Spectroscopic and Computational Study of the Protonation Equilibria of Amino-Substituted benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazoles as Novel pH-Sensing Materials

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Methods for Synthesis
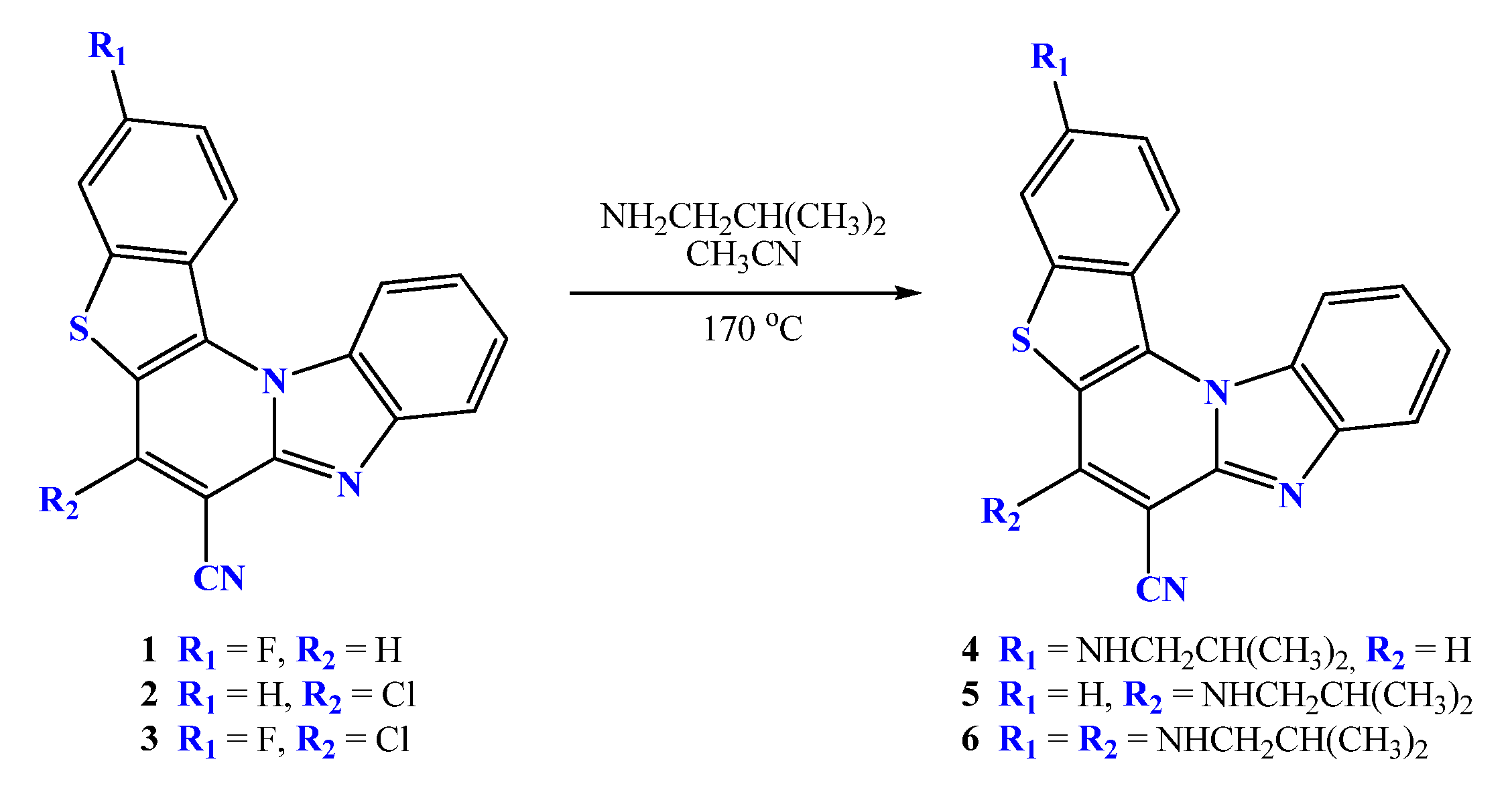
2.2. General Method for Preparation of Compounds 4–6
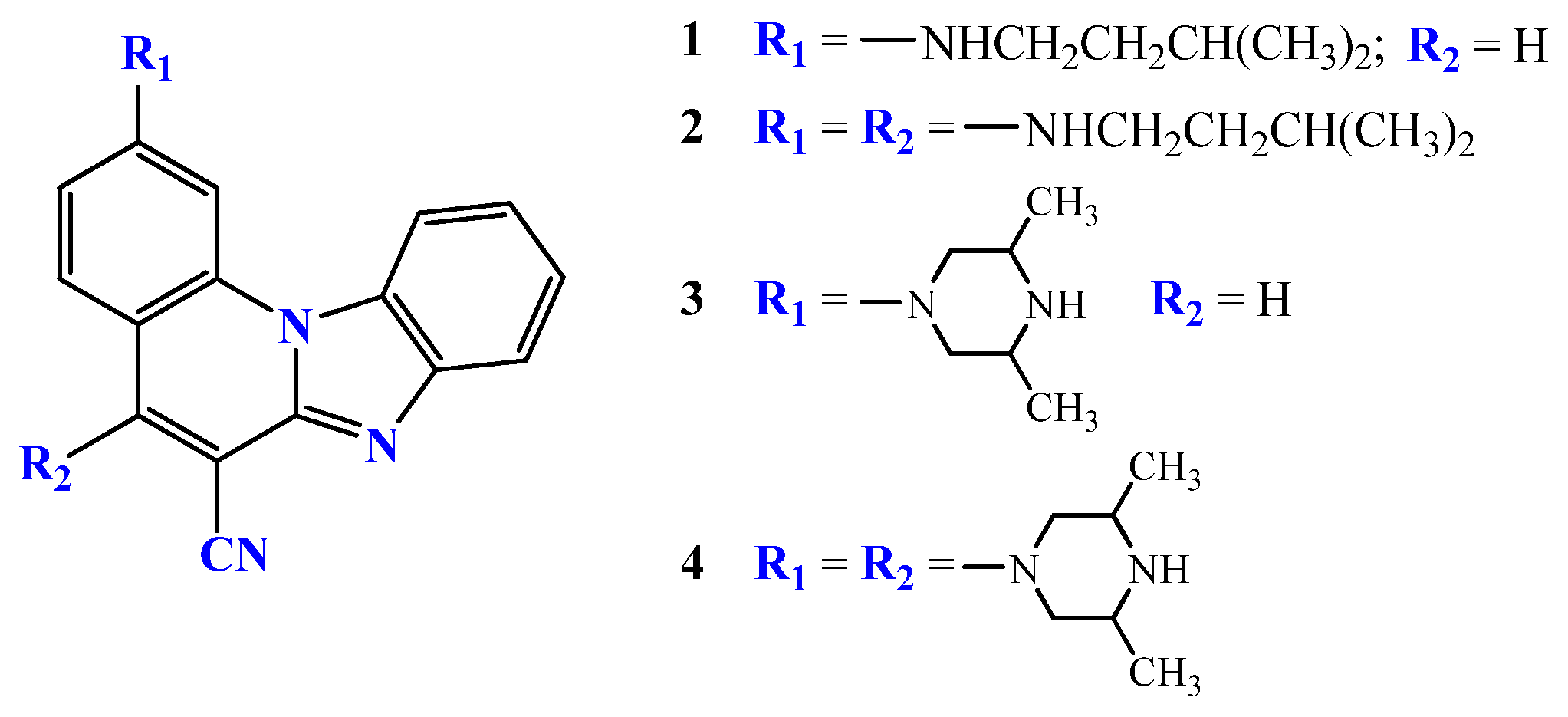
2.2.1. 3-N-i-butylaminobenzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazole-7-carbonitrile 4
2.2.2. 6-N-i-butylaminobenzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazole-7-carbonitrile 5
2.2.3. 3,6-di-(N-i-butylamino)benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazole-7-carbonitrile 6
2.3. Spectroscopic Characterization
2.4. pH Titrations
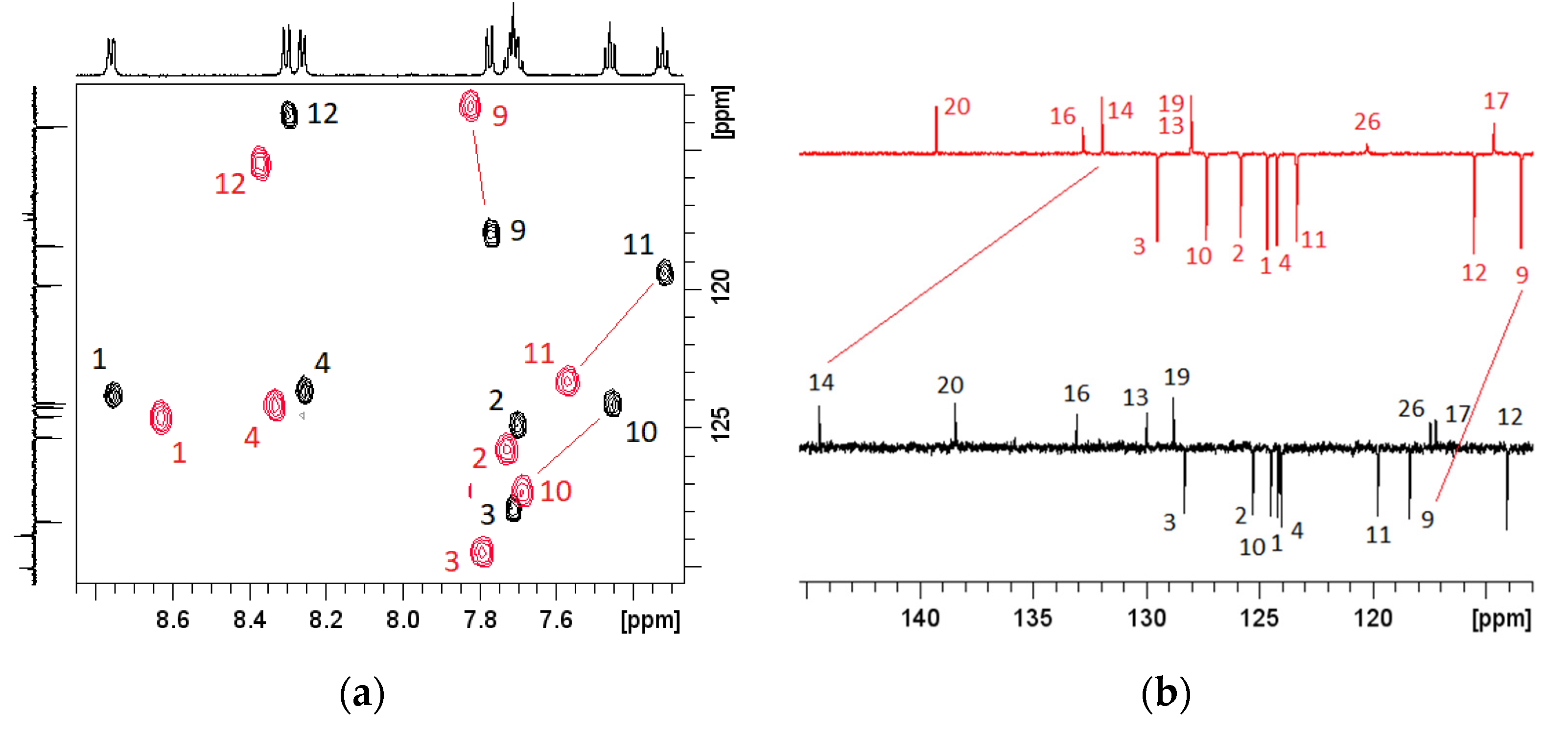
2.5. Analysis of Perturbations in NMR Chemical Shifts
2.6. Computational Details
3. Results and Discussion
3.1. Chemistry
3.2. Spectroscopic Characterization
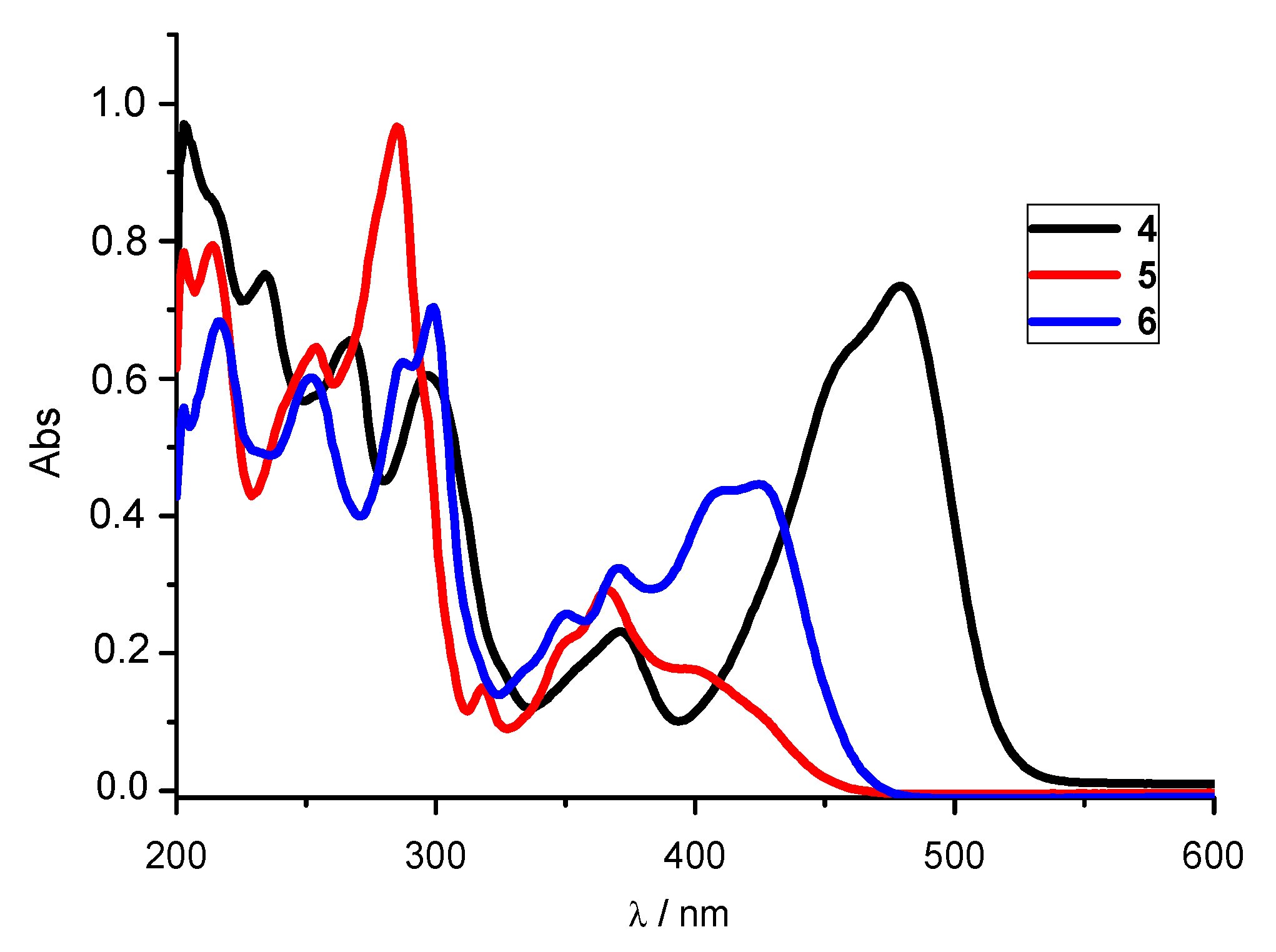
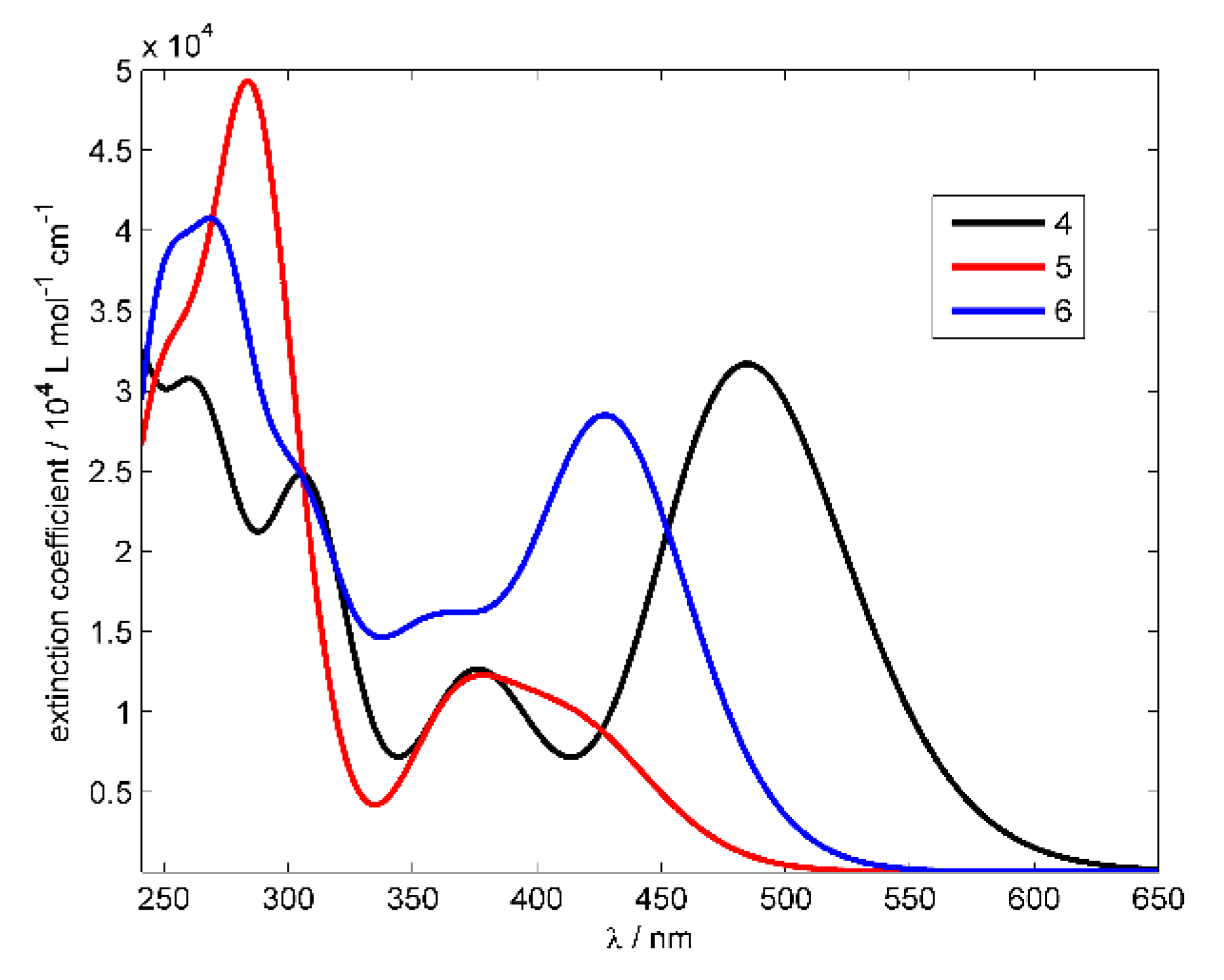
3.2.1. UV-Vis Absorption Spectra
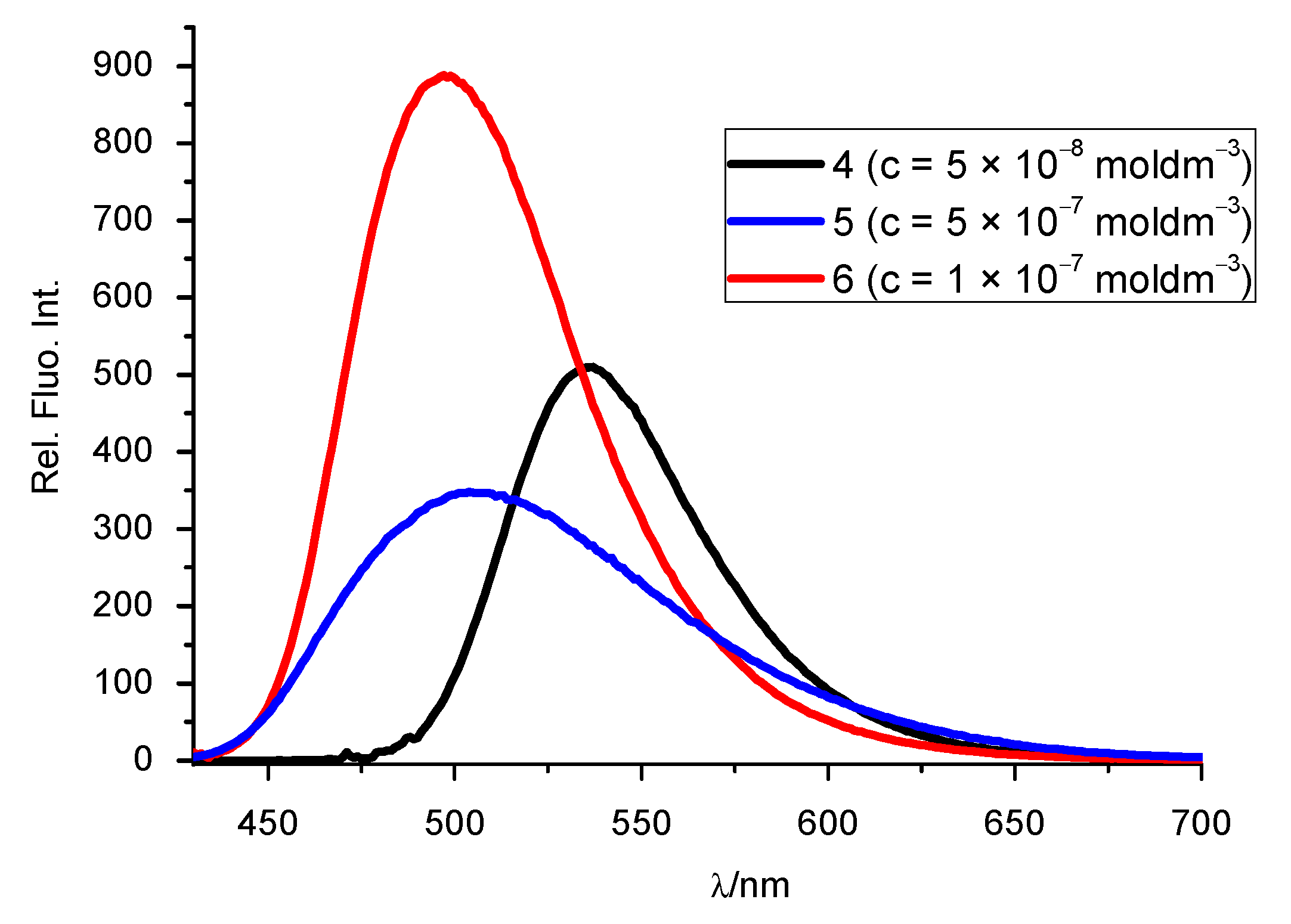
3.2.2. Fluorescence Emission Spectra
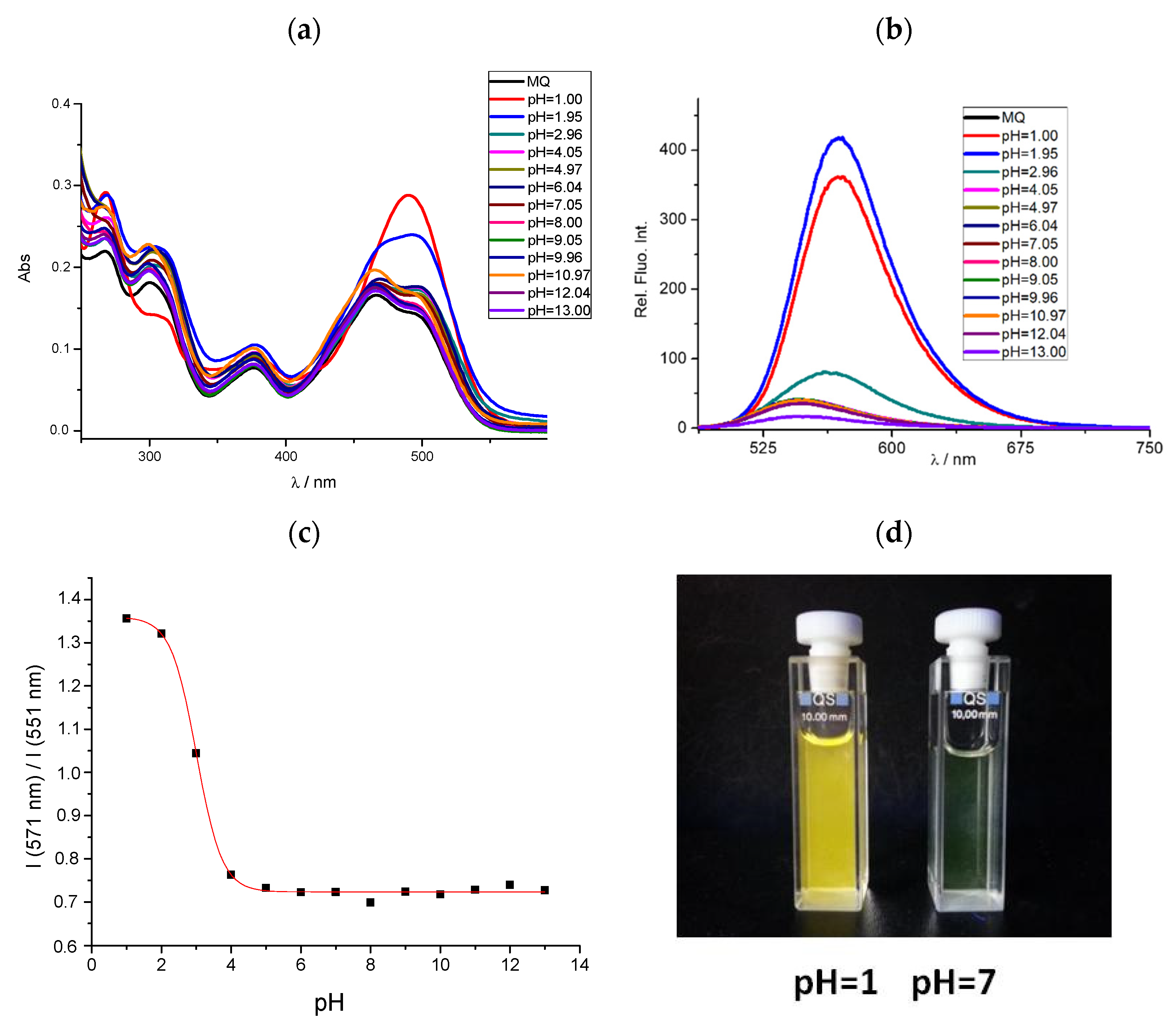
3.3. Effects of pH on Spectral Properties
3.4. Effects of pH on NMR Spectra
3.5. Computational Analysis
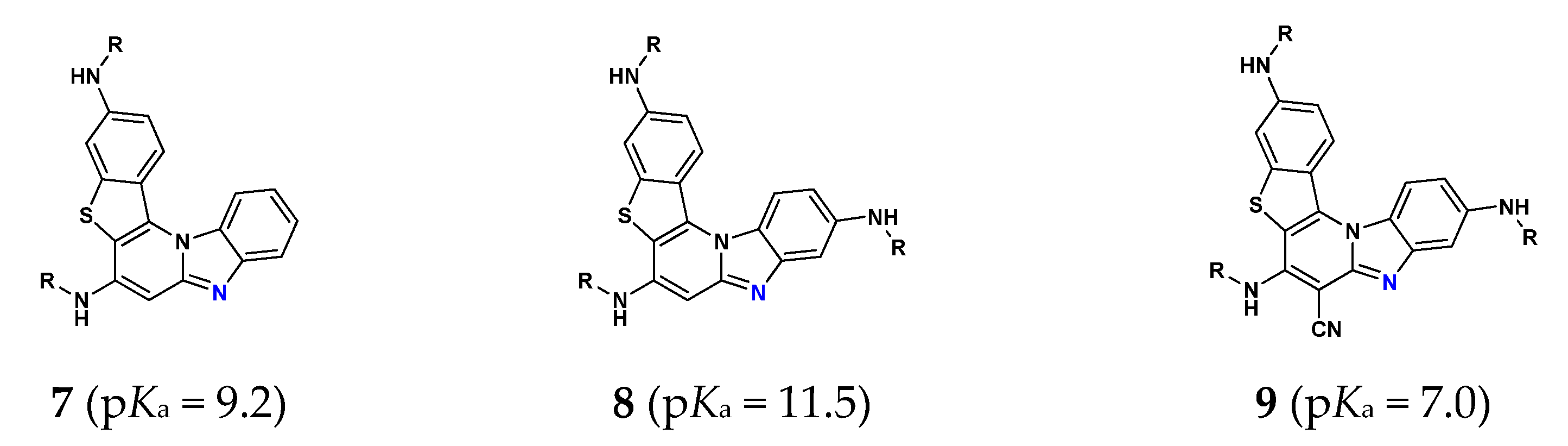
3.5.1. Acid-Base Properties
3.5.2. Absorption and Emission Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, B.; Anslyn, E.V. Chemosensors: Principles, Strategies and Applications; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Valeur, B.; Leray, I. Design principles of fluorescent molecular sensors for cation recognition. Coord. Chem. Rev. 2000, 205, 3–40. [Google Scholar] [CrossRef]
- Wu, D.; Chen, L.; Lee, W.; Ko, G.; Yin, J.; Yoon, J. Recent progress in the development of organic dye based near-infrared fluorescence probes for metal ions. Coord. Chem. Rev. 2018, 354, 74–97. [Google Scholar] [CrossRef]
- Wencel, D.; Abel, T.; McDonagh, C. Optical chemical pH sensors. Anal. Chem. 2014, 86, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Bodedla, G.B.; Thomas, K.R.J.; Kumar, S.; Jou, J.H.; Li, C.J. Phenothiazine-based bipolar green-emitters containing benzimidazole units: Synthesis, photophysical and electroluminescence properties. RSC Adv. 2015, 5, 87416–87428. [Google Scholar] [CrossRef]
- Kim, T.D. D–π–A Conjugated molecules for optoelectronic applications. Macromol. Rapid Commun. 2015, 36, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Debia, N.P.; Rodríguez, J.J.P.; da Silveira, C.H.; Chaves, O.A.; Iglesias, B.A.; Rodembusch, F.S.; Lüdtke, D.S. Synthesis and photophysics of benzazole based triazoles with amino acid-derived pendant units. Multiparametric optical sensors for BSA and CT-DNA in solution. J. Mol. Liq. 2020, 309, 113092. [Google Scholar] [CrossRef]
- Burke, C.S.; McGaughey, O.; Sabattie, J.M.; Barry, H.; McEvoy, A.K.; McDonagh, C.; MacCraith, B.D. Development of an integrated optic oxygen sensor using a novel, generic platform. Analyst 2005, 130, 41–45. [Google Scholar] [CrossRef]
- Horak, E.; Vianello, R.; Murković Steinberg, I. Optical sensing (nano)materials based on benzimidazole derivatives. In Chemistry and Applications of Benzimidazole and Its Derivatives; Marinescu, M., Ed.; IntechOpen: London, UK, 2019; pp. 184–205. [Google Scholar]
- Pfeifer, D.; Klimant, I.; Borisov, S.M. Ultrabright red-emitting photostable perylene bisimide dyes: New indicators for ratiometric sensing of high pH or carbon dioxide. Chem. Eur. J. 2018, 24, 10711–10720. [Google Scholar] [CrossRef]
- Silverman, R.B. The organic Chemistry of Drug Design and Drug Action, 2nd ed.; Elsevier Academic Press: London, UK, 2004. [Google Scholar]
- Keri, R.S.; Hiremathad, A.; Budagumpi, S.; Nagaraja, B.M. Comprehensive review in current developments of benzimidazole-based medicinal chemistry. Chem. Biol. Drug Des. 2015, 86, 19–65. [Google Scholar] [CrossRef]
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Shahar Yar, M. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef]
- Kumar, G.; Gupta, N.; Paul, K.; Luxami, V. Acrylonitrile embedded benzimidazole-anthraquinone based chromofluorescent sensor for ratiometric detection of CN− ions in bovine serum albumin. Sens. Actuators B Chem. 2018, 267, 549–558. [Google Scholar] [CrossRef]
- Staneva, D.; Betcheva, R. Synthesis and functional properties of new optical pH sensor based on benzo[de]anthracen-7-one immobilized on the viscose. Dyes Pigm. 2007, 74, 148–153. [Google Scholar] [CrossRef]
- Chen, W.; Ma, X.; Chen, H.; Liu, S.H.; Yin, J. Fluorescent probes for pH and alkali metal ions. Coord. Chem. Rev. 2021, 427, 213584. [Google Scholar] [CrossRef]
- Steinegger, A.; Wolfbeis, O.S.; Borisov, S.M. Optical sensing and imaging of pH values: Spectroscopies, materials, and applications. Chem. Rev. 2020, 120, 12357–12489. [Google Scholar] [CrossRef] [PubMed]
- Shamsipur, M.; Barati, A.; Nematifar, Z. Fluorescent pH nanosensors: Design strategies and applications. J. Photochem. Photobiol. C 2019, 39, 76–141. [Google Scholar] [CrossRef]
- Nawaz, H.; Tian, W.; Zhang, J.; Jia, R.; Yang, T.; Yu, J.; Zhang, J. Visual and precise detection of pH values under extreme acidic and strong basic environments by cellulose-based superior sensor. Anal. Chem. 2019, 91, 3085–3092. [Google Scholar] [CrossRef]
- Hranjec, M.; Horak, E.; Tireli, M.; Pavlović, G.; Karminski-Zamola, G. Synthesis, crystal structure and spectroscopic study of novel benzimidazoles and benzimidazo[1,2-a]quinolines as potential chemosensors for different cations. Dyes Pigment. 2012, 95, 644–656. [Google Scholar] [CrossRef]
- Hranjec, M.; Horak, E.; Babić, D.; Plavljanin, S.; Srdović, Z.; Murković Steinberg, I.; Vianello, R.; Perin, N. Fluorescent benzimidazo[1,2-a]quinolines: Synthesis, spectroscopic and computationalstudies of protonation equilibria and metal ion sensitivity. New J. Chem. 2017, 41, 358–371. [Google Scholar] [CrossRef] [Green Version]
- Carey, W.P.; DeGrandpre, M.D.; Jorgensen, B.S. Polymer-coated cylindrical waveguide absorption sensor for high acidities. Anal. Chem. 1989, 61, 1674–1678. [Google Scholar] [CrossRef]
- Perin, N.; Bobanović, K.; Zlatar, I.; Jelić, D.; Kelava, V.; Koštrun, S.; Gabelica Marković, V.; Brajša, K.; Hranjec, M. Antiproliferative activity of amino substituted benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazoles explored by 2D and 3D cell culture system. Eur. J. Med. Chem. 2017, 125, 722–735. [Google Scholar] [CrossRef]
- Horak, E.; Kassal, P.; Hranjec, M.; Murković Steinberg, I. Benzimidazole functionalized Schiff bases: Novel pH sensitive fluorescence turn-on chromoionophores for ion-selective optodes. Sens. Actuators B Chem. 2018, 258, 415–423. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Hok, L.; Vianello, R. Direct metal-free transformation of alkynes to nitriles: Computational evidence for the precise reaction mechanism. Int. J. Mol. Sci. 2021, 22, 3193. [Google Scholar] [CrossRef]
- Rowlands, G.J.; Severinsen, R.J.; Buchanan, J.K.; Shaffer, K.J.; Jameson, H.T.; Thennakoon, N.; Leito, I.; Lõkov, M.; Kütt, A.; Vianello, R.; et al. Synthesis and basicity studies of quinoline[7,8-h]quinoline derivatives. J. Org. Chem. 2020, 85, 11297–11308. [Google Scholar] [CrossRef]
- Tissandier, M.D.; Cowen, K.A.; Feng, W.Y.; Gundlach, E.; Cohen, M.H.; Earhart, A.D.; Coe, J.V.; Tuttle, T.R. The proton’s absolute aqueous enthalpy and Gibbs free energy of solvation from cluster-ion solvation data. J. Phys. Chem. A 1998, 102, 7787–7794. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Demchenko, A.P. Introduction to fluorescence sensing. Anal. Bioanal. Chem. 2009, 395, 1195–1196. [Google Scholar]
- Benassi, R.; Grandi, R.; Pagnoni, U.M.; Taddei, F. Study of the effect of N-protonation and N-methylation on the 1H and 13C chemical shifts of the six-membered ring in benzazoles and 2-substituted N,N-dimethylamino derivatives. Magn. Reson. Chem. 1986, 24, 415–420. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the basicity of organic bases in different media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Vianello, R.; Maksić, Z.B. Towards highly powerful neutral organic superacids—A DFT study of some polycyano derivatives of planar hydrocarbons. Tetrahedron 2005, 61, 9381–9390. [Google Scholar] [CrossRef]
- Valadbeigi, Y. Design of strongest organic Broensted acids in gas phase. Chem. Phys. Lett. 2017, 681, 50–55. [Google Scholar] [CrossRef]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Kluwer: New York, NY, USA, 1999. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | λmax (nm) | ε × 103 (dm3 mol−1 cm−1) | λemis (nm) | Rel. Fluorescence Int. |
|---|---|---|---|---|
| 4 | 479 459 371 297 267 234 | 36.6 32.3 11.45 30.05 32.7 37.4 | 536 | 512 |
| 5 | 401 367 319 285 253 | 8.8 14.5 7.2 48.1 32.1 | 505 | 347 |
| 6 | 425 412 370 350 230 287 252 | 22.2 21.9 16.35 12.85 35.2 31.15 30.0 | 498 | 885 |
| λmax (nm) | ε × 103 (dm3 mol−1 cm−1) | λemis (nm) | Stokes Shift (nm) | pKa | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acidic | Neutral | Basic | Acidic | Neutral | Basic | Acidic | Neutral | Basic | Acidic | Neutral | Basic | ||
| 4 | 491 378 | 499 469 377 | 494 466 | 14.40 4.60 | 8.30 9.05 4.45 | 7.40 8.55 4.05 | 571 | 548 | 551 | 80 | 49 | 57 | 3.01 |
| 5 | 356 | 447 385 313 | 443 380 311 | 16.05 | 11.30 12.05 15.35 | 12.05 13.55 18.80 | 511 | 524 | 517 | 155 | 77 | 74 | 3.18 |
| 6 | 416 361 | 444 415 377 | 446 416 378 | 5.90 10.75 | 10.70 9.85 8.95 | 12.25 11.15 10.15 | 527 | 513 | 512 | 111 | 69 | 66 | 4.73 |
 | ||||||
|---|---|---|---|---|---|---|
| Position | Proton Chemical Shifts (ppm) | Carbon Chemical Shifts (ppm) | ||||
| pH = 7.5 | pH = 2.4 | ∆ | pH = 7.5 | pH = 2.4 | ∆ | |
| 1 | 8.76 | 8.62 | 0.13 | 124.63 | 125.10 | 0.47 |
| 2 | 7.70 | 7.72 | 0.02 | 125.72 | 126.27 | 0.55 |
| 3 | 7.20 | 7.79 | 0.07 | 128.75 | 129.95 | 1.20 |
| 4 | 8.26 | 8.33 | 0.07 | 124.47 | 124.68 | 0.21 |
| 6 | 148.67 | 150.71 | 2.05 | |||
| 7 | 71.75 | 68.55 | −3.20 | |||
| 9 | 7.77 | 7.82 | 0.05 | 118.80 | 113.90 | −4.90 |
| 10 | 7.46 | 7.68 | 0.22 | 124.95 | 127.78 | 2.82 |
| 11 | 7.32 | 7.57 | 0.25 | 120.22 | 123.80 | 3.58 |
| 12 | 8.30 | 8.37 | 0.06 | 114.51 | 115.97 | 1.46 |
| 13 | 130.41 | 128.47 | −1.94 | |||
| 14 | 144.86 | 132.40 | −12.46 | |||
| 16 | 133.50 | 133.23 | −0.27 | |||
| 17 | 117.65 | 115.09 | −2.55 | |||
| 18 | 151.53 | 148.53 | −3.01 | |||
| 19 | 129.23 | 128.45 | −0.78 | |||
| 20 | 138.87 | 139.70 | 0.83 | |||
| 22 | 3.62 | 3.66 | 0.05 | 50.94 | 51.17 | 0.23 |
| 23 | 2.12 | 2.13 | 0.01 | 28.73 | 28.99 | 0.26 |
| 24,25 | 0.97 | 0.98 | 0.02 | 19.71 | 19.67 | −0.04 |
| 26 | 117.89 | 120.70 | 2.80 | |||
| System | Protonation State | pKa(calc) | Protonation Reaction |
|---|---|---|---|
 | 4− | 24.2 | N2− → N2–H |
| 4 | 3.2 [3.01] | N1 → N1–H+ | |
| 4+ | −1.3 | N2 → N2–H+ | |
 | 5− | 17.0 | N2− → N2–H |
| 5 | 3.9 [3.18] | N1 → N1–H+ | |
| 5+ | −14.1 | N2 → N2–H+ | |
 | 62− | 28.5 | N3− → N3–H |
| 6− | 18.4 | N2− → N2–H | |
| 6 | 5.0 [4.73] | N1 → N1–H+ | |
| 6+ | −1.0 | N3 → N3–H+ | |
| 62+ | −13.9 | N2 → N2–H+ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perin, N.; Babić, D.; Kassal, P.; Čikoš, A.; Hranjec, M.; Vianello, R. Spectroscopic and Computational Study of the Protonation Equilibria of Amino-Substituted benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazoles as Novel pH-Sensing Materials. Chemosensors 2022, 10, 21. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors10010021
Perin N, Babić D, Kassal P, Čikoš A, Hranjec M, Vianello R. Spectroscopic and Computational Study of the Protonation Equilibria of Amino-Substituted benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazoles as Novel pH-Sensing Materials. Chemosensors. 2022; 10(1):21. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors10010021
Chicago/Turabian StylePerin, Nataša, Darko Babić, Petar Kassal, Ana Čikoš, Marijana Hranjec, and Robert Vianello. 2022. "Spectroscopic and Computational Study of the Protonation Equilibria of Amino-Substituted benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazoles as Novel pH-Sensing Materials" Chemosensors 10, no. 1: 21. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors10010021