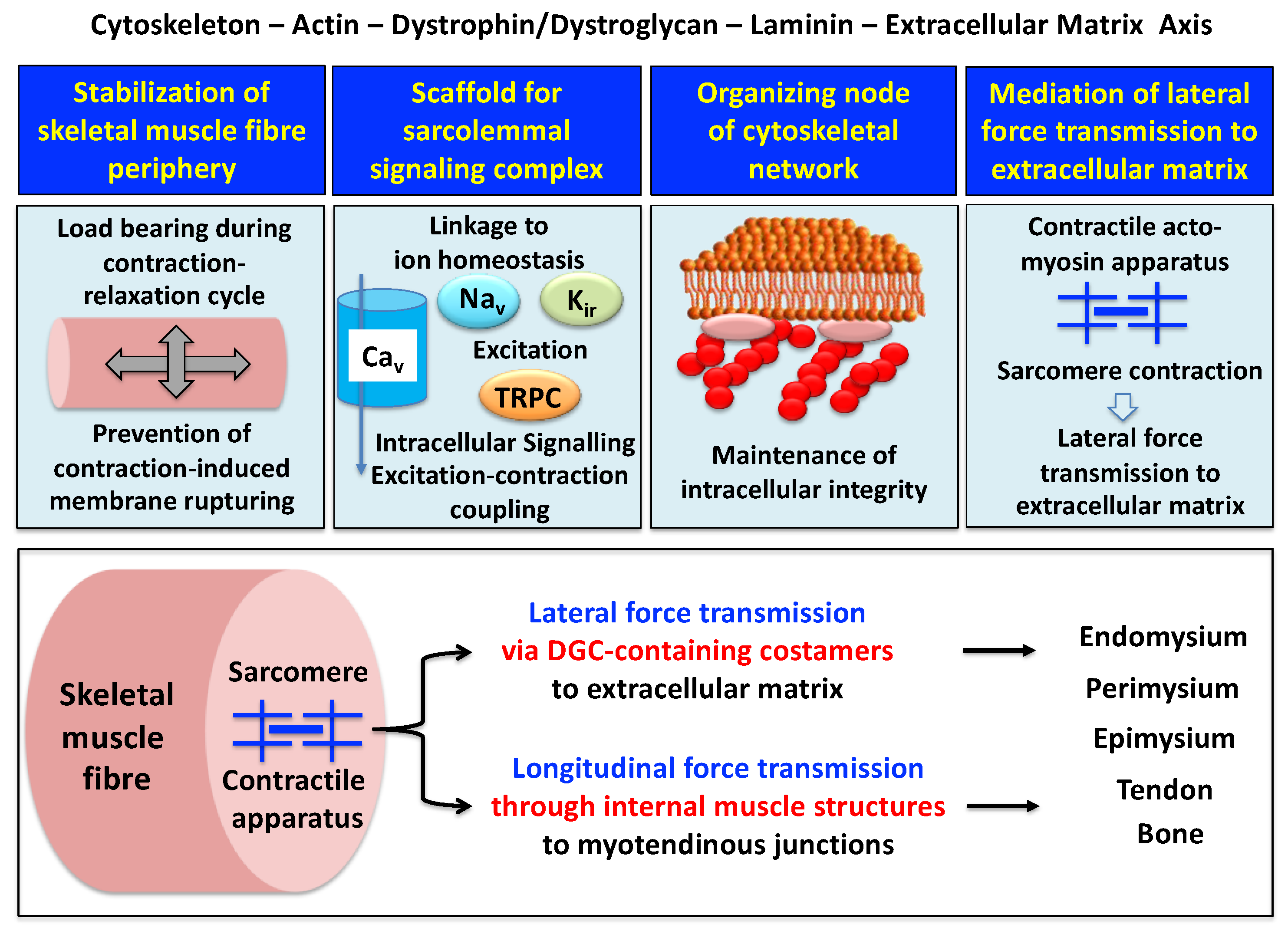{kind=link}
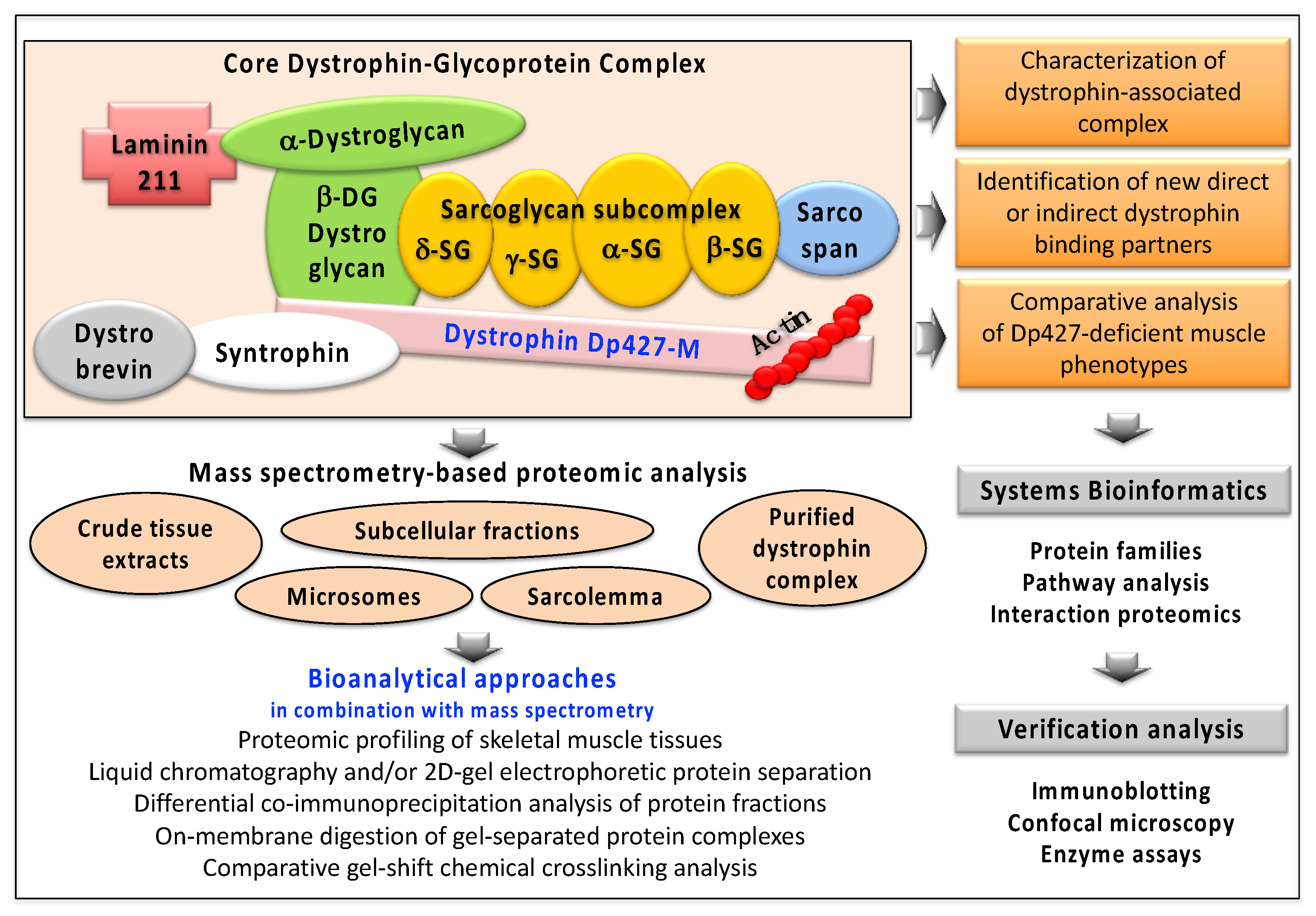{kind=link}
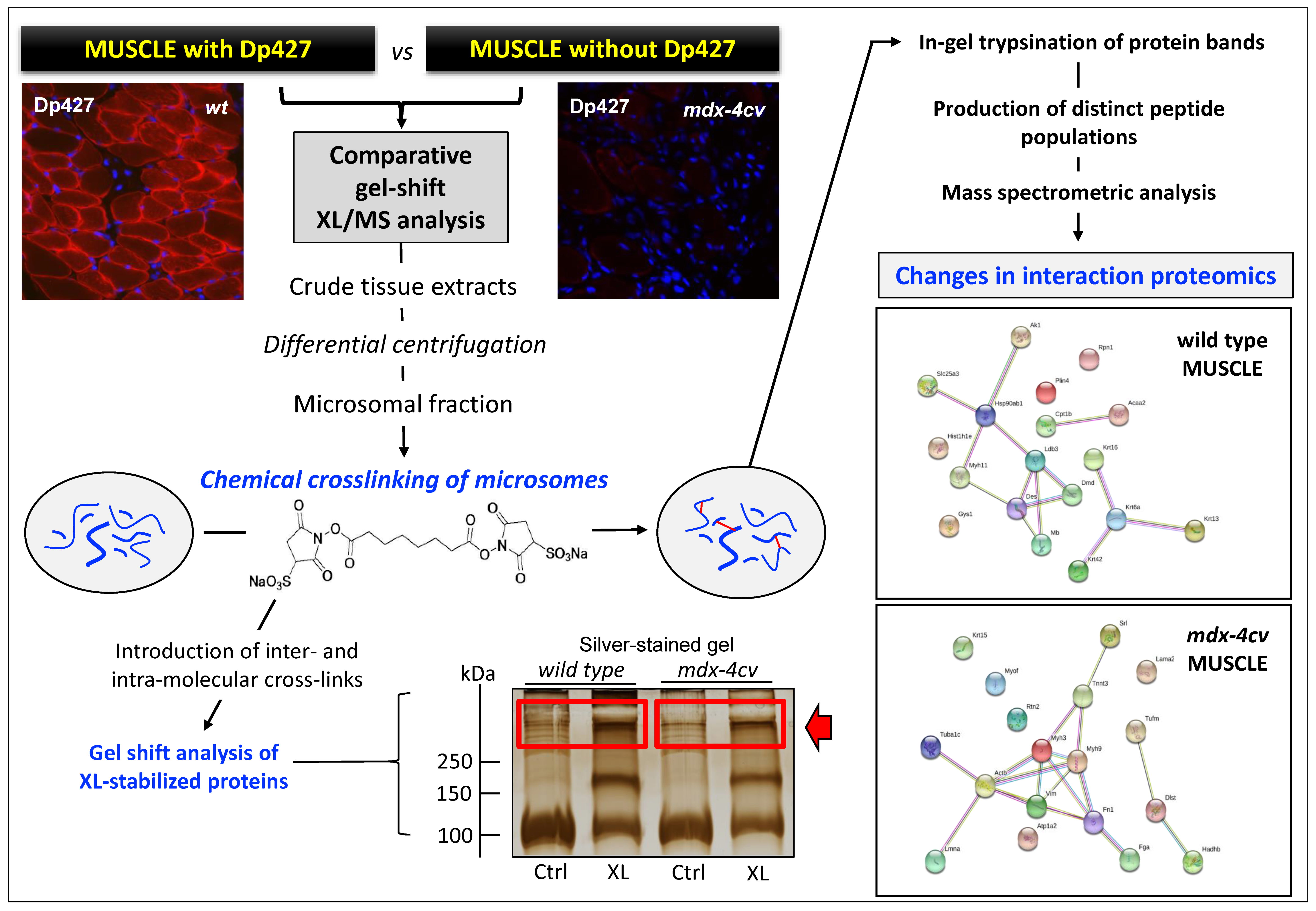{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Core Dystrophin Complex in Skeletal Muscle
2.1. The Dystrophin Node in Skeletal Muscle
2.2. The Sarcolemmal Dystrophin Complex and Lateral Force Transmission
2.3. Muscle Dystrophin Dp427-M and Its Associated Glycoprotein Complex
3. Proteomic and Biochemical Characterization of the Dystrophin Network in Skeletal Muscle
3.1. Proteomics of the Dystrophin Complex from Skeletal Muscle
3.2. The Dystrophin Complex as a Cellular Signaling Node in Skeletal Muscle
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Koenig, M.; Monaco, A.P.; Kunkel, L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988, 53, 219–228. [Google Scholar] [CrossRef]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Petkova, M.V.; Morales-Gonzales, S.; Relizani, K.; Gill, E.; Seifert, F.; Radke, J.; Stenzel, W.; Garcia, L.; Amthor, H.; Schuelke, M. Characterization of a Dmd (EGFP) reporter mouse as a tool to investigate dystrophin expression. Skelet. Muscle 2016, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.P.; Kahl, S.D. Association of dystrophin and an integral membrane glycoprotein. Nature 1989, 338, 259–262. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef]
- Yoshida, M.; Ozawa, E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990, 108, 748–752. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hagiwara, Y.; Mizuno, Y.; Yoshida, M.; Ozawa, E. Heterogeneity of dystrophin-associated proteins. J. Biochem. 1993, 114, 132–139. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K.P. Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell 1995, 80, 675–679. [Google Scholar] [CrossRef] [Green Version]
- Ohlendieck, K. Towards an understanding of the dystrophin-glycoprotein complex: Linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur. J. Cell Biol. 1996, 69, 1–10. [Google Scholar] [PubMed]
- Murphy, S.; Ohlendieck, K. Mass spectrometric identification of dystrophin, the protein product of the Duchenne muscular dystrophy gene, in distinct muscle surface membranes. Int. J. Mol. Med. 2017, 40, 1078–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlendieck, K.; Ervasti, J.M.; Snook, J.B.; Campbell, K.P. Dystrophin-glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J. Cell Biol. 1991, 112, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, E.; Samitt, C.E.; Miranda, A.F.; Hays, A.P.; Salviati, G.; DiMauro, S.; Kunkel, L.M.; Hoffman, E.P.; Rowland, L.P. Duchenne muscular dystrophy: Deficiency of dystrophin at the muscle cell surface. Cell 1988, 54, 447–452. [Google Scholar] [CrossRef]
- Watkins, S.C.; Hoffman, E.P.; Slayter, H.S.; Kunkel, L.M. Immunoelectron microscopic localization of dystrophin in myofibres. Nature 1988, 333, 863–866. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Sonnemann, K.J. Biology of the striated muscle dystrophin-glycoprotein complex. Int. Rev. Cytol. 2008, 265, 191–225. [Google Scholar]
- Murphy, S.; Ohlendieck, K. The biochemical and mass spectrometric profiling of the dystrophin complexome from skeletal muscle. Comput. Struct. Biotechnol. J. 2015, 14, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.; Ohlendieck, K. Mass spectrometric identification of dystrophin isoform Dp427 by on-membrane digestion of sarcolemma from skeletal muscle. Anal. Biochem. 2010, 404, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Johnson, E.; Xu, R.; Martin, L.T.; Martin, P.T.; Montanaro, F. Comparative proteomic profiling of dystroglycan-associated proteins in wild type, mdx, and Galgt2 transgenic mouse skeletal muscle. J. Proteome Res. 2012, 11, 4413–4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.K.; Li, B.; Yoon, J.H.; Flanigan, K.M.; Martin, P.T.; Ervasti, J.; Montanaro, F. Identification of new dystroglycan complexes in skeletal muscle. PLoS ONE 2013, 8, e73224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, R.; Hsiao, J.J.; Smits, M.M.; Ng, B.H.; Pospisil, T.C.; Jones, K.S.; Campbell, K.P.; Wright, M.E. Molecular signatures of membrane protein complexes underlying muscular dystrophy. Mol. Cell. Proteom. 2016, 15, 2169–2185. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.; Brinkmeier, H.; Krautwald, M.; Henry, M.; Meleady, P.; Ohlendieck, K. Proteomic profiling of the dystrophin complex and membrane fraction from dystrophic mdx muscle reveals decreases in the cytolinker desmoglein and increases in the extracellular matrix stabilizers biglycan and fibronectin. J. Muscle Res. Cell. Motil. 2017, 38, 251–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlendieck, K.; Matsumura, K.; Ionasescu, V.V.; Towbin, J.A.; Bosch, E.P.; Weinstein, S.L.; Sernett, S.W.; Campbell, K.P. Duchenne muscular dystrophy: Deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 1993, 43, 795–800. [Google Scholar] [CrossRef]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.J.; Kunkel, L.M. The pathogenesis and therapy of muscular dystrophies. Annu. Rev. Genom. Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef] [Green Version]
- Doorenweerd, N. Combining genetics, neuropsychology and neuroimaging to improve understanding of brain involvement in Duchenne muscular dystrophy—A narrative review. Neuromuscul. Disord. 2020, 30, 437–442. [Google Scholar] [CrossRef]
- Goemans, N.; Buyse, G. Current treatment and management of dystrophinopathies. Curr. Treat. Options Neurol. 2014, 16, 287. [Google Scholar] [CrossRef]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [Green Version]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, K.; Faelan, C.; Patterson-Kane, J.C.; Rudmann, D.G.; Moore, S.A.; Frank, D.; Charleston, J.; Tinsley, J.; Young, G.D.; Milici, A.J. Duchenne and becker muscular dystrophies: A review of animal models, clinical end points, and biomarker quantification. Toxicol. Pathol. 2017, 45, 961–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mázala, D.A.; Novak, J.S.; Hogarth, M.W.; Nearing, M.; Adusumalli, P.; Tully, C.B.; Habib, N.F.; Gordish-Dressman, H.; Chen, Y.W.; Jaiswal, J.K.; et al. TGF-β-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight 2020, 5, e135703. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, A.; Murphy, S.; Dowling, P.; Ohlendieck, K. Pathoproteomic profiling of the skeletal muscle matrisome in dystrophinopathy associated myofibrosis. Proteomics 2016, 16, 345–366. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of inherited muscular dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar]
- Liewluck, T.; Milone, M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018, 58, 167–177. [Google Scholar] [CrossRef]
- Pradhan, B.S.; Prószyński, T.J. A role for caveolin-3 in the pathogenesis of muscular dystrophies. Int. J. Mol. Sci. 2020, 21, 8736. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic serum biomarkers for neuromuscular diseases. Expert Rev. Proteom. 2018, 15, 277–291. [Google Scholar] [CrossRef]
- Dowling, P.; Murphy, S.; Zweyer, M.; Raucamp, M.; Swandulla, D.; Ohlendieck, K. Emerging proteomic biomarkers of X-linked muscular dystrophy. Expert Rev. Mol. Diagn. 2019, 19, 739–755. [Google Scholar] [CrossRef] [Green Version]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalili Szigyarto, C. Duchenne Muscular Dystrophy: Recent advances in protein biomarkers and the clinical application. Expert Rev. Proteom. 2020, 17, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Spendiff, S.; Roos, A.; Bourque, P.R.; Warman Chardon, J.; Kirschner, J.; Horvath, R.; Lochmüller, H. Advances in the diagnosis of inherited neuromuscular diseases and implications for therapy development. Lancet Neurol. 2020, 19, 522–532. [Google Scholar] [CrossRef]
- Partridge, T.A. Impending therapies for Duchenne muscular dystrophy. Curr. Opin. Neurol. 2011, 24, 415–422. [Google Scholar] [CrossRef]
- Spinazzola, J.M.; Kunkel, L.M. Pharmacological therapeutics targeting the secondary defects and downstream pathology of Duchenne muscular dystrophy. Expert Opin. Orphan Drugs 2016, 4, 1179–1194. [Google Scholar] [CrossRef] [Green Version]
- Waldrop, M.A.; Flanigan, K.M. Update in Duchenne and Becker muscular dystrophy. Curr. Opin. Neurol. 2019, 32, 722–727. [Google Scholar] [CrossRef]
- Werneck, L.C.; Lorenzoni, P.J.; Ducci, R.D.; Fustes, O.H.; Kay, C.S.K.; Scola, R.H. Duchenne muscular dystrophy: An historical treatment review. Arq. Neuropsiquiatr. 2019, 77, 579–589. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Jafari Abarghan, Y.; Bozorg Qomi, S.; Bayat, H.; Yousefi, M.; Azhdari, S.; Talebi, S.; Mojarrad, M. Common therapeutic advances for Duchenne muscular dystrophy (DMD). Int. J. Neurosci. 2020. [Google Scholar] [CrossRef]
- Murphy, S.; Dowling, P.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of giant skeletal muscle proteins. Expert Rev. Proteom. 2019, 16, 241–256. [Google Scholar] [CrossRef]
- Koenig, M.; Kunkel, L.M. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J. Biol. Chem. 1990, 265, 4560–4566. [Google Scholar] [CrossRef]
- Fealey, M.E.; Horn, B.; Coffman, C.; Miller, R.; Lin, A.Y.; Thompson, A.R.; Schramel, J.; Groth, E.; Hinderliter, A.; Cembran, A.; et al. Dynamics of dystrophin’s actin-binding domain. Biophys. J. 2018, 115, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullah, M.; Hassan, A.; Rashid, S.; Naeem, M. Tyrosine phosphorylation as a regulator of dystrophin and beta-dystroglycan interaction: A molecular insight. J. Mol. Graph Model. 2020, 99, 107623. [Google Scholar] [CrossRef] [PubMed]
- Delalande, O.; Molza, A.E.; Dos Santos Morais, R.; Chéron, A.; Pollet, É.; Raguenes-Nicol, C.; Tascon, C.; Giudice, E.; Guilbaud, M.; Nicolas, A.; et al. Dystrophin’s central domain forms a complex filament that becomes disorganized by in-frame deletions. J. Biol. Chem. 2018, 293, 6637–6646. [Google Scholar] [CrossRef] [Green Version]
- Rybakova, I.N.; Ervasti, J.M. Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J. Biol. Chem. 1997, 272, 28771–28778. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.Q.; McNally, E.M. The dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar]
- Gawor, M.; Prószyński, T.J. The molecular cross talk of the dystrophin-glycoprotein complex. Ann. NY Acad. Sci. 2018, 1412, 62–72. [Google Scholar] [CrossRef]
- Bhat, H.F.; Mir, S.S.; Dar, K.B.; Bhat, Z.F.; Shah, R.A.; Ganai, N.A. ABC of multifaceted dystrophin glycoprotein complex (DGC). J. Cell. Physiol. 2018, 233, 5142–5159. [Google Scholar] [CrossRef]
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr. Protein Pept. Sci. 2013, 14, 680–697. [Google Scholar] [CrossRef]
- Leyva-Leyva, M.; Sandoval, A.; Felix, R.; González-Ramírez, R. Biochemical and functional interplay between Ion channels and the components of the dystrophin-associated glycoprotein complex. J. Membr. Biol. 2018, 251, 535–550. [Google Scholar] [CrossRef]
- Constantin, B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 2014, 1838, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybakova, I.N.; Amann, K.J.; Ervasti, J.M. A new model for the interaction of dystrophin with F-actin. J. Cell Biol. 1996, 135, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybakova, I.N.; Patel, J.R.; Ervasti, J.M. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 2000, 150, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.K.; Cheng, H.; Ross, R.S.; Knowlton, K.U.; Chen, J. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog. Pediatr. Cardiol. 2011, 31, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, K.S.; Palmer, M.L.; van der Meulen, J.H.; Renoux, A.; Kostrominova, T.Y.; Michele, D.E.; Faulkner, J.A. Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J. Physiol. 2011, 589, 1195–1208. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P.; Stull, J.T. Skeletal muscle basement membrane-sarcolemma-cytoskeleton interaction minireview series. J. Biol. Chem. 2003, 278, 12599–12600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishmarev, D. Excitation-contraction coupling in skeletal muscle: Recent progress and unanswered questions. Biophys. Rev. 2020, 12, 143–153. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [Green Version]
- Sarkis, J.; Vié, V.; Winder, S.J.; Renault, A.; Le Rumeur, E.; Hubert, J.F. Resisting sarcolemmal rupture: Dystrophin repeats increase membrane-actin stiffness. FASEB J. 2013, 27, 359–367. [Google Scholar] [CrossRef]
- Rader, E.P.; Turk, R.; Willer, T.; Beltrán, D.; Inamori, K.; Peterson, T.A.; Engle, J.; Prouty, S.; Matsumura, K.; Saito, F.; et al. Role of dystroglycan in limiting contraction-induced injury to the sarcomeric cytoskeleton of mature skeletal muscle. Proc. Natl. Acad. Sci. USA 2016, 113, 10992–10997. [Google Scholar] [CrossRef] [Green Version]
- Ervasti, J.M. Costameres: The Achilles’ heel of Herculean muscle. J. Biol. Chem. 2003, 278, 13591–13594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, D.C.; Wallace, M.A.; Baar, K. Effects of aging, exercise, and disease on force transfer in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E1–E10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaka, O.; Casas-Fraile, L.; López de Munain, A.; Sáenz, A. Costamere proteins and their involvement in myopathic processes. Expert Rev. Mol. Med. 2015, 17, e12. [Google Scholar] [CrossRef] [PubMed]
- Purslow, P.P. The Structure and Role of Intramuscular Connective Tissue in Muscle Function. Front. Physiol. 2020, 11, 495. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.C.; Young, P.W. The actinin family of actin cross-linking proteins—A genetic perspective. Cell. Biosci. 2015, 5, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlendieck, K.; Campbell, K.P. Dystrophin constitutes 5% of membrane cytoskeleton in skeletal muscle. FEBS Lett. 1991, 283, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Ohlendieck, K. Characterisation of the dystrophin-related protein utrophin in highly purified skeletal muscle sarcolemma vesicles. Biochim. Biophys. Acta 1996, 1283, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, S.A.; Duncan, N.M.; Rash, S.M.; Sadeghi, A.; Dewan, A.K.; Pillers, D.A. Redefinition of dystrophin isoform distribution in mouse tissue by RT-PCR implies role in nonmuscle manifestations of duchenne muscular dystrophy. Mol. Genet. Metab. 1998, 65, 272–281. [Google Scholar] [CrossRef]
- Ueda, H.; Baba, T.; Ohno, S. Current knowledge of dystrophin and dystrophin-associated proteins in the retina. Histol. Histopathol. 2000, 15, 753–760. [Google Scholar]
- Lidov, H.G.; Kunkel, L.M. Dystrophin and Dp140 in the adult rodent kidney. Lab. Invest. 1998, 78, 1543–1551. [Google Scholar]
- Mahyoob Rani, A.Q.; Maeta, K.; Kawaguchi, T.; Awano, H.; Nagai, M.; Nishio, H.; Matsuo, M. Schwann cell-specific Dp116 is expressed in glioblastoma cells, revealing two novel DMD gene splicing patterns. Biochem. Biophys. Rep. 2019, 20, 100703. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the neuropathophysiology of duchenne muscular dystrophy. Mol. Neurobiol. 2020, 57, 1748–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteome-wide changes in the mdx-4cv spleen due to pathophysiological crosstalk with dystrophin-deficient skeletal muscle. iScience 2020, 23, 101500. [Google Scholar] [CrossRef] [PubMed]
- Romo-Yáñez, J.; Rodríguez-Martínez, G.; Aragón, J.; Siqueiros-Márquez, L.; Herrera-Salazar, A.; Velasco, I.; Montanez, C. Characterization of the expression of dystrophins and dystrophin-associated proteins during embryonic neural stem/progenitor cell differentiation. Neurosci. Lett. 2020, 736, 135247. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Pinzan, E. Advances in imaging of brain abnormalities in neuromuscular disease. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419845567. [Google Scholar] [CrossRef] [PubMed]
- Waite, A.; Brown, S.C.; Blake, D.J. The dystrophin-glycoprotein complex in brain development and disease. Trends Neurosci. 2012, 35, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Kahl, S.D.; Campbell, K.P. Purification of dystrophin from skeletal muscle. J. Biol. Chem. 1991, 266, 9161–9165. [Google Scholar] [CrossRef]
- Finn, D.M.; Ohlendieck, K. Oligomerization of beta-dystroglycan in rabbit diaphragm and brain as revealed by chemical crosslinking. Biochim. Biophys. Acta 1998, 1370, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Chemical crosslinking analysis of β-dystroglycan in dystrophin-deficient skeletal muscle. HRB Open Res. 2018, 1, 17. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Suzuki, A.; Yamamoto, H.; Noguchi, S.; Mizuno, Y.; Ozawa, E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl beta-D-glucoside. Eur. J. Biochem. 1994, 222, 1055–1061. [Google Scholar] [CrossRef]
- Sadoulet-Puccio, H.M.; Feener, C.A.; Schaid, D.J.; Thibodeau, S.N.; Michels, V.V.; Kunkel, L.M. The genomic organization of human dystrobrevin. Neurogenetics 1997, 1, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, M.; Takahashi, M.P. The role of alpha-dystrobrevin in striated muscle. Int. J. Mol. Sci. 2011, 12, 1660–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, S.H.; Gee, R.; Madhaven, S.H.; Levinson, S.R.; Caldwell, J.H.; Sealock, R.; Froehner, S.C. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J. Neurosci. 1998, 18, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.S.; Ali, R.; Khanday, F.A. Syntrophins entangled in cytoskeletal meshwork: Helping to hold it all together. Cell Prolif. 2019, 52, e12562. [Google Scholar] [CrossRef] [Green Version]
- Roberds, S.L.; Anderson, R.D.; Ibraghimov-Beskrovnaya, O.; Campbell, K.P. Primary structure and muscle-specific expression of the 50-kDa dystrophin-associated glycoprotein (adhalin). J. Biol. Chem. 1993, 268, 23739–23742. [Google Scholar] [CrossRef]
- Anastasi, G.; Cutroneo, G.; Rizzo, G.; Favaloro, A. Sarcoglycan subcomplex in normal and pathological human muscle fibers. Eur. J. Histochem. 2007, 51, 29–33. [Google Scholar] [PubMed]
- Tarakci, H.; Berger, J. The sarcoglycan complex in skeletal muscle. Front. Biosci. 2016, 21, 744–756. [Google Scholar]
- Crosbie, R.H.; Heighway, J.; Venzke, D.P.; Lee, J.C.; Campbell, K.P. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J. Biol. Chem. 1997, 272, 31221. [Google Scholar] [CrossRef] [Green Version]
- Crosbie, R.H.; Lim, L.E.; Moore, S.A.; Hirano, M.; Hays, A.P.; Maybaum, S.W.; Collin, H.; Dovico, S.A.; Stolle, C.A.; Fardeau, M.; et al. Molecular and genetic characterization of sarcospan: Insights into sarcoglycan-sarcospan interactions. Hum. Mol. Genet. 2000, 9, 2019–2027. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.L.; Crosbie-Watson, R.H. Sarcospan: A small protein with large potential for Duchenne muscular dystrophy. Skelet. Muscle 2013, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Carmignac, V.; Durbeej, M. Cell-matrix interactions in muscle disease. J. Pathol. 2012, 226, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, M.; Morlacchi, S.; Bigotti, M.G.; Sciandra, F.; Brancaccio, A. Functional diversity of dystroglycan. Matrix Biol. 2009, 28, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Barresi, R.; Campbell, K.P. Dystroglycan: From biosynthesis to pathogenesis of human disease. J. Cell Sci. 2006, 119, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mias-Lucquin, D.; Dos Santos Morais, R.; Chéron, A.; Lagarrigue, M.; Winder, S.J.; Chenuel, T.; Pérez, J.; Appavou, M.S.; Martel, A.; Alviset, G.; et al. How the central domain of dystrophin acts to bridge F-actin to sarcolemmal lipids. J. Struct. Biol. 2020, 209, 107411. [Google Scholar] [CrossRef]
- Ibraghimov-Beskrovnaya, O.; Milatovich, A.; Ozcelik, T.; Yang, B.; Koepnick, K.; Francke, U.; Campbell, K.P. Human dystroglycan: Skeletal muscle cDNA, genomic structure, origin of tissue specific isoforms and chromosomal localization. Hum. Mol. Genet. 1993, 2, 1651–1657. [Google Scholar] [CrossRef]
- Miller, G.; Moore, C.J.; Terry, R.; La Riviere, T.; Mitchell, A.; Piggott, R.; Dear, T.N.; Wells, D.J.; Winder, S.J. Preventing phosphorylation of dystroglycan ameliorates the dystrophic phenotype in mdx mouse. Hum. Mol. Genet. 2012, 21, 4508–4520. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.J.; Winder, S.J. The inside and out of dystroglycan post-translational modification. Neuromuscul. Disord. 2012, 22, 959–965. [Google Scholar] [CrossRef]
- Swiderski, K.; Shaffer, S.A.; Gallis, B.; Odom, G.L.; Arnett, A.L.; Scott Edgar, J.; Baum, D.M.; Chee, A.; Naim, T.; Gregorevic, P.; et al. Phosphorylation within the cysteine-rich region of dystrophin enhances its association with β-dystroglycan and identifies a potential novel therapeutic target for skeletal muscle wasting. Hum. Mol. Genet. 2014, 23, 6697–6711. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the mouse diaphragm and refined mass spectrometric analysis of the dystrophic phenotype. J. Muscle Res. Cell. Motil. 2019, 40, 9–28. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of the sarcolemma-enriched fraction from dystrophic mdx-4cv skeletal muscle. J. Proteom. 2019, 191, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Comparative gel-based proteomic analysis of chemically crosslinked complexes in dystrophic skeletal muscle. Electrophoresis 2018, 39, 1735–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobucci, C.; Götze, M.; Sinz, A. Cross-linking/mass spectrometry to get a closer view on protein interaction networks. Curr. Opin. Biotechnol. 2020, 63, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Belsom, A.; Rappsilber, J. Anatomy of a crosslinker. Curr. Opin. Chem. Biol. 2020, 60, 39–46. [Google Scholar] [CrossRef]
- Hill, M.M.; Bastiani, M.; Luetterforst, R.; Kirkham, M.; Kirkham, A.; Nixon, S.J.; Walser, P.; Abankwa, D.; Oorschot, V.M.; Martin, S.; et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 2008, 132, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Lo, H.P.; Hall, T.E.; Parton, R.G. Mechanoprotection by skeletal muscle caveolae. Bioarchitecture 2016, 6, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, E.; Fischbeck, K.; Schotland, D.L. Freeze-fracture studies of muscle caveolae in human muscular dystrophy. Am. J. Pathol. 1981, 104, 167–173. [Google Scholar]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef]
- Harsini, F.M.; Bui, A.A.; Rice, A.M.; Chebrolu, S.; Fuson, K.L.; Turtoi, A.; Bradberry, M.; Chapman, E.R.; Sutton, R.B. Structural basis for the distinct membrane binding activity of the homologous C2A domains of myoferlin and dysferlin. J. Mol. Biol. 2019, 431, 2112–2126. [Google Scholar] [CrossRef]
- Seungjin, N.; Paek, E. Computational methods in mass spectrometry-based structural proteomics for studying protein structure, dynamics, and interactions. Comput. Struct. Biotechnol. J. 2020, 18, 1391–1402. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, J.G.; Bernstein, M.A.; Boudreau-Larivière, C. Plakins in striated muscle. Muscle Nerve 2010, 41, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Ursitti, J.A.; Lee, P.C.; Resneck, W.G.; McNally, M.M.; Bowman, A.L.; O’Neill, A.; Stone, M.R.; Bloch, R.J. Cloning and characterization of cytokeratins 8 and 19 in adult rat striated muscle. Interaction with the dystrophin glycoprotein complex. J. Biol. Chem. 2004, 279, 41830–41838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, M.R.; O’Neill, A.; Catino, D.; Bloch, R.J. Specific interaction of the actin-binding domain of dystrophin with intermediate filaments containing keratin 19. Mol. Biol. Cell 2005, 16, 4280–4293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhosle, R.C.; Michele, D.E.; Campbell, K.P.; Li, Z.; Robson, R.M. Interactions of intermediate filament protein synemin with dystrophin and utrophin. Biochem. Biophys. Res. Commun. 2006, 346, 768–777. [Google Scholar] [CrossRef]
- Hijikata, T.; Murakami, T.; Ishikawa, H.; Yorifuji, H. Plectin tethers desmin intermediate filaments onto subsarcolemmal dense plaques containing dystrophin and vinculin. Histochem. Cell Biol. 2003, 119, 109–123. [Google Scholar] [CrossRef]
- Rezniczek, G.A.; Konieczny, P.; Nikolic, B.; Reipert, S.; Schneller, D.; Abrahamsberg, C.; Davies, K.E.; Winder, S.J.; Wiche, G. Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J. Cell Biol. 2007, 176, 965–977. [Google Scholar] [CrossRef] [Green Version]
- Hijikata, T.; Nakamura, A.; Isokawa, K.; Imamura, M.; Yuasa, K.; Ishikawa, R.; Kohama, K.; Takeda, S.; Yorifuji, H. Plectin 1 links intermediate filaments to costameric sarcolemma through beta-synemin, alpha-dystrobrevin and actin. J. Cell Sci. 2008, 121, 2062–2074. [Google Scholar] [CrossRef] [Green Version]
- Prins, K.W.; Humston, J.L.; Mehta, A.; Tate, V.; Ralston, E.; Ervasti, J.M. Dystrophin is a microtubule-associated protein. J. Cell Biol. 2009, 186, 363–369. [Google Scholar] [CrossRef]
- Eid Mutlak, Y.; Aweida, D.; Volodin, A.; Ayalon, B.; Dahan, N.; Parnis, A.; Cohen, S. A signaling hub of insulin receptor, dystrophin glycoprotein complex and plakoglobin regulates muscle size. Nat. Commun. 2020, 11, 1381. [Google Scholar] [CrossRef] [Green Version]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and 1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Gorza, L.; Sorge, M.; Seclì, L.; Brancaccio, M. Master Regulators of Muscle Atrophy: Role of Costamere Components. Cells 2021, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Tasca, E.; Nascimbeni, A.C.; Angelini, C. Sarcolemmal neuronal nitric oxide synthase defect in limb-girdle muscular dystrophy: An adverse modulating factor in the disease course? J. Neuropathol. Exp. Neurol. 2009, 68, 383–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, C.; Tasca, E.; Nascimbeni, A.C.; Fanin, M. Muscle fatigue, nNOS and muscle fiber atrophy in limb girdle muscular dystrophy. Acta Myol. 2014, 33, 119–126. [Google Scholar]
- Stamler, J.S.; Meissner, G. Physiology of nitric oxide in skeletal muscle. Physiol. Rev. 2001, 81, 209–237. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; LaFond, K.M.; Bonini, M.G. Nitric oxide in cellular adaptation and disease. Redox Biol. 2020, 34, 101550. [Google Scholar] [CrossRef]
- Sato, K.; Yokota, T.; Ichioka, S.; Shibata, M.; Takeda, S. Vasodilation of intramuscular arterioles under shear stress in dystrophin-deficient skeletal muscle is impaired through decreased nNOS expression. Acta Myol. 2008, 27, 30–36. [Google Scholar]
- Tidball, J.G.; Wehling-Henricks, M. Nitric oxide synthase deficiency and the pathophysiology of muscular dystrophy. J. Physiol. 2014, 592, 4627–4638. [Google Scholar] [CrossRef]
- Nichols, B.; Takeda, S.; Yokota, T. Nonmechanical roles of dystrophin and associated proteins in exercise, neuromuscular junctions, and brains. Brain Sci. 2015, 5, 275–298. [Google Scholar] [CrossRef] [Green Version]
- Wehling-Henricks, M.; Oltmann, M.; Rinaldi, C.; Myung, K.H.; Tidball, J.G. Loss of positive allosteric interactions between neuronal nitric oxide synthase and phosphofructokinase contributes to defects in glycolysis and increased fatigability in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 3439–3451. [Google Scholar] [CrossRef] [Green Version]
- Di Mauro, S.; Angelini, C.; Catani, C. Enzymes of the glycogen cycle and glycolysis in various human neuromuscular disorders. J. Neurol. Neurosurg. Psychiatry 1967, 30, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinet, A.E.; Even, P.C.; Decrouy, A. Dystrophin-dependent efficiency of metabolic pathways in mouse skeletal muscles. Experientia 1994, 50, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Amenta, A.R.; Yilmaz, A.; Bogdanovich, S.; McKechnie, B.A.; Abedi, M.; Khurana, T.S.; Fallon, J.R. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc. Natl. Acad. Sci. USA 2011, 108, 762–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowe, M.A.; Mendis, D.B.; Fallon, J.R. The small leucine-rich repeat proteoglycan biglycan binds to alpha-dystroglycan and is upregulated in dystrophic muscle. J. Cell Biol. 2000, 148, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Nastase, M.V.; Young, M.F.; Schaefer, L. Biglycan: A multivalent proteoglycan providing structure and signals. J. Histochem. Cytochem. 2012, 60, 963–975. [Google Scholar] [CrossRef]
- Mercado, M.L.; Amenta, A.R.; Hagiwara, H.; Rafii, M.S.; Lechner, B.E.; Owens, R.T.; McQuillan, D.J.; Froehner, S.C.; Fallon, J.R. Biglycan regulates the expression and sarcolemmal localization of dystrobrevin, syntrophin, and nNOS. FASEB J. 2006, 20, 1724–1726. [Google Scholar] [CrossRef]
- Rafii, M.S.; Hagiwara, H.; Mercado, M.L.; Seo, N.S.; Xu, T.; Dugan, T.; Owens, R.T.; Hook, M.; McQuillan, D.J.; Young, M.F.; et al. Biglycan binds to alpha- and gamma-sarcoglycan and regulates their expression during development. J. Cell Physiol. 2006, 209, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.; Henry, M.; Meleady, P.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Simultaneous pathoproteomic evaluation of the dystrophin-glycoprotein complex and secondary changes in the mdx-4cv mouse model of duchenne muscular dystrophy. Biology 2015, 4, 397–423. [Google Scholar] [CrossRef]
- Shu, C.; Parfenova, L.; Mokhonova, E.; Collado, J.R.; Damoiseaux, R.; Campagna, J.; John, V.; Crosbie, R.H. High-throughput screening identifies modulators of sarcospan that stabilize muscle cells and exhibit activity in the mouse model of Duchenne muscular dystrophy. Skelet. Muscle 2020, 10, 26. [Google Scholar] [CrossRef]
- Miller, G.; Wang, E.L.; Nassar, K.L.; Peter, A.K.; Crosbie, R.H. Structural and functional analysis of the sarcoglycan-sarcospan subcomplex. Exp. Cell Res. 2007, 313, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.L.; Oh, J.; Chou, E.; Lee, J.A.; Holmberg, J.; Burkin, D.J.; Crosbie-Watson, R.H. Sarcospan integration into laminin-binding adhesion complexes that ameliorate muscular dystrophy requires utrophin and alpha7 integrin. Hum. Mol. Genet. 2015, 24, 2011–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Rumeur, E.; Winder, S.J.; Hubert, J.F. Dystrophin: More than just the sum of its parts. Biochim. Biophys. Acta 2010, 1804, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Yang, B.; Meyer, J.; Chamberlain, J.S.; Campbell, K.P. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J. Biol. Chem. 1995, 270, 27305–27310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Jung, D.; Motto, D.; Meyer, J.; Koretzky, G.; Campbell, K.P. SH3 domain-mediated interaction of dystroglycan and Grb2. J. Biol. Chem. 1995, 270, 11711–11714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonoudakis, D.; Conti, L.R.; Anderson, S.; Radeke, C.M.; McGuire, L.M.; Adams, M.E.; Froehner, S.C.; Yates, J.R., 3rd; Vandenberg, C.A. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J. Biol. Chem. 2004, 279, 22331–22346. [Google Scholar] [CrossRef] [Green Version]
- Sabourin, J.; Lamiche, C.; Vandebrouck, A.; Magaud, C.; Rivet, J.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J. Biol. Chem. 2009, 284, 36248–36261. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.E.; Mueller, H.A.; Froehner, S.C. In vivo requirement of the alpha-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J. Cell Biol. 2001, 155, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Neely, J.D.; Amiry-Moghaddam, M.; Ottersen, O.P.; Froehner, S.C.; Agre, P.; Adams, M.E. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14108–14113. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, O.; von Wegner, F.; Chamberlain, J.S.; Fink, R.H.; Rohrbach, P. L-type Ca2+ channel function is linked to dystrophin expression in mammalian muscle. PLoS ONE 2008, 3, e1762. [Google Scholar] [CrossRef] [Green Version]
- Sotgia, F.; Lee, H.; Bedford, M.T.; Petrucci, T.; Sudol, M.; Lisanti, M.P. Tyrosine phosphorylation of beta-dystroglycan at its WW domain binding motif, PPxY, recruits SH2 domain containing proteins. Biochemistry 2001, 40, 14585–14592. [Google Scholar] [CrossRef]
- Thompson, O.; Kleino, I.; Crimaldi, L.; Gimona, M.; Saksela, K.; Winder, S.J. Dystroglycan, Tks5 and Src mediated assembly of podosomes in myoblasts. PLoS ONE 2008, 3, e3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramovici, H.; Hogan, A.B.; Obagi, C.; Topham, M.K.; Gee, S.H. Diacylglycerol kinase-zeta localization in skeletal muscle is regulated by phosphorylation and interaction with syntrophins. Mol. Biol. Cell. 2003, 14, 4499–4511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramovici, H.; Gee, S.H. Morphological changes and spatial regulation of diacylglycerol kinase-zeta, syntrophins, and Rac1 during myoblast fusion. Cell Motil. Cytoskelet. 2007, 64, 549–567. [Google Scholar] [CrossRef]
- Lumeng, C.; Phelps, S.; Crawford, G.E.; Walden, P.D.; Barald, K.; Chamberlain, J.S. Interactions between beta 2-syntrophin and a family of microtubule-associated serine/threonine kinases. Nat. Neurosci. 1999, 2, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Cuenda, A.; Spillantini, M.G.; Thomas, G.M.; Buée-Scherrer, V.; Cohen, P.; Goedert, M. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J. Biol. Chem. 1999, 274, 12626–12631. [Google Scholar] [CrossRef] [Green Version]
- Boppart, M.D.; Mahmassani, Z.S. Integrin signaling: Linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C629–C641. [Google Scholar] [CrossRef]
- Ilha, J.; do Espírito-Santo, C.C.; de Freitas, G.R. mTOR Signaling Pathway and Protein Synthesis: From Training to Aging and Muscle Autophagy. Adv. Exp. Med. Biol. 2018, 1088, 139–151. [Google Scholar]
- Luo, S.; Chen, Y.; Lai, K.O.; Arévalo, J.C.; Froehner, S.C.; Adams, M.E.; Chao, M.V.; Ip, N.Y. {alpha}-Syntrophin regulates ARMS localization at the neuromuscular junction and enhances EphA4 signaling in an ARMS-dependent manner. J. Cell Biol. 2005, 169, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Joe, M.K.; Kee, C.; Tomarev, S.I. Myocilin interacts with syntrophins and is member of dystrophin-associated protein complex. J. Biol. Chem. 2012, 287, 13216–13227. [Google Scholar] [CrossRef] [Green Version]
- Lyssand, J.S.; Whiting, J.L.; Lee, K.S.; Kastl, R.; Wacker, J.L.; Bruchas, M.R.; Miyatake, M.; Langeberg, L.K.; Chavkin, C.; Scott, J.D.; et al. Alpha-dystrobrevin-1 recruits alpha-catulin to the alpha1D-adrenergic receptor/dystrophin-associated protein complex signalosome. Proc. Natl. Acad. Sci. USA 2010, 107, 21854–21859. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.J.; Abraham, L.S.; van Hengel, J.; Stove, C.; Proszynski, T.J.; Gevaert, K.; DiMario, J.X.; Sanes, J.R.; van Roy, F.; Kim, H. Interaction of α-catulin with dystrobrevin contributes to integrity of dystrophin complex in muscle. J. Biol. Chem. 2012, 287, 21717–21728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingras, J.; Gawor, M.; Bernadzki, K.M.; Grady, R.M.; Hallock, P.; Glass, D.J.; Sanes, J.R.; Proszynski, T.J. Α-Dystrobrevin-1 recruits Grb2 and α-catulin to organize neurotransmitter receptors at the neuromuscular junction. J. Cell Sci. 2016, 129, 898–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernadzki, K.M.; Gawor, M.; Pęziński, M.; Mazurek, P.; Niewiadomski, P.; Rędowicz, M.J.; Prószyński, T.J. Liprin-α-1 is a novel component of the murine neuromuscular junction and is involved in the organization of the postsynaptic machinery. Sci. Rep. 2017, 7, 9116. [Google Scholar] [CrossRef] [Green Version]
- Bernadzki, K.M.; Daszczuk, P.; Rojek, K.O.; Pęziński, M.; Gawor, M.; Pradhan, B.S.; de Cicco, T.; Bijata, M.; Bijata, K.; Włodarczyk, J.; et al. Arhgef5 binds α-dystrobrevin 1 and regulates neuromuscular junction integrity. Front. Mol. Neurosci. 2020, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Karpati, G.; Carpenter, S.; Morris, G.E.; Davies, K.E.; Guerin, C.; Holland, P. Localization and quantitation of the chromosome 6-encoded dystrophin-related protein in normal and pathological human muscle. J. Neuropathol. Exp. Neurol. 1993, 52, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Ervasti, J.M.; Matsumura, K.; Kahl, S.D.; Leveille, C.J.; Campbell, K.P. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron 1991, 7, 499–508. [Google Scholar] [CrossRef]
- Blake, D.J.; Tinsley, J.M.; Davies, K.E. Utrophin: A structural and functional comparison to dystrophin. Brain Pathol. 1996, 6, 37–47. [Google Scholar] [CrossRef]
- Matsumura, K.; Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Campbell, K.P. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature 1992, 360, 588–591. [Google Scholar] [CrossRef]
- Belhasan, D.C.; Akaaboune, M. The role of the dystrophin glycoprotein complex on the neuromuscular system. Neurosci. Lett. 2020, 722, 134833. [Google Scholar] [CrossRef]
- Ng, S.Y.; Ljubicic, V. Recent insights into neuromuscular junction biology in Duchenne muscular dystrophy: Impacts, challenges, and opportunities. EBioMedicine 2020, 61, 103032. [Google Scholar] [CrossRef]
- Smith, L.R.; Meyer, G.; Lieber, R.L. Systems analysis of biological networks in skeletal muscle function. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013, 5, 55–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dowling, P.; Gargan, S.; Murphy, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes 2021, 9, 9. https://0-doi-org.brum.beds.ac.uk/10.3390/proteomes9010009
Dowling P, Gargan S, Murphy S, Zweyer M, Sabir H, Swandulla D, Ohlendieck K. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes. 2021; 9(1):9. https://0-doi-org.brum.beds.ac.uk/10.3390/proteomes9010009
Chicago/Turabian StyleDowling, Paul, Stephen Gargan, Sandra Murphy, Margit Zweyer, Hemmen Sabir, Dieter Swandulla, and Kay Ohlendieck. 2021. "The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle" Proteomes 9, no. 1: 9. https://0-doi-org.brum.beds.ac.uk/10.3390/proteomes9010009