
Efficient Oxidase Biosensors Based on Bioelectrocatalytic Surfaces of Electrodeposited Ferrocenyl Polycyclosiloxanes—Pt Nanoparticles


Abstract
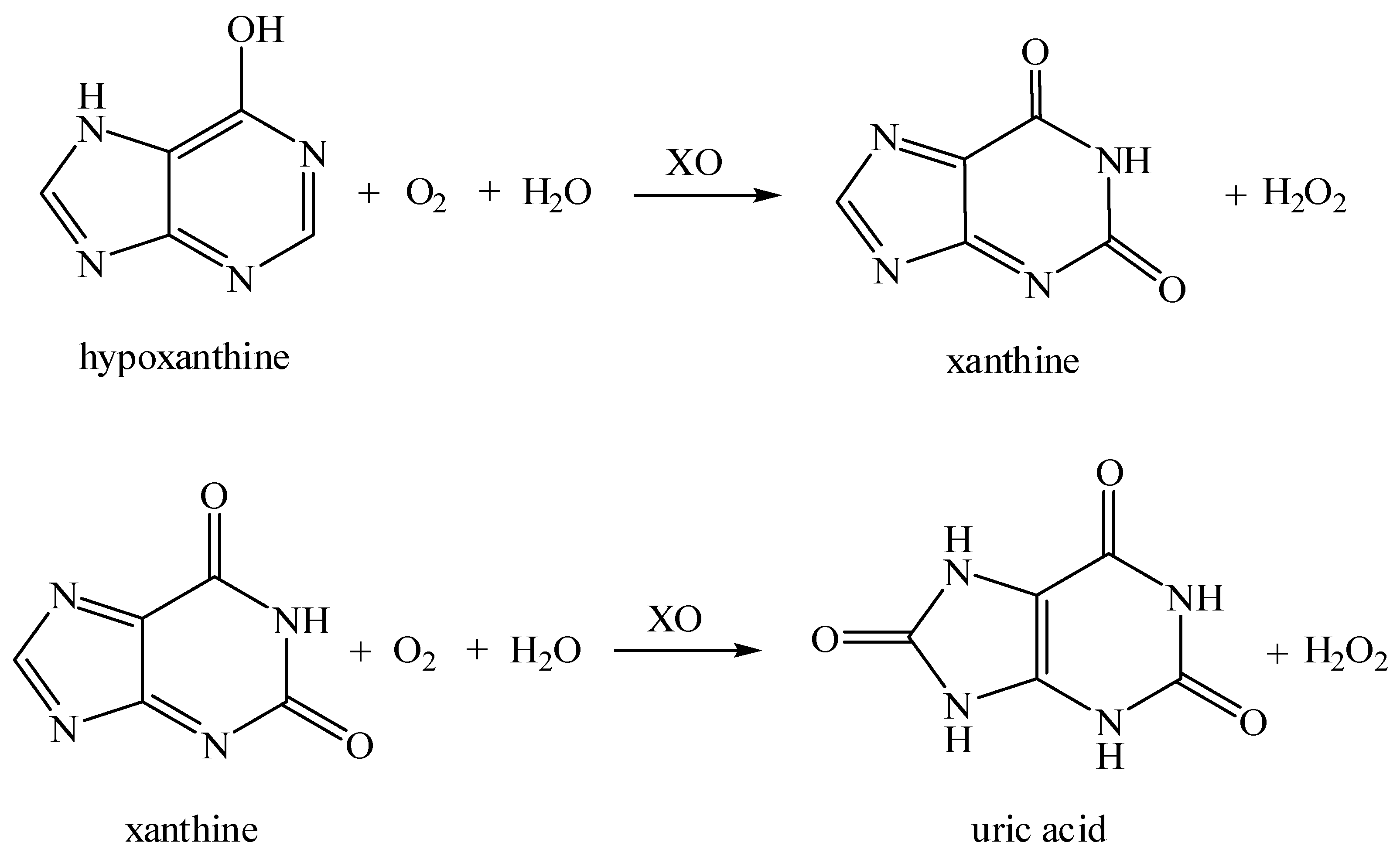
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments
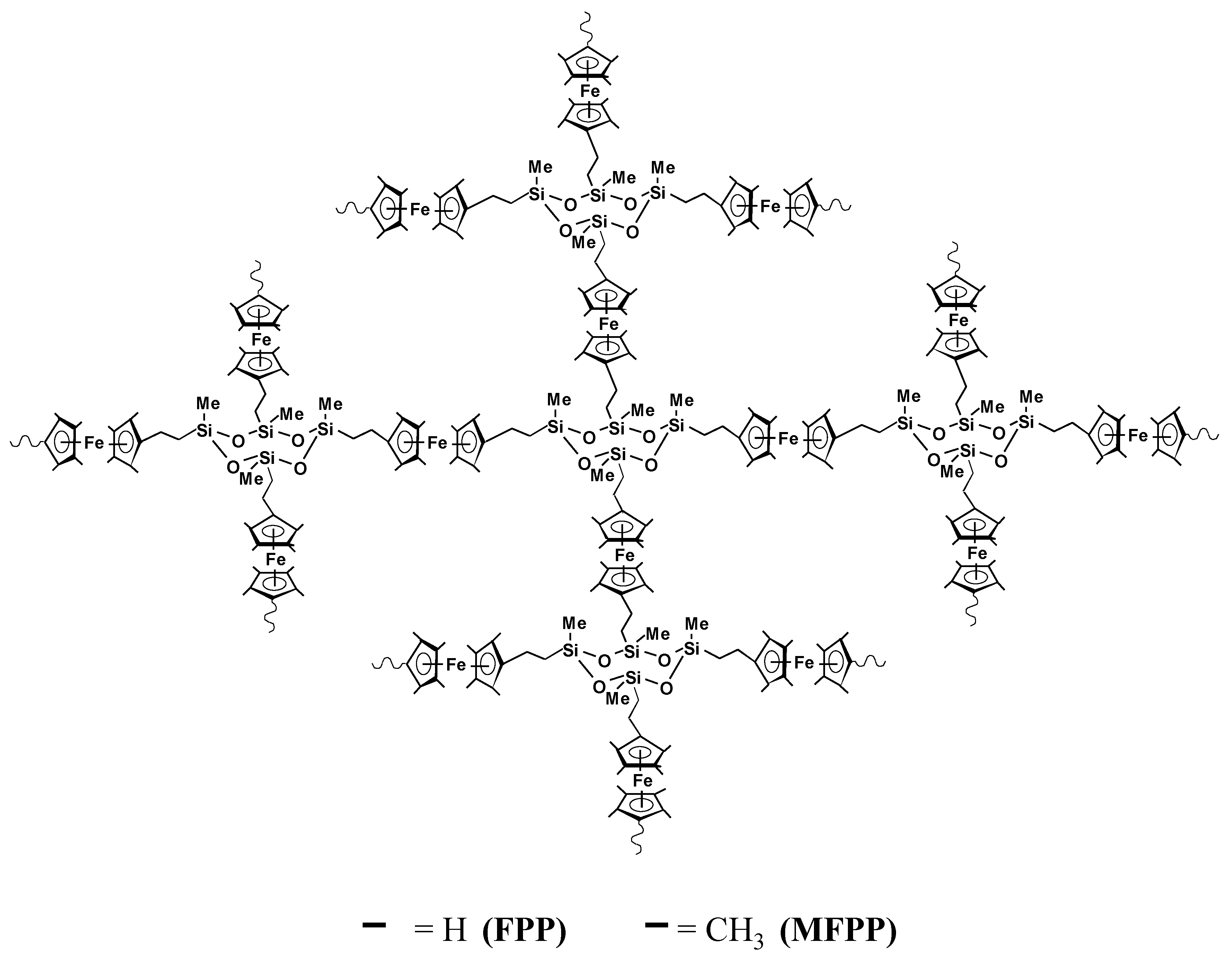
2.3. Preparation of Modified Electrodes
2.4. Preparation of Fish Samples
3. Results and Discussion
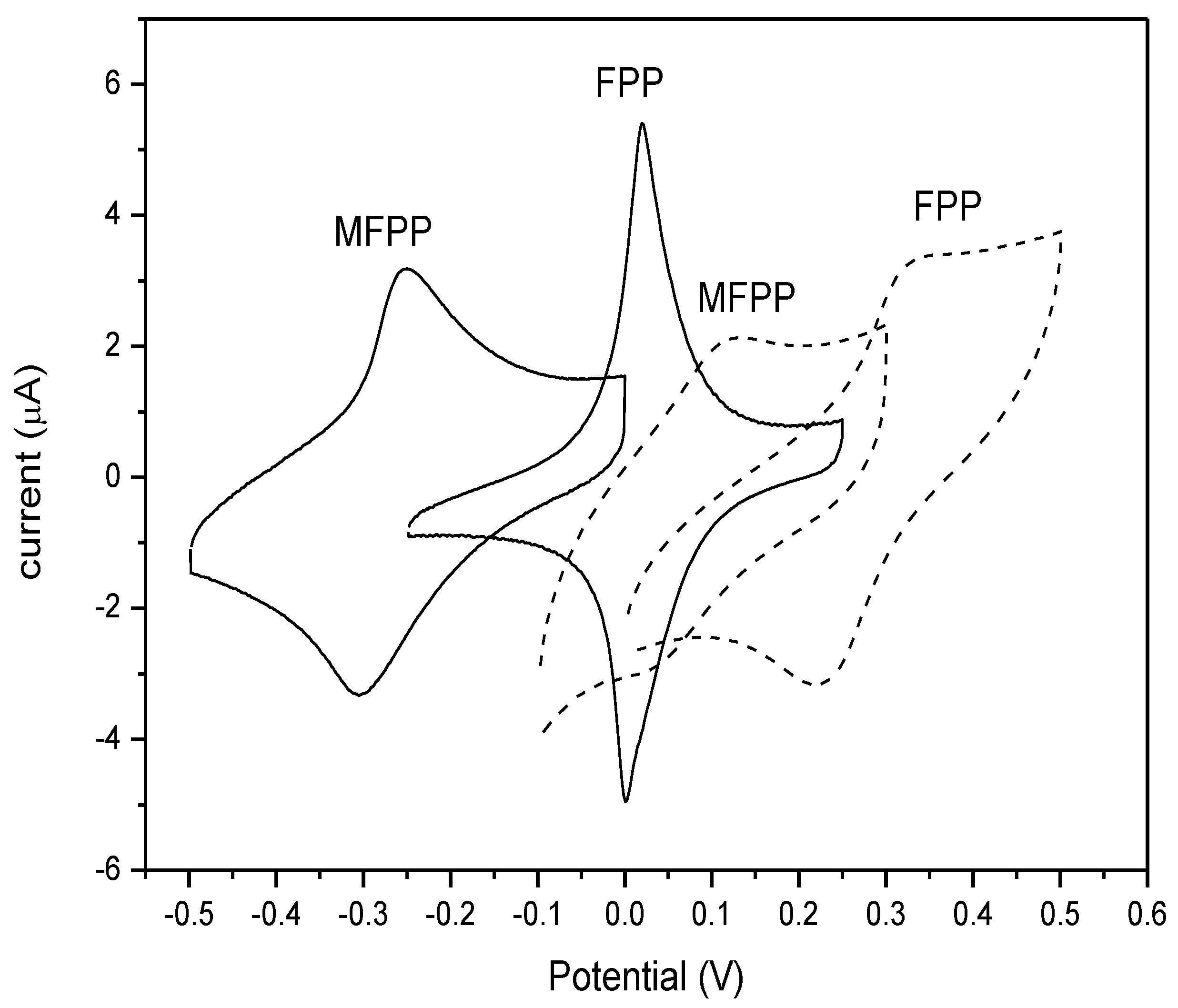
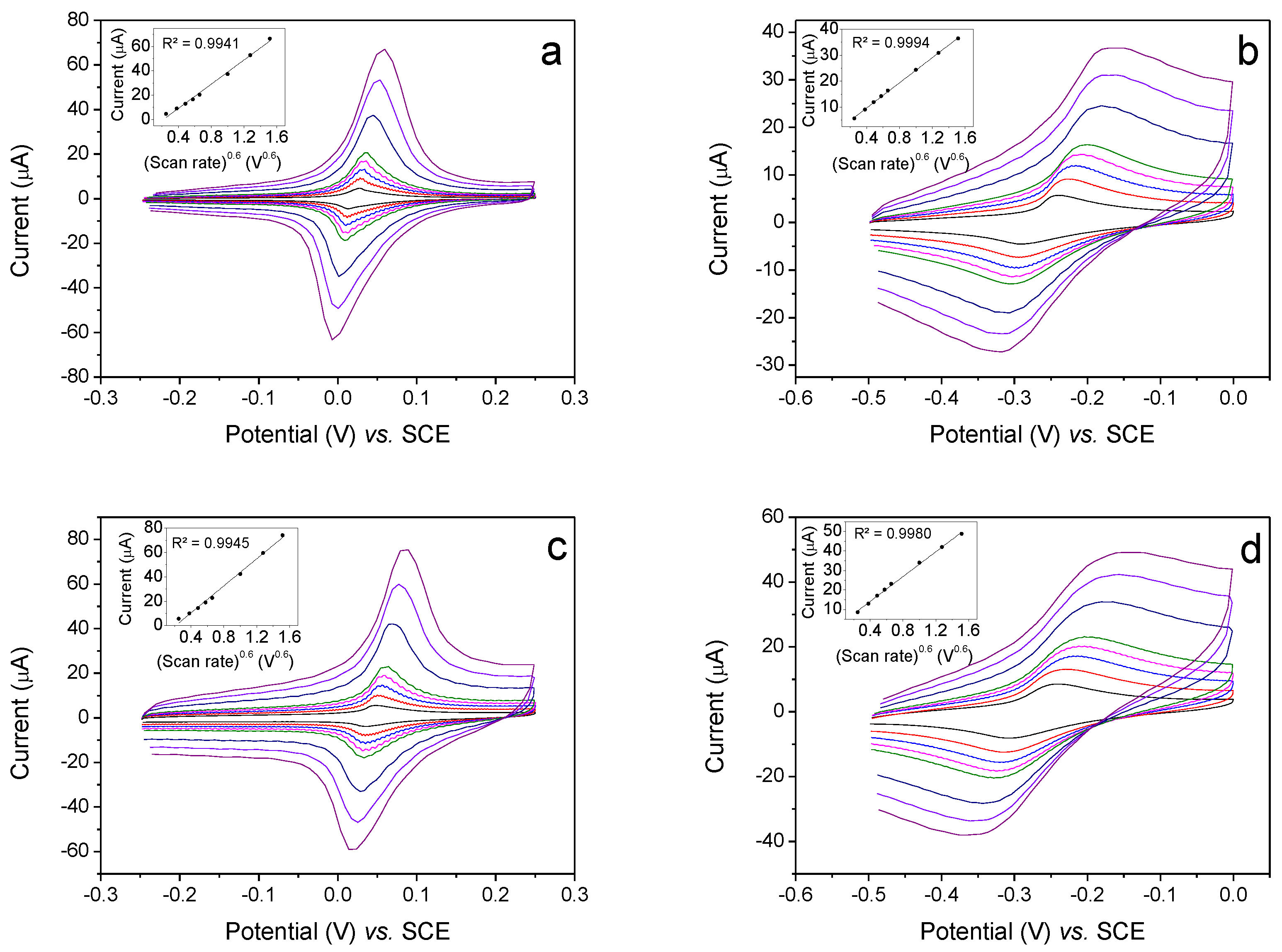
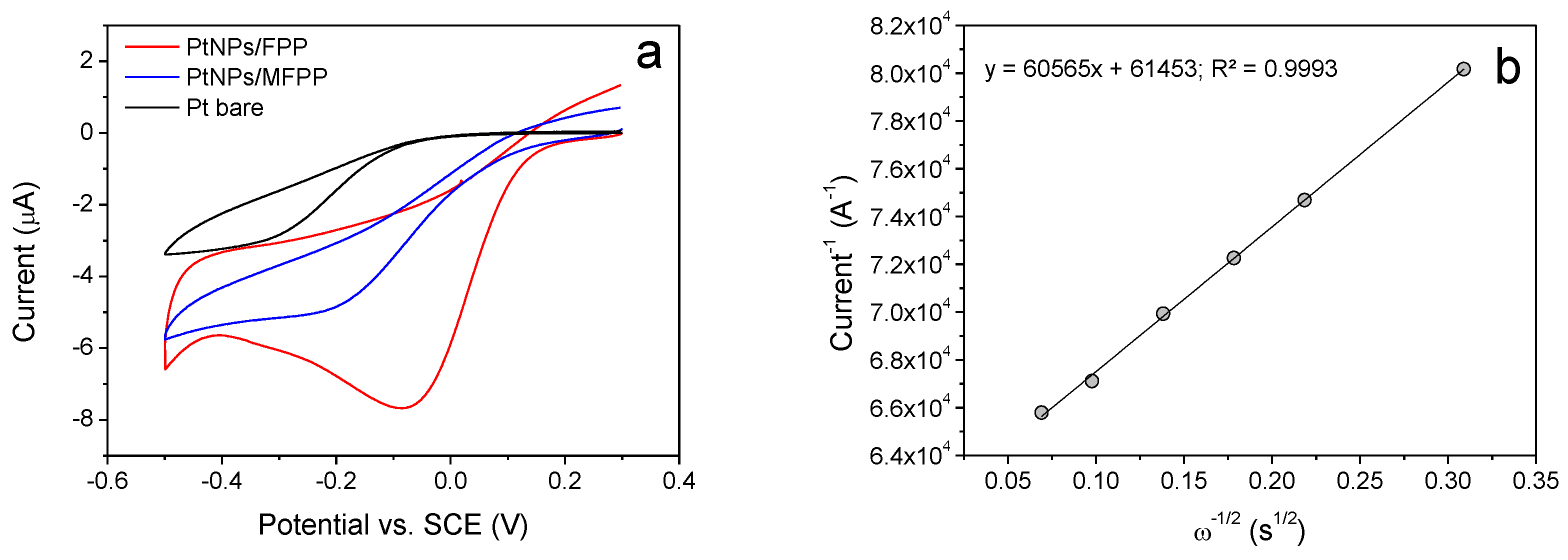
3.1. Electrochemistry, Kinetics and Morphology of Modified Electrodes
3.2. Hydrogen Peroxide Determination
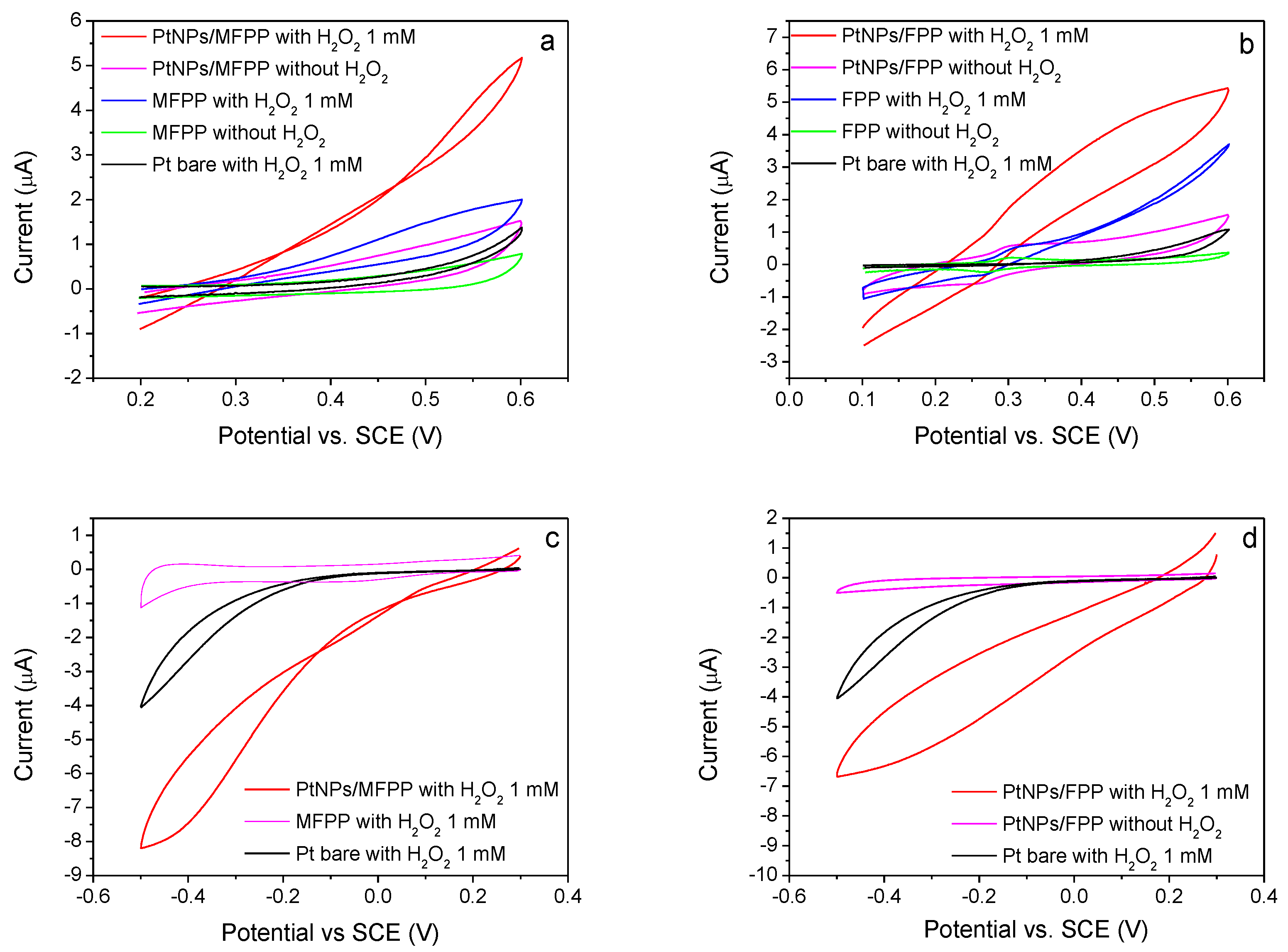
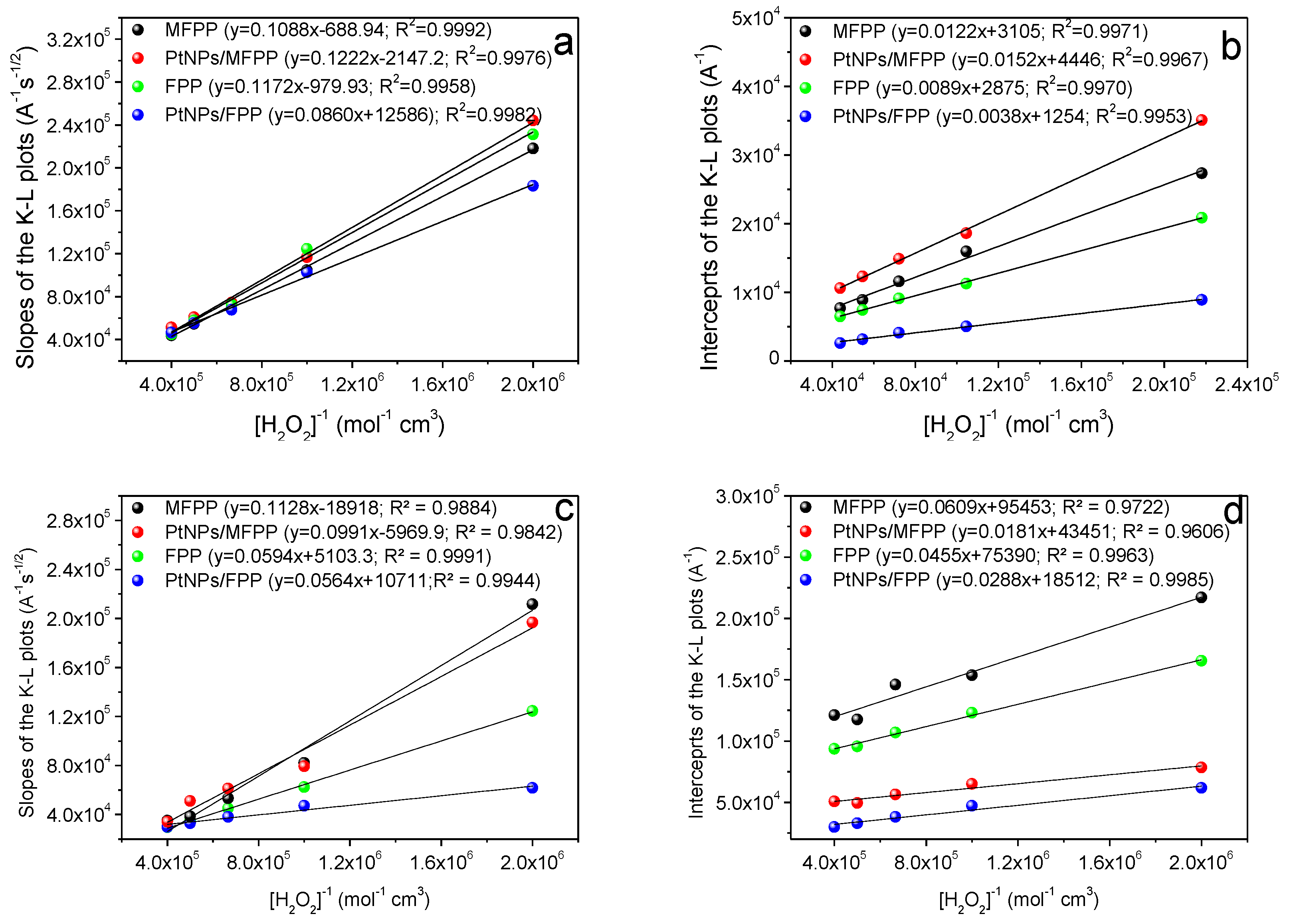
3.3. Oxygen Reduction Catalysis
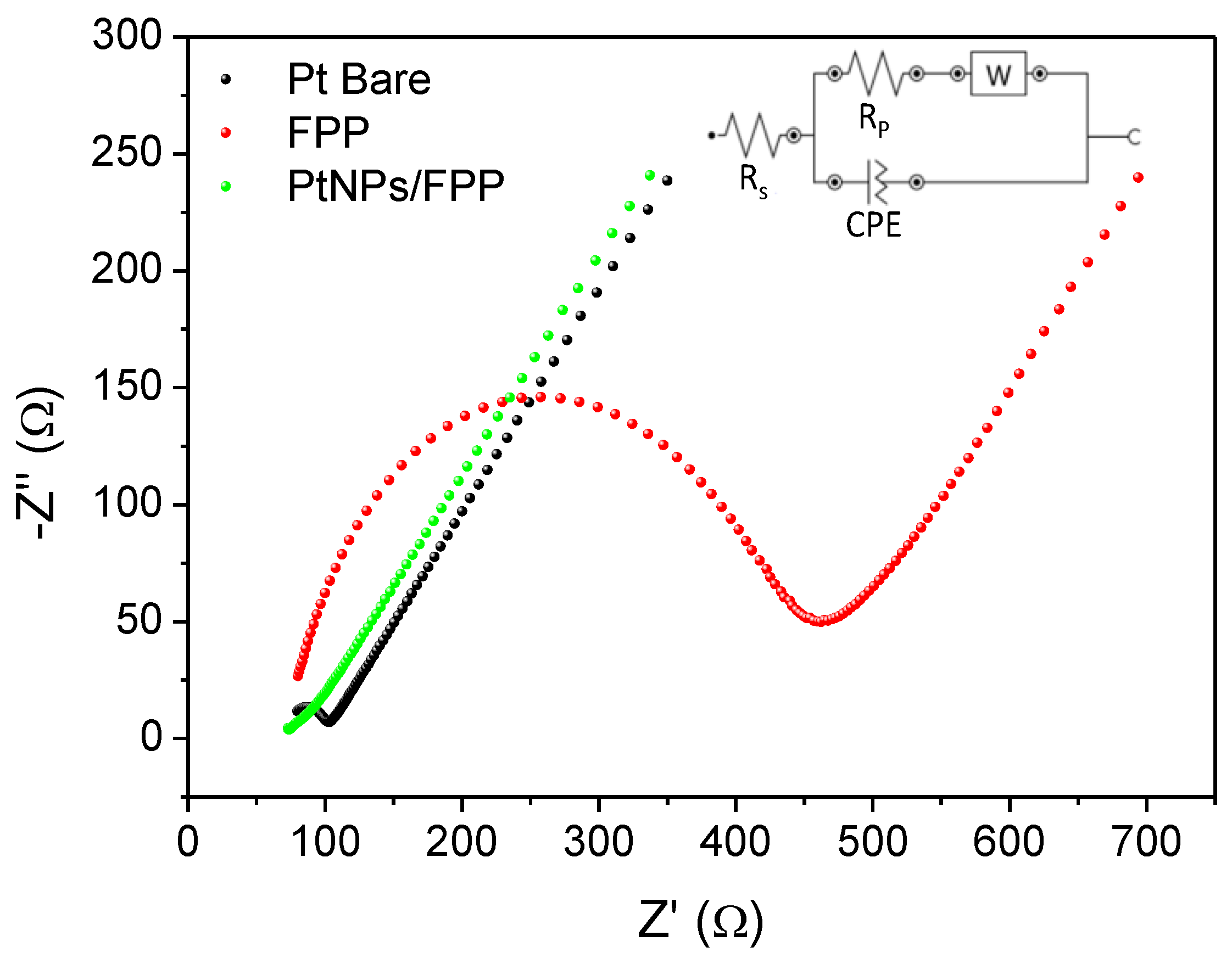
3.4. Electrochemical Impedance Spectroscopy (EIS)
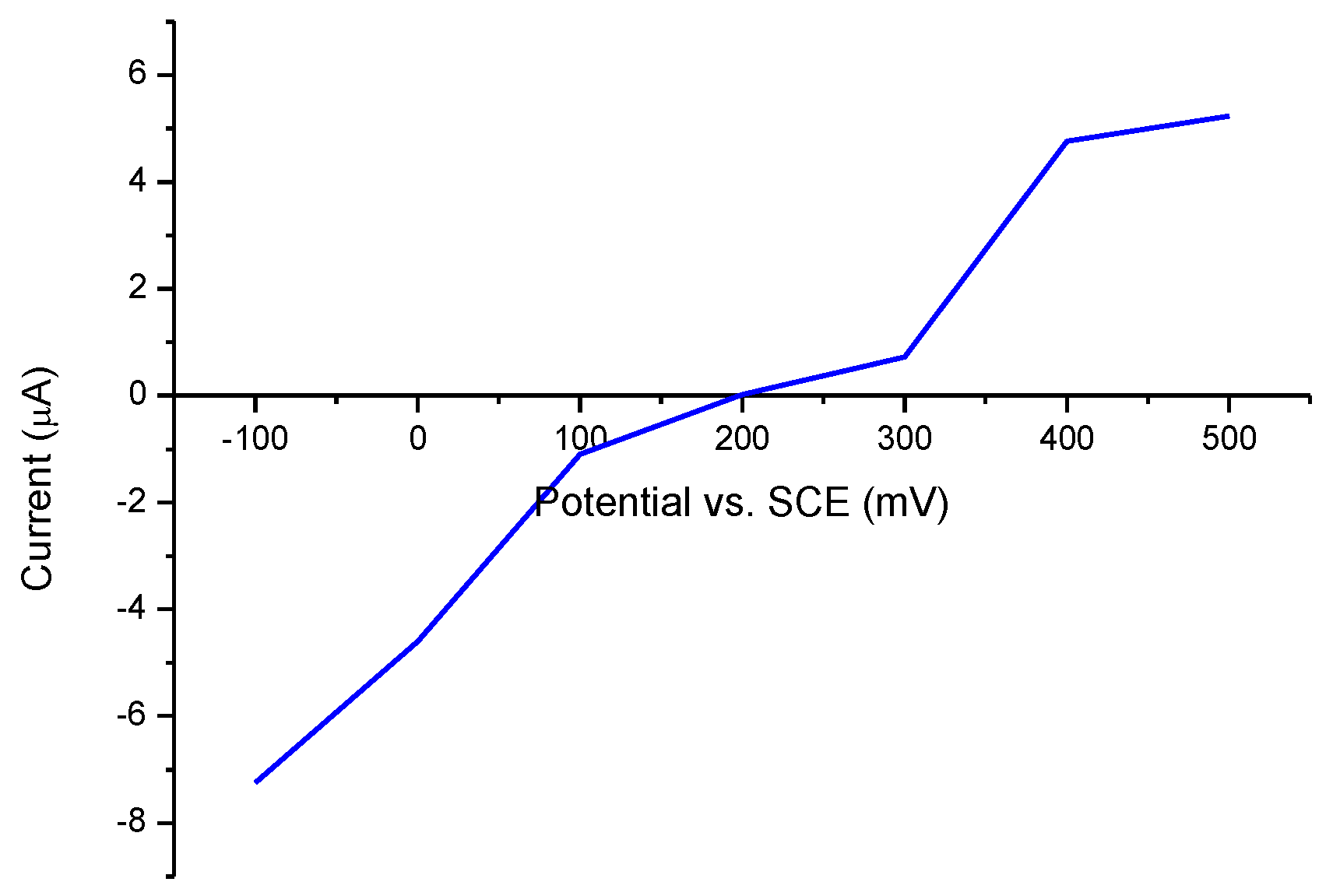
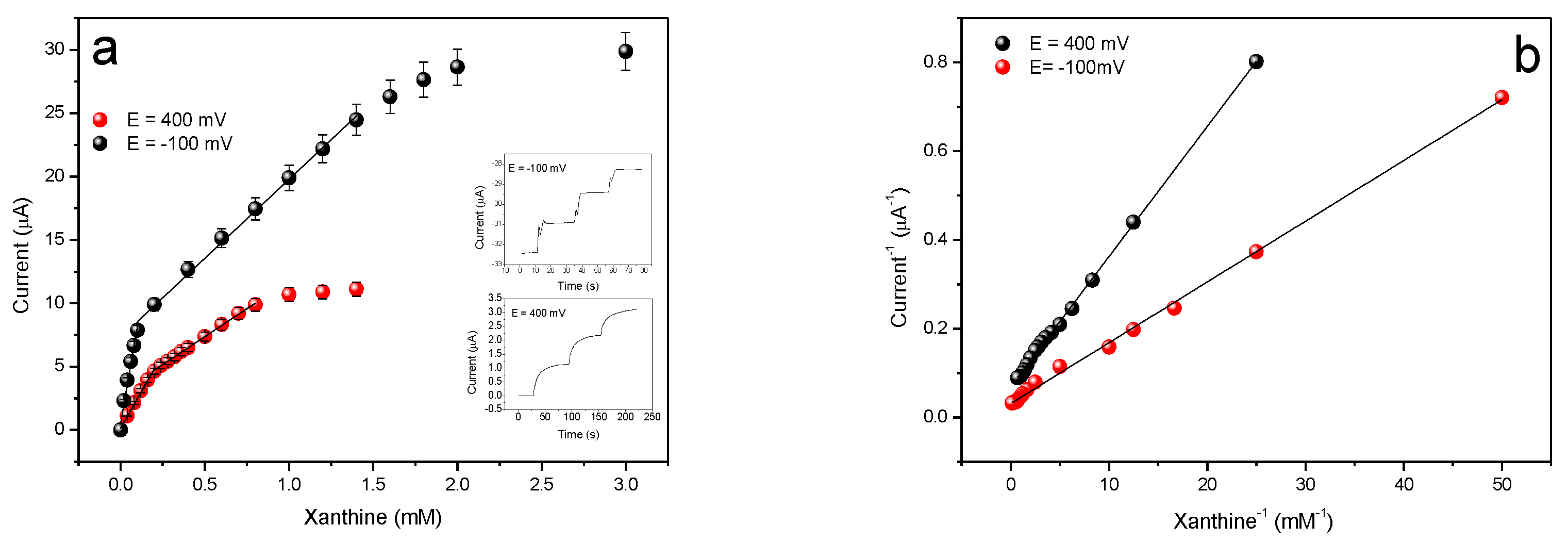
3.5. Xanthine Biosensor Characterization
3.6. Interferences, Reproducibility, and Stability
3.7. Application to Fish Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dervisevic, M.; Dervisevic, E.; Senel, M.; Cevik, E.; Abasiyanik, F.M. Novel Amperometric Xanthine Biosensors Based on REGO-NP (Pt, Pd, and Au) Bionanocomposite Film. Food Anal. Methods 2017, 10, 1252–1263. [Google Scholar] [CrossRef]
- Sen, S.; Sarkar, P. A simple electrochemical approach to fabricate functionalized MWCNT-nanogold decorated PEDOT nanohybrid for simultaneous quantification of uric acid, xanthine and hypoxanthine. Anal. Chim. Acta 2020, 1114, 15–28. [Google Scholar] [CrossRef]
- Chena, J.; Lua, Y.; Yana, F.; Wua, Y.; Huanga, D.; Weng, Z. A fluorescent biosensor based on catalytic activity of platinum nanoparticles for freshness evaluation of aquatic products. Food Chem. 2020, 310, 125922. [Google Scholar] [CrossRef]
- Xue, G.; Yu, W.; Yutong, L.; Qiang, Z.; Xiuying, L.; Yiweia, T.; Jianrong, L. Construction of a novel xanthine biosensor using zinc oxide (ZnO) and the biotemplate method for detection of fish freshness. Anal. Methods 2019, 11, 1021–1026. [Google Scholar] [CrossRef]
- Dervisevic, M.; Dervisevic, E.; Şenel, M. Recent progress in nanomaterial-based electrochemical and optical sensors for hypoxanthine and xanthine. A review. Microchim. Acta 2019, 186, 749. [Google Scholar] [CrossRef] [PubMed]
- Wua, D.; Zhanga, M.; Chena, H.; Bhandari, B. Freshness monitoring technology of fish products in intelligent packaging. Crit. Rev. Food Sci. 2020, 61, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lina, Y.; Ma, X.; Guo, L.; Qiu, B.; Chena, G.; Lin, Z. Multicolor biosensor for fish freshness assessment with the naked eye. Sens. Actuators B 2017, 252, 201–208. [Google Scholar] [CrossRef]
- Borisova, B.; Sánchez, A.; Jiménez-Falcao, S.; Martín, M.; Salazar, P.; Parrado, C.; Pingarrón, J.M.; Villalonga, R. Reduced graphene oxide-carboxymethylcellulose layered withplatinum nanoparticles/PAMAM dendrimer/magnetic nanoparticleshybrids. Application to the preparation of enzyme electrochemicall biosensors. Sens. Actuators B 2016, 232, 84–90. [Google Scholar] [CrossRef]
- Chen, H.; Xiang, H.; Lei, H.; Zhang, D.; Liu, Y.; Qiu, Y.; Xu, L. Inhibition and molecular mechanism of diosmetin against xanthine oxidase by multiple spectroscopies and molecular docking. New J. Chem. 2020, 44, 6799–6809. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, B.; Shen, C.; La, O.-M.; Tan, C.-P.; Cheong, L.-Z. Electrochemical Biosensing of Chilled Seafood Freshness by Xanthine Oxidase Immobilized on Copper-Based Metal–Organic Framework Nanofiber Film. Food Anal. Method. 2019, 12, 1715–1724. [Google Scholar] [CrossRef]
- Shan, D.; Wang, Y.; Xue, H.; Cosnier, S. Sensitive and selective xanthine amperometric sensors based on calcium carbonate nanoparticles. Sensor. Actuators B 2009, 136, 510–515. [Google Scholar] [CrossRef]
- Yazdanparast, S.; Benvidi, A.; Abbasi, S.; Rezaeinasab, M. Enzyme-based ultrasensitive electrochemical biosensor using poly(L-aspartic acid)/MWCNT bio-nanocomposite for xanthine detection: A meat freshness marker. Microchem. J. 2019, 149, 104000. [Google Scholar] [CrossRef]
- Pundir, C.S.; Devi, R. Biosensing methods for xanthine determination: A review. Enzyme Microb. Technol. 2014, 57, 55–62. [Google Scholar] [CrossRef]
- Yam, Y.; Kataho, N.; Watanabe, M.; Nakamura, T.; Asano, Y. Evaluation of beef aging by determination of hypoxanthine and xanthine contents: Application of a xanthine sensor. Food Chem. 1995, 52, 439–445. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Umasankar, Y.; Bhushan, P.; Hirt, P.A.; MacQuhae, F.E.; Borda, L.J.; Lev-Tov, H.A.; Kirsner, R.; Bhansali, S. Nanocomposite Bienzymatic Sensor for Monitoring Xanthine in Wound Diagnostics. J. Electrochem. Soc. 2019, 166, B3295–B3301. [Google Scholar] [CrossRef]
- Sahyar, B.Y.; Kaplanb, M.; Ozsoz, M.; Celik, E.; Otles, S. Electrochemical xanthine detection by enzymatic method based on Ag doped ZnO nanoparticles by using polypyrrole. Bioelectrochemistry 2019, 130, 107327. [Google Scholar] [CrossRef] [PubMed]
- Casado, C.M.; Alonso, B.; Losada, J.; García-Armada, M.P.; Campagna, S.; Ceroni, P.; Puntoriero, F. (Eds.) Wiley Book on Designing Dendrimers; Wiley-VCH: Weinheim, Germany, 2012; pp. 219–262. ISBN 978-0-470-43355-3. [Google Scholar]
- Alonso, B.; García Armada, M.P.; Losada, J.; Cuadrado, I.; González, B.; Casado, C.M. Amperometric Enzyme Electrodes for Aerobic and Anaerobic Glucose Monitoring Prepared by Glucose Oxidase Immobilized in Mixed Ferrocene-Cobaltocenium Dendrimers. Biosens. Bioelectron. 2004, 19, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- García Armada, M.P.; Losada, J.; Zamora, M.; Alonso, B.; Cuadrado, I.; Casado, C.M. Electrocatalytical properties of polymethylferrocenyl dendrimers and their applications in biosensing. Bioelectrochemistry 2006, 69, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ospina, E.; García Armada, M.P.; Losada, J.; Alonso, B.; Casado, C.M. Polyferrocenyl Polycyclosiloxane/Gold Nanoparticles: An Efficient Electrocatalytic Platform for Immobilization and Direct Electrochemistry of HRP. J. Electrochem. Soc. 2016, 63, H826–H833. [Google Scholar] [CrossRef]
- García Armada, M.P.; Vallejo, E.; Villena, C.; Losada, J.; Casado, C.M.; Alonso, B. New acetaminophen amperometric sensor based on ferrocenyl dendrimers deposited onto Pt nanoparticles. J. Solid State Electrochem. 2016, 20, 1551–1563. [Google Scholar] [CrossRef]
- Villena, C.; Punjabi, D.; Casado, C.M.; Alonso, B.; Losada, J.; García Armada, M.P. Monodispersed Size-Controlled Gold Nanoparticles from Electrodeposited Aminoferrocenyl Dendrimer-Templates and Their Application as Efficient Hydrogen Peroxide Electrocatalyst. J. Electrochem. Soc. 2018, 165, B310–B322. [Google Scholar] [CrossRef]
- Casado, C.M.; Cuadrado, I.; Morán, M.; Alonso, B.; Lobete, F.; Losada, J. Synthesis of the first redox-active organometallic polymers containing cyclosiloxanes as frameworks. Organometallics 1995, 14, 2618–2620. [Google Scholar] [CrossRef]
- Casado, C.M.; Cuadrado, I.; Morán, M.; Alonso, B.; Barranco, M.; Losada, J. Cyclic siloxanes and silsesquioxanes as cores and frameworks for the construction of ferrocenyl dendrimers and polymers. Appl. Organomet. Chem. 1999, 13, 245–259. [Google Scholar] [CrossRef]
- Losada, J.; García Armada, M.P.; Cuadrado, I.; Alonso, B.; González, B.; Casado, C.M.; Zhang, J. Ferrocenyl and permethylferrocenyl cyclic and polyhedral siloxane polymers as mediators in amperometric biosensors. J. Organomet. Chem. 2004, 689, 2799–2807. [Google Scholar] [CrossRef]
- Murray, R.W. Molecular Design of Electrode Surfaces; Wiley: New York, NY, USA, 1992; ISBN 978-0-471-55773-9. [Google Scholar]
- Abruña, H. Electrode Modification with Polymeric Reagents. In Electro-Responsive Molecular and Polymeric Systems; Skotheim, T.A., Ed.; Marcel Dekker: New York, NY, USA, 1988; Volume 1, Chapter 3. [Google Scholar]
- Laviron, E. A multilayer model for the study of space distributed Redox modified electrodes. Part II. Theory and application of linear potential sweep voltammetry for a simple reaction. J. Electroanal. Chem. 1980, 112, 11–23. [Google Scholar] [CrossRef]
- Laviron, E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 1979, 101, 19–28. [Google Scholar] [CrossRef]
- Levich, V.G. Physicochemical Hydrodynamics; Prentice Hall: Englewood Cliffs, NJ, USA, 1982. [Google Scholar]
- Koutecky, J.; Levich, V.G. The application of the rotating disc electrode to studies of kinetic and catalytic processes. Zh. Fiz. Khim. 1958, 32, 1565. [Google Scholar]
- Munteanu, F.D.; Kubota, L.T.; Gorton, L. Effect of pH on the catalytic electrooxidation of NADH using different two-electron mediators immobilised on zirconium phosphate. J. Electroanal. Chem. 2001, 509, 2–10. [Google Scholar] [CrossRef]
- Peña, R.C.; Gamboa, J.C.M.; Bertotti, M.; Paixao, T.R.L.C. Studies on the electrocatalytic reduction of hydrogen peroxide on a glassy carbon electrode modified with a ruthenium oxide hexacyanoferrate film. Int. J. Electrochem. Sci. 2011, 6, 394–403. [Google Scholar]
- Gomez-Marin, A.M.; Feliu, J.M.; Ticianelli, E.A. Oxygen reduction on platinum surfaces in acid media: Experimental evidence of a CECE/DISP initial reaction path. ACS Catal. 2019, 9, 2238–2251. [Google Scholar] [CrossRef]
- Ma, R.; Lin, G.; Zhou, Y.; Liu, Q.; Zhang, T.; Shan, G.; Yang, M.; Wang, J. A review of oxygen reduction mechanisms for metal-free carbon-based electrocatalysts. NPJ Comput. Mater. 2019, 5, 78. [Google Scholar] [CrossRef]
- Lide, R. (Ed.) “Diffusion of gases in water” and “Solubility of selected gases in water”. In CRC Handbook of Chemistry and Physics; Internet Version 2005; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Katz, E.; Willner, I. Probing Biomolecular Interactions at Conductive and Semiconductive Surfaces by Impedance Spectroscopy: Routes to Impedimetric Immunosensors, DNA-Sensors, and Enzyme Biosensors. Electroanalysis 2003, 15, 913–947. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods. Fundamentals and Applications, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2001; p. 381. [Google Scholar]
- Dalkıran, B.; Erden, P.E.; Kılıç, E. Amperometric biosensors based on carboxylated multiwalled carbon nanotubes-metal oxide nanoparticles-7,7,8,8-tetracyanoquinodimethane composite for the determination of xanthine. Talanta 2017, 167, 286–295. [Google Scholar] [CrossRef]
- Chen, C.J.; Liu, C.C.; Savinell, R.F. Polymeric redox mediator enzyme electrodes for anaerobic glucose monitoring. J. Electroanal. Chem. 1993, 348, 317–338. [Google Scholar] [CrossRef]
- Khan, M.Z.H.; Ahommedb, M.S.; Daizy, M. Detection of xanthine in food samples with an electrochemical biosensor based on PEDOT:PSS and functionalized gold nanoparticles. RSC Adv. 2020, 10, 36147. [Google Scholar] [CrossRef]
- Dodevska1, T.; Horozova, E.; Dimcheva, N. Design of an amperometric xanthine biosensor based on a graphite transducer patterned with noble metal microparticles. Cent. Eur. J. Chem. 2010, 8, 19–27. [Google Scholar] [CrossRef]
- Bas, S.Z.; Gülce, H.; Yıldız, S.; Gülce, A. Amperometric biosensors based on deposition of gold and platinum nanoparticles on polyvinylferrocene modified electrode for xanthine detection. Talanta 2011, 87, 189–196. [Google Scholar] [CrossRef]
- Magro, M.; Baratella, D.; Venerando, A.; Nalotto, G.; Basso, C.R.; Molinari, S.; Salviulo, G.; Ugolotti, J.; Pedrosa, V.A.; Vianello, F. Enzyme immobilization on maghemite nanoparticles with improved catalytic activity: An electrochemical study for xanthine. Materials 2020, 13, 1776. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.B.; Labban, N.; Conway, G.E.; Pollock, J.A.; Leopold, M.C. Adaptable xerogel-layered amperometric biosensor platforms on wire electrodes for clinically relevant measurements. Sensors 2019, 19, 2584. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode. | E0′ (V) | α | k1 (s−1) |
|---|---|---|---|
| FPP | 0.016 | 0.60 [20] | 2.5 × 105 [20] |
| PtNPs/FPP | 0.044 | 0.54 | 3.7 × 105 |
| MFPP | −0.255 | 0.53 | 2.6 × 105 |
| PtNPs/MFPP | −0.270 | 0.52 | 1.6 × 105 |
| Electrode | H2O2 Oxidation | H2O2 Reduction | O2 Reduction | |||
|---|---|---|---|---|---|---|
| D (cm2 s−1) | kobs (M−1 s−1) | D (cm2 s−1) | kobs (M−1 s−1) | n | kobs (M−1 s−1) | |
| FPP | 1.1 × 10−5 | 1.8 × 104 | 2.9 × 10−5 | 4.5 × 103 | ||
| PtNPs/FPP | 1.6 × 10−5 | 5.9 × 104 | 3.1 × 10−5 | 7.1 × 103 | 1.8 | 2.7 × 104 |
| MFPP | 1.2 × 10−5 | 2.0 × 104 | 1.1 × 10−5 | 2.2 × 103 | ||
| PtNPs/MFPP | 9.7 × 10−6 | 2.6 × 104 | 1.3 × 10−5 | 9.0 × 103 | ||
| Electrode | RCT (Ω) | CPE (μF) | n | i0 (μA) | k0 (cm s−1) |
|---|---|---|---|---|---|
| Pt Bare | 32 | 4.29 | 0.829 | 791 | 1.17 × 10−2 |
| FPP | 379 | 4.35 | 0.821 | 68 | 1.01 × 10−3 |
| PtNPs/FPP | 30 | 1246.5 | 0.455 | 859 | 1.27 × 10−2 |
| Electrode | E (V) | Method | LOD (μM) | LR (μM) | Sensitivity (A M−1 cm−2) |
|---|---|---|---|---|---|
| XOD/nanoAg-ZnO/Ppy/PGE [18] | +0.7 | Amp | 0.07 | 0.06–0.6 | 0.03 * |
| P(Vfc-GMA)/REGO-Pt [1] | +0.35 | Amp | 0.003 | 1–40 | 21.98 * |
| MWCNT/Au/XO/HRP [2] | −0.44 | Amp | 1.3 | 22–400 | 0.156 |
| XOD/c-MWCNTs/Fe3O4/TCNQ/CHIT/GCE [40] | +0.30 | Amp | 0.2 | 1.9–230 | 0.025 |
| XO/MNP-PAMAM_PtNP/rGO-CMC/GCE [8] | +0.60 | Amp | 0.013 | 0.05–12 | 0.140 |
| GCE/PEDOT:PSS-AuNPs [41] | +0.78 | DPV | 0.03 | 0.05–10 | 0.583 * |
| XOD@Cu-MOF/SA/GCE [11] | +0.65 | DPV | 0.064 | 0.01–10 | 6.74 |
| (70%Pd, 30% Pt)/Graphite [42] | −0.05 | Amp | 1.5 | 1.5–70 | 0.23 * |
| XO/Pt/PVF/Pt [43] | +0.40 | Amp | 0.6 | 2–660 | 0.021 |
| SAMN@XO [44] | +0.10 | DPV | 0.1 | 1–10 | 0.0094 * |
| Pt/PtMS(XOx)/100%HPU [45] | +0.35 | Amp | 1.7 | 1.7–50 | 0.0022 * |
| XO/PtNPs/FPP [this work] | +0.40 −0.10 | Amp Amp | 0.030 0.045 | 30–200 200–800 10–100 100–1400 | 0.33 0.13 1.10 0.18 |
| Fish Type | mg Xanthine/g Fish | ||
|---|---|---|---|
| Day 1 | Day 6 | Day 11 | |
| Hake | 0.09 | 0.86 | 1.10 |
| Salmon | 0.52 | 0.81 | 0.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boluda, A.; Casado, C.M.; Alonso, B.; García Armada, M.P. Efficient Oxidase Biosensors Based on Bioelectrocatalytic Surfaces of Electrodeposited Ferrocenyl Polycyclosiloxanes—Pt Nanoparticles. Chemosensors 2021, 9, 81. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9040081
Boluda A, Casado CM, Alonso B, García Armada MP. Efficient Oxidase Biosensors Based on Bioelectrocatalytic Surfaces of Electrodeposited Ferrocenyl Polycyclosiloxanes—Pt Nanoparticles. Chemosensors. 2021; 9(4):81. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9040081
Chicago/Turabian StyleBoluda, Alvaro, Carmen M. Casado, Beatriz Alonso, and M. Pilar García Armada. 2021. "Efficient Oxidase Biosensors Based on Bioelectrocatalytic Surfaces of Electrodeposited Ferrocenyl Polycyclosiloxanes—Pt Nanoparticles" Chemosensors 9, no. 4: 81. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9040081