
Anodic Stripping Voltammetry with the Hanging Mercury Drop Electrode for Trace Metal Detection in Soil Samples


Abstract
:1. Introduction
2. Experimental
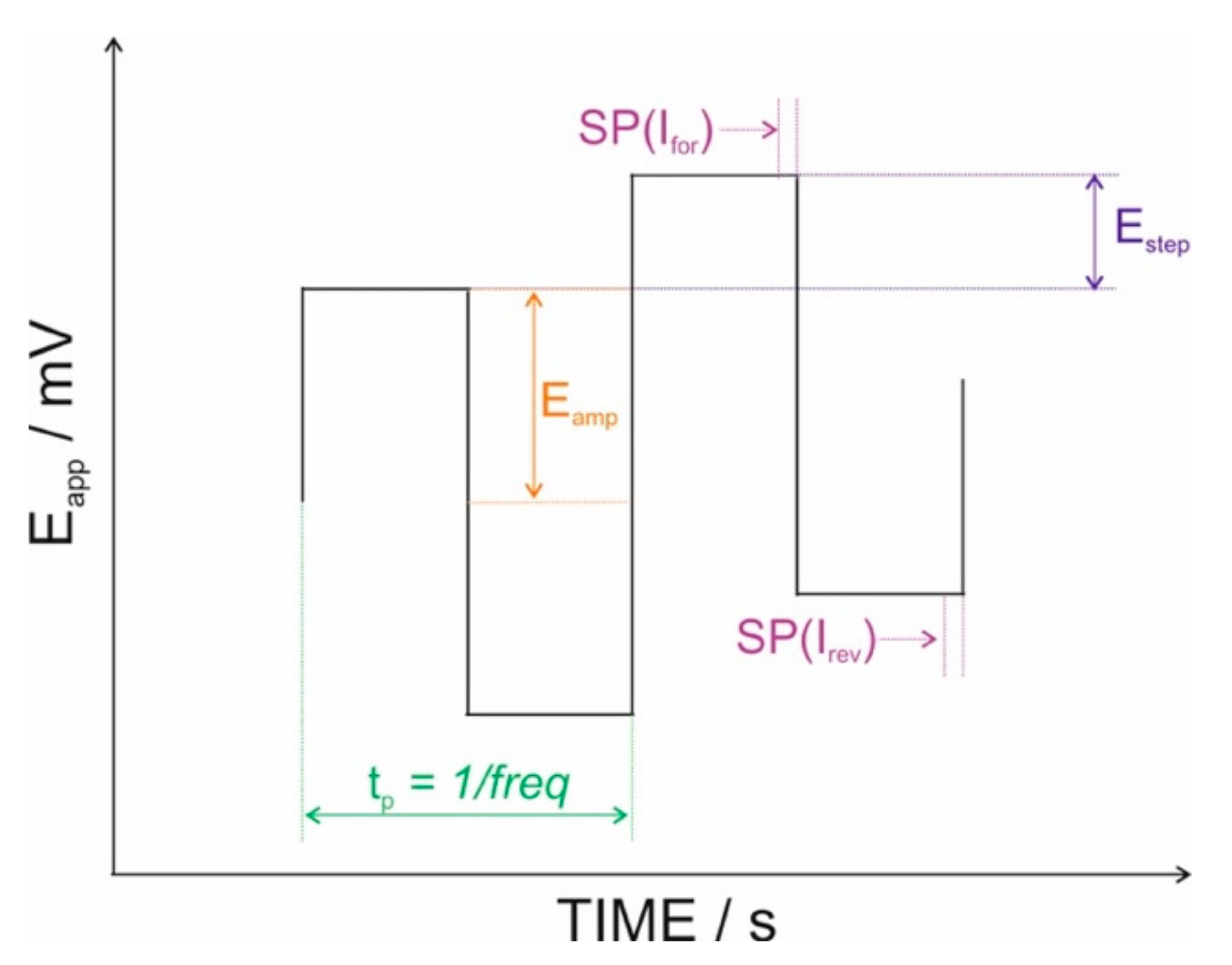
2.1. Reagents and Instrumentation
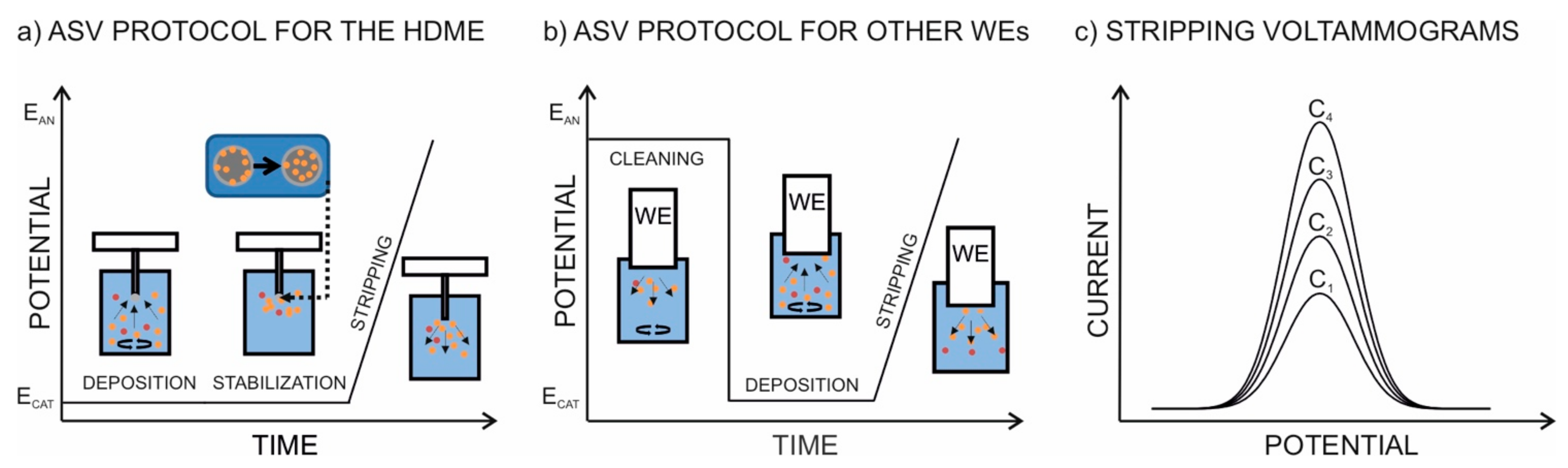
2.2. Measurements with the HMDE
2.3. Protocol for the Analysis of Soil Samples
3. Results and Discussion
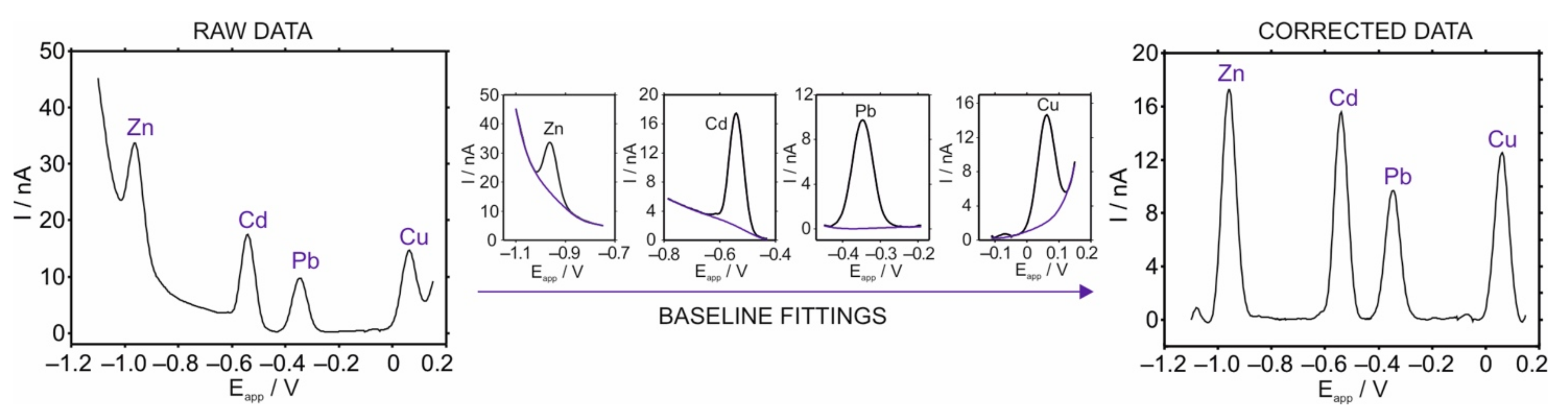
3.1. Fundaments of ASV Measurements
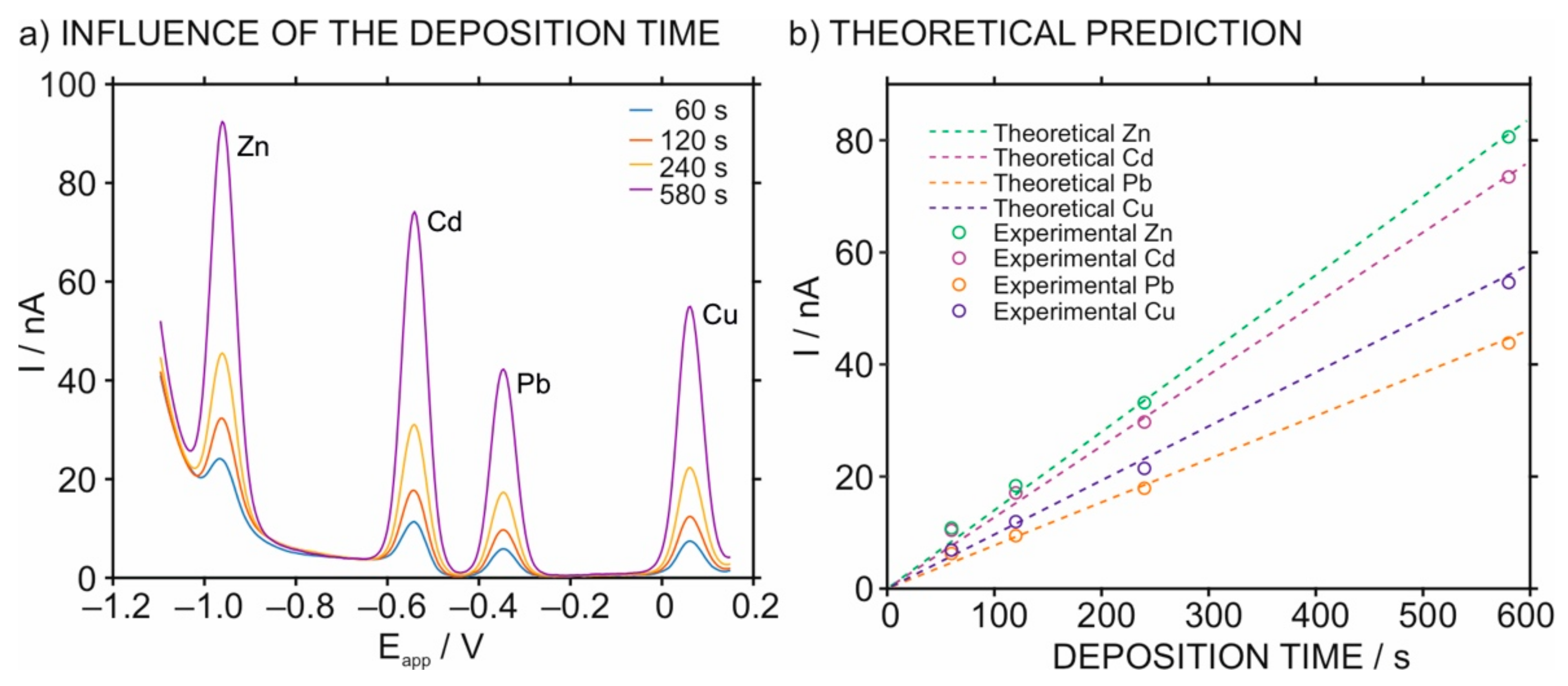
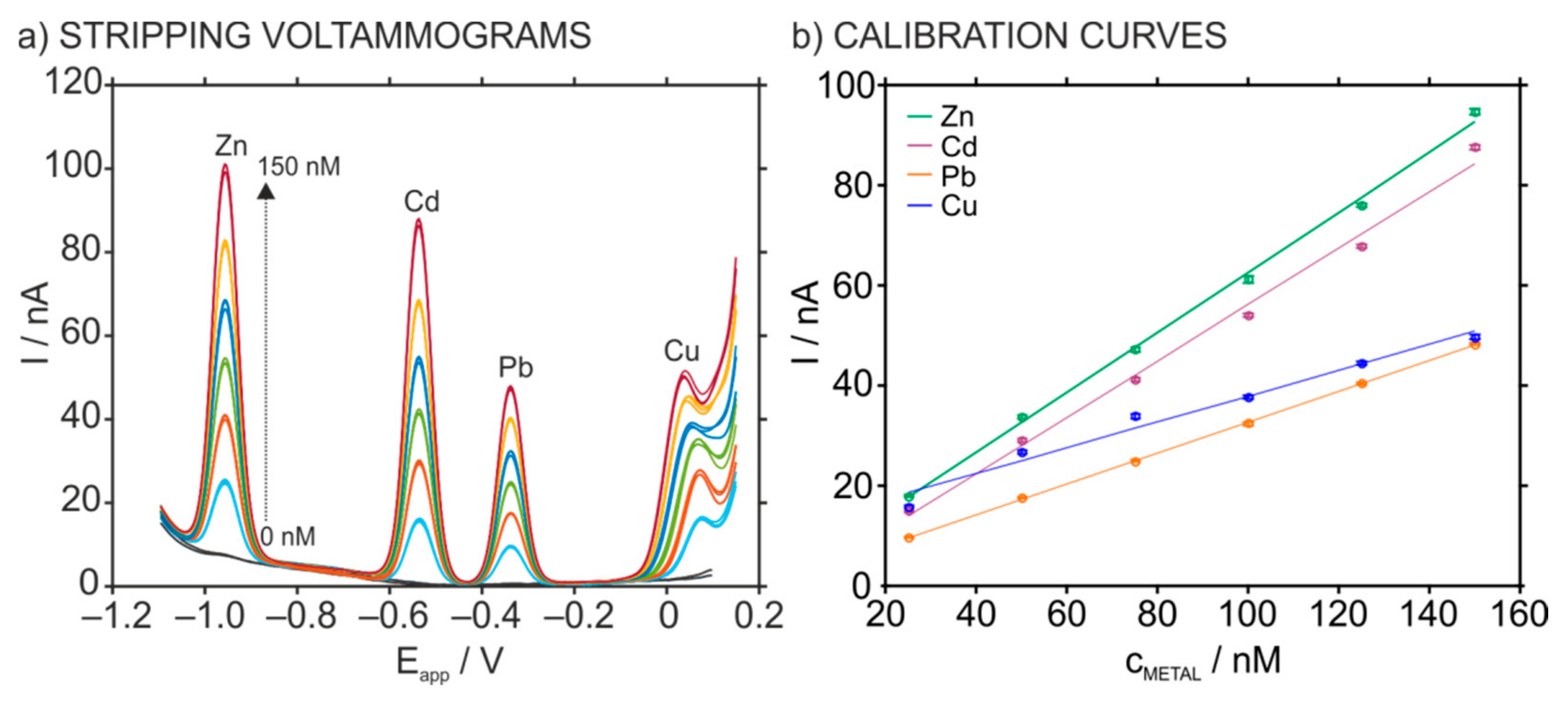
3.2. Optimization of the Stripping Peaks in the HMDE for Multi-Ion Detection
3.3. The Analytical Characteristics of the HMDE for Multi-Ion Analysis
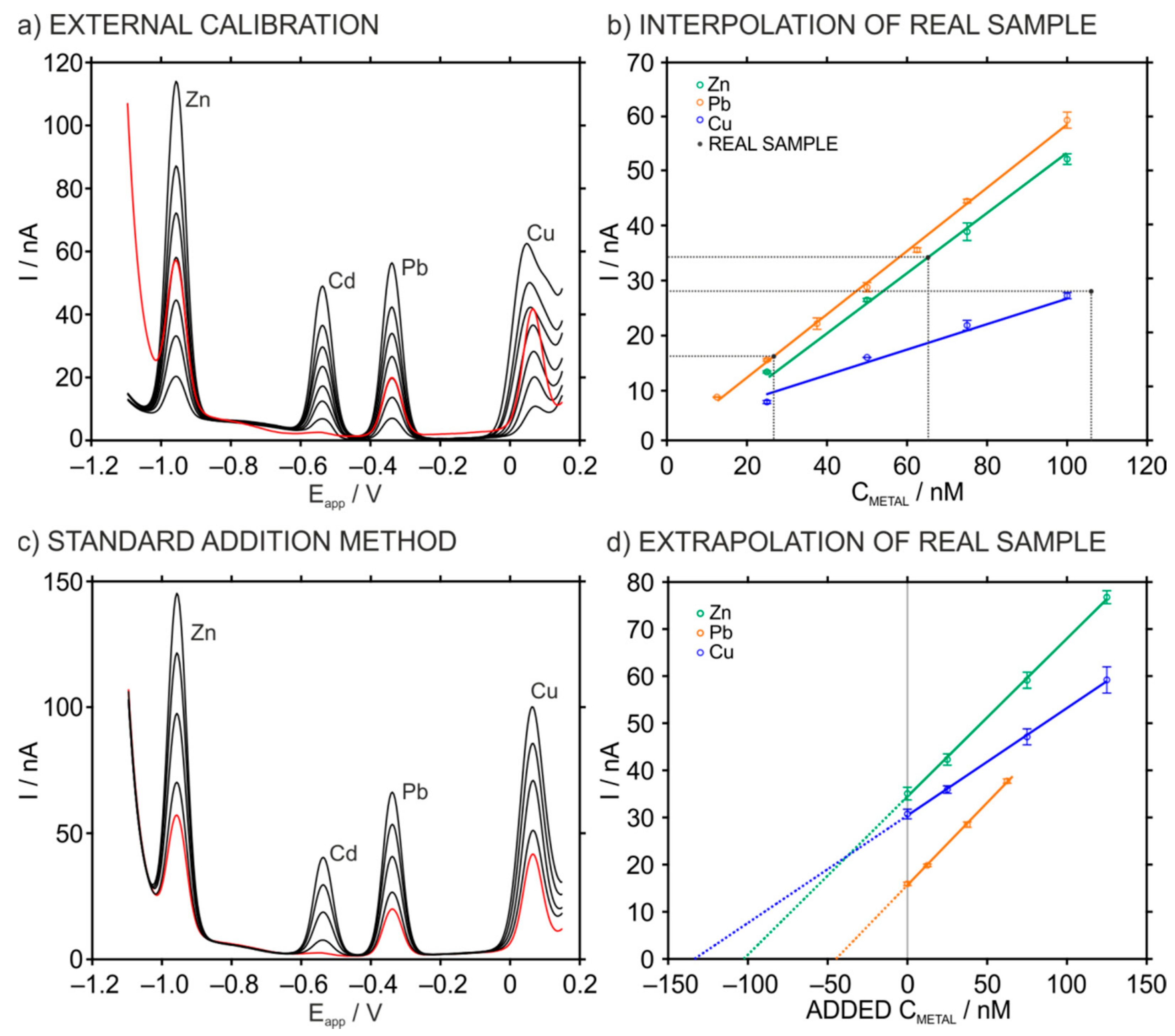
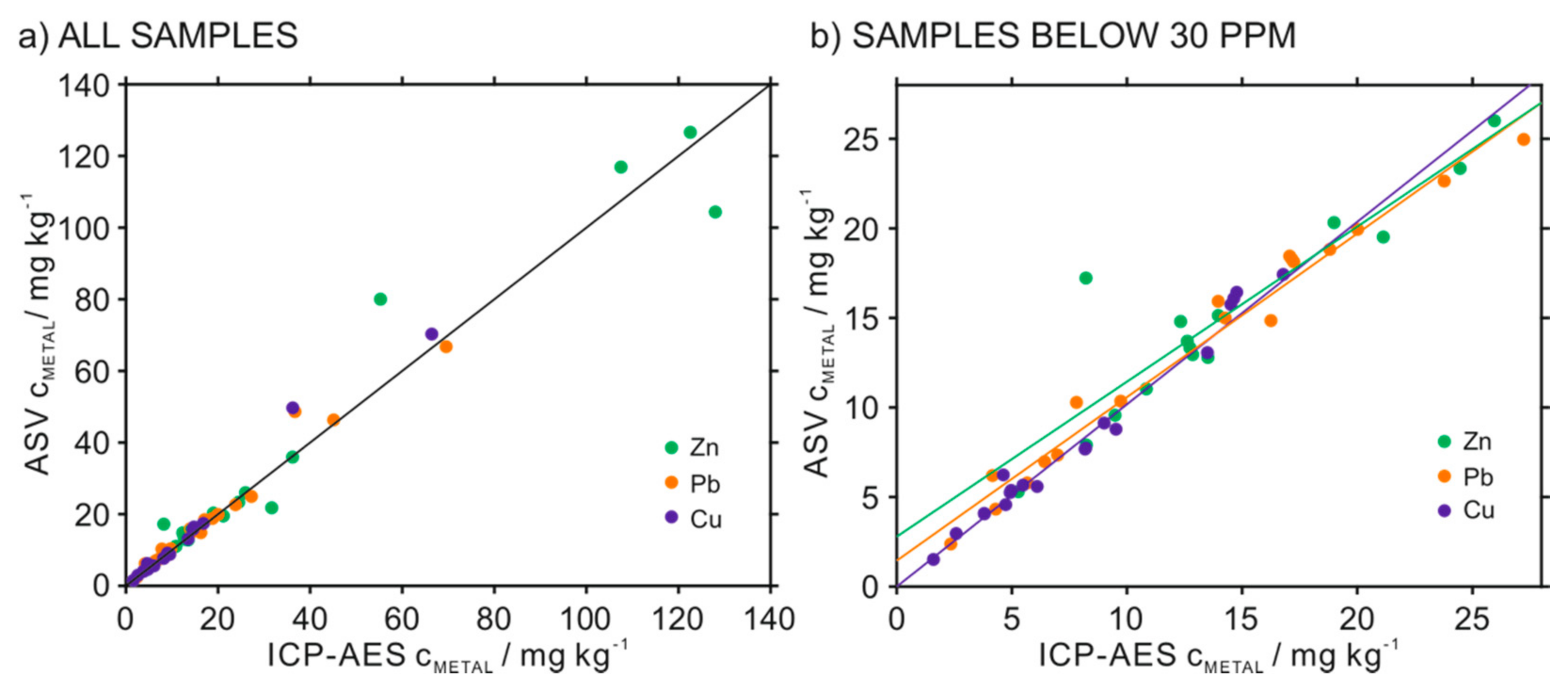
3.4. Simultaneous Determination of Zn2+, Cd2+, Pb2+ and Cu2+ in Soil Samples Using the HMDE
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, J.B.; Larry, R.F. Electrochemical Methods Fundamentals and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Copeland, T.; Skogerboe, R. Anodic stripping voltammetry. Anal. Chem. 1974, 46, 1257A–1268A. [Google Scholar] [CrossRef]
- Rodrigues, J.A.; Rodrigues, C.M.; Almeida, P.J.; Valente, I.M.; Gonçalves, L.M.; Compton, R.G.; Barros, A.A. Increased sensitivity of anodic stripping voltammetry at the hanging mercury drop electrode by ultracathodic deposition. Anal. Chim. Acta 2011, 701, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Jia, M.; Wang, Z.; Zhang, W.; Zhang, Q.; Liu, G.; Zhang, Z.; Li, P. Simultaneous voltammetric determination of cadmium (II), lead (II), mercury (II), zinc (II), and copper (II) using a glassy carbon electrode modified with magnetite (Fe3O4) nanoparticles and fluorinated multiwalled carbon nanotubes. Microchim. Acta 2019, 186, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Serrano, N.; González-Calabuig, A.; Del Valle, M. Crown ether-modified electrodes for the simultaneous stripping voltammetric determination of Cd(II), Pb(II) and Cu(II). Talanta 2015, 138, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Ràfols, C.; Rosal, M.; Serrano, N.; Ariño, C.; Esteban, M.; Díaz-Cruz, J.M. Expanding the possibilities of electrografting modification of voltammetric sensors through two complementary strategies. Electrochim. Acta 2019, 319, 878–884. [Google Scholar] [CrossRef]
- Saha, S.; Sarkar, P. Differential pulse anodic stripping voltammetry for detection of As (III) by Chitosan-Fe(OH)3 modified glassy carbon electrode: A new approach towards speciation of arsenic. Talanta 2016, 158, 235–245. [Google Scholar] [CrossRef]
- Torres-Rivero, K.; Pérez-Ràfols, C.; Bastos-Arrieta, J.; Florido, A.; Martí, V.; Serrano, N. Direct As(V) determination using screen-printed electrodes modified with silver nanoparticles. Nanomaterials 2020, 10, 1280. [Google Scholar] [CrossRef]
- Wang, R.; Chakrabarti, C.L. Copper speciation by competing ligand exchange method using differential pulse anodic stripping voltammetry with ethylenediaminetetraacetic acid (EDTA) as competing ligand. Anal. Chim. Acta 2008, 614, 153–160. [Google Scholar] [CrossRef]
- Landner, L.; Reuther, R. A critical review of current knowledge on fluxes, speciation, bioavailability and risk for adverse effects of copper, chromium, nickel and zinc. In Metals in Society and in the Environment; Kluwer Academic Publisher: Dordrecht, The Netherlands, 2004. [Google Scholar]
- Ariño, C.; Serrano, N.; Díaz-Cruz, J.M.; Esteban, M. Voltammetric determination of metal ions beyond mercury electrodes. A review. Anal. Chim. Acta 2017, 990, 11–53. [Google Scholar] [CrossRef] [Green Version]
- Barek, J.; Zima, J. Eighty years of polarography–history and future. Electroanalysis 2003, 15, 467–472. [Google Scholar] [CrossRef]
- Barek, J.; Fogg, A.G.; Muck, A.; Zima, J. Polarography and voltammetry at mercury electrodes. Crit. Rev. Anal. Chem. 2001, 31, 291–309. [Google Scholar] [CrossRef]
- Serrano, N.; Díaz-Cruz, J.M.; Ariño, C.; Esteban, M. Antimony-based electrodes for analytical determinations. TrAC Trends Anal. Chem. 2016, 77, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Serrano, N.; Alberich, A.; Díaz-Cruz, J.M.; Ariño, C.; Esteban, M. Coating methods, modifiers and applications of bismuth screen-printed electrodes. TrAC Trends Anal. Chem. 2013, 46, 15–29. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Hart, J.P. Screen-printed electrochemical sensors for monitoring metal pollutants. TrAC Trends Anal. Chem. 2003, 22, 456–469. [Google Scholar] [CrossRef]
- Sawan, S.; Maalouf, R.; Errachid, A.; Jaffrezic-Renault, N. Metal and metal oxide nanoparticles in the voltammetric detection of heavy metals: A review. TrAC Trends Anal. Chem. 2020, 131, 116014. [Google Scholar] [CrossRef]
- Hub, E.S. European Union Reference Laboratories. Available online: https://ec.europa.eu/jrc/en/eurls (accessed on 29 March 2021).
- Metrohm 663 VA Stand. Available online: https://www.metrohm-autolab.com/Products/Echem/Accessories/663VAStand.html (accessed on 3 May 2021).
- Bard, A.J.; Faulkner, L.R. Fundamentals and applications. Electrochem. Methods 2001, 2, 580–632. [Google Scholar]
- Krulic, D.; Fatouros, N. Peak heights and peak widths at half-height in square wave voltammetry without and with ohmic potential drop for reversible and irreversible systems. J. Electroanal. Chem. 2011, 652, 26–31. [Google Scholar] [CrossRef]
- Galus, Z. Diffusion coefficients of metals in mercury. Pure Appl. Chem. 1984, 56, 635–644. [Google Scholar] [CrossRef]
- Wang, J. Analytical Electrochemistry; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Daneshgar, P.; Norouzi, P.; Ganjali, M.R. Application of a continuous square-wave potential program for sub nano molar determination of ketotifen. Chem. Pharm. Bull. 2009, 57, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Mirceski, V.; Gulaboski, R.; Lovric, M.; Bogeski, I.; Kappl, R.; Hoth, M. Square-wave voltammetry: A review on the recent progress. Electroanalysis 2013, 25, 2411–2422. [Google Scholar] [CrossRef] [Green Version]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 89th ed; Taylor & Francis: Abingdon, UK, 2008. [Google Scholar]
- Sosa, V.; Barceló, C.; Serrano, N.; Ariño, C.; Díaz-Cruz, J.M.; Esteban, M. Antimony film screen-printed carbon electrode for stripping analysis of Cd(II), Pb(II), and Cu(II) in natural samples. Anal. Chim. Acta 2015, 855, 34–40. [Google Scholar] [CrossRef]
- Sang, S.; Li, D.; Zhang, H.; Sun, Y.; Jian, A.; Zhang, Q.; Zhang, W. Facile synthesis of AgNPs on reduced graphene oxide for highly sensitive simultaneous detection of heavy metal ions. RSC Adv. 2017, 7, 21618–21624. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Gao, C.; Meng, F.-L.; Li, H.-H.; Wang, L.; Liu, J.-H.; Huang, X.-J. SnO2/reduced graphene oxide nanocomposite for the simultaneous electrochemical detection of cadmium (II), lead (II), copper (II), and mercury (II): An interesting favorable mutual interference. J. Phys. Chem. C 2012, 116, 1034–1041. [Google Scholar] [CrossRef]
- Veerakumar, P.; Veeramani, V.; Chen, S.-M.; Madhu, R.; Liu, S.-B. Palladium nanoparticle incorporated porous activated carbon: Electrochemical detection of toxic metal ions. ACS Appl. Mater. Interfaces 2016, 8, 1319–1326. [Google Scholar] [CrossRef]
- Alberich, A.; Díaz-Cruz, J.M.; Ariño, C.; Esteban, M. Potential shift correction in multivariate curve resolution of voltammetric data. General formulation and application to some experimental systems. Analyst 2008, 133, 112–125. [Google Scholar] [CrossRef]
- Xu, X.; Duan, G.; Li, Y.; Liu, G.; Wang, J.; Zhang, H.; Dai, Z.; Cai, W. Fabrication of gold nanoparticles by laser ablation in liquid and their application for simultaneous electrochemical detection of Cd2+, Pb2+, Cu2+, Hg2+. ACS Appl. Mater. Interfaces 2014, 6, 65–71. [Google Scholar] [CrossRef]
- Van Staden, J.; Matoetoe, M. Simultaneous determination of copper, lead, cadmium and zinc using differential pulse anodic stripping voltammetry in a flow system. Anal. Chim. Acta 2000, 411, 201–207. [Google Scholar] [CrossRef]
- Kokab, T.; Shah, A.; Iftikhar, F.J.; Nisar, J.; Akhter, M.S.; Khan, S.B. Amino acid-fabricated glassy carbon electrode for efficient simultaneous sensing of zinc (II), cadmium (II), copper (II), and mercury (II) ions. ACS Omega 2019, 4, 22057–22068. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Deng, Y.; Luo, Y.; Lv, J.; Li, T.; Xu, J.; Chen, S.-W.; Wang, J. Graphene aerogel–metal–organic framework-based electrochemical method for simultaneous detection of multiple heavy-metal ions. Anal. Chem. 2018, 91, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sun, G.; Pan, J.; Pan, Y.; Wang, S.; Zhai, H. A nanocomposite consisting of ionic liquid-functionalized layered Mg(II)/Al(III) double hydroxides for simultaneous electrochemical determination of cadmium (II), copper (II), mercury (II) and lead (II). Microchim. Acta 2019, 186, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kitte, S.A.; Li, S.; Nsabimana, A.; Gao, W.; Lai, J.; Liu, Z.; Xu, G. Stainless steel electrode for simultaneous stripping analysis of Cd(II), Pb(II), Cu(II) and Hg(II). Talanta 2019, 191, 485–490. [Google Scholar] [CrossRef]
- Rehman, A.U.; Ikram, M.; Kan, K.; Zhao, Y.; Zhang, W.J.; Zhang, J.; Liu, Y.; Wang, Y.; Du, L.; Shi, K. 3D interlayer nanohybrids composed of reduced graphenescheme oxide/SnO2/PPy grown from expanded graphite for the detection of ultra-trace Cd2+, Cu2+, Hg2+ and Pb2+ ions. Sens. Actuators B Chem. 2018, 274, 285–295. [Google Scholar] [CrossRef]
- Hassan, K.M.; Gaber, S.E.; Altahan, M.F.; Azzem, M.A. Novel sensor based on poly (1, 2-diaminoanthraquinone) for individual and simultaneous anodic stripping voltammetry of Cd2+, Pb2+, Cu2+ and Hg2+. Electroanalysis 2018, 30, 1155–1162. [Google Scholar] [CrossRef]
- Fomo, G.; Nwaji, N.; Nyokong, T. Low symmetric metallophthalocyanine modified electrode via click chemistry for simultaneous detection of heavy metals. J. Electroanal. Chem. 2018, 813, 58–66. [Google Scholar] [CrossRef]
- Thiruppathi, A.R.; Sidhureddy, B.; Keeler, W.; Chen, A. Facile one-pot synthesis of fluorinated graphene oxide for electrochemical sensing of heavy metal ions. Electrochem. Commun. 2017, 76, 42–46. [Google Scholar] [CrossRef]
- Kumar, R.; Bhuvana, T.; Sharma, A. Nickel tungstate–graphene nanocomposite for simultaneous electrochemical detection of heavy metal ions with application to complex aqueous media. RSC Adv. 2017, 7, 42146–42158. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Gao, N.; Nishitani, A.; Tanaka, H. Rod-like hydroxyapatite and Nafion nanocomposite as an electrochemical matrix for simultaneous and sensitive detection of Hg2+, Cu2+, Pb2+ and Cd2+. J. Electroanal. Chem. 2016, 775, 212–218. [Google Scholar] [CrossRef]
- Xie, Y.-L.; Zhao, S.-Q.; Ye, H.-L.; Yuan, J.; Song, P.; Hu, S.-Q. Graphene/CeO2 hybrid materials for the simultaneous electrochemical detection of cadmium (II), lead (II), copper (II), and mercury (II). J. Electroanal. Chem. 2015, 757, 235–242. [Google Scholar] [CrossRef]
- Muralikrishna, S.; Sureshkumar, K.; Varley, T.S.; Nagaraju, D.H.; Ramakrishnappa, T. In situ reduction and functionalization of graphene oxide with L-cysteine for simultaneous electrochemical determination of cadmium (II), lead (II), copper (II), and mercury (II) ions. Anal. Methods 2014, 6, 8698–8705. [Google Scholar] [CrossRef]
- Pesavento, M.; Alberti, G.; Biesuz, R. Analytical methods for determination of free metal ion concentration, labile species fraction and metal complexation capacity of environmental waters: A review. Anal. Chim. Acta 2009, 631, 129–141. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, X.; Niyungeko, C.; Zhou, J.; Xu, J.; Tian, G. A review of the identification and detection of heavy metal ions in the environment by voltammetry. Talanta 2018, 178, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Brainina, K.Z.; Malakhova, N.; Stojko, N.Y. Stripping voltammetry in environmental and food analysis. Fresenius’ J. Anal. Chem. 2000, 368, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Uslu, B.; Ozkan, S.A. Electroanalytical methods for the determination of pharmaceuticals: A review of recent trends and developments. Anal. Lett. 2011, 44, 2644–2702. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Peak Potential (V) | Observations | LRR a | Ref | ||||
|---|---|---|---|---|---|---|---|---|
| Zn2+ | Cd2+ | Pb2+ | Cu2+ | Hg2+ | ||||
| HMDE | −0.96 | −0.54 | −0.35 | 0.06 | – | Well-resolved peaks | Zn2+: 14−150 nM Cd2+: 21–150 nM Pb2+: 7–150 nM Cu2+: 40–150 nM | This work |
| PdNPs − PACs modified GCE | − | −0.79 | −0.54 | −0.07 | 0.27 | Peak shoulders appear as concentration increases | Cd2+: 0.5–5.5 µM Pb2+: 0.5–8.9 µM Cu2+: 0.5–5.0 µM Hg2+: 0.24–7.5 µM | [30] |
| AuNPs modified GCE | − | −0.78 | −0.56 | 0.00 | 0.25 | Double peaks | Cd2+: 0.3–1.4 µM Pb2+: 0.3–1.4 µM Cu2+: 0.3–1.4 µM Hg2+: 0.3–1.4 µM | [32] |
| GCE | −1.06 | −0.73 | −0.47 | −0.04 | – | Epeak is pH dependent and electrode can only be used for one scan | Zn2+: 1.530–6.12 µM Cd2+: 0.445–4.45 µM Pb2+: 0.242–2.42 µM Cu2+: 0.787–7.87 µM | [33] |
| Amino Acid modified GCE | −1.1 | −0.7 | – | −0.1 | 0.3 | Well-resolved peaks | Zn2+: 5–100 nM Cd2+: 5–100 nM Cu2+: 5–100 nM Hg2+: 5–100 nM | [34] |
| GA-MOF modified GCE | – | −0.82 | −0.61 | −0.25 | 0.10 | Epeak shifts with increasing concentration | Cd2+: 0.01–1.5 µM Pb2+: 0.001–2 µM Cu2+: 0.01–1.6 µM Hg2+: 0.001–2.2 µM | [35] |
| SnO2/rGO modified GCE | – | −0.77 | −0.58 | −0.11 | 0.24 | Peak shoulder for Pb(II) | Cd2+: 0.3–1.2 µM Pb2+: 0.3–1.2 µM Cu2+: 0.3–1.2 µM Hg2+: 0.3–1.2 µM | [29] |
| IL dopped with Mg(II)/Al(III) LDHs modified GCE | – | −0.77 | −0.54 | −0.04 | 0.30 | Epeak shifts with increasing concentration. Peak shoulder for Pb(II) | Cd2+: 4.45–177.94 nM Pb2+: 0.24–96.60 nM Cu2+: 0.79–314.96 nM Hg2+: 2.49–99.70 nM | [36] |
| Stainless steel | – | −0.70 | −0.41 | −0.05 | 0.30 | Epeak shifts with increasing concentration. Peak shoulder for Pb(II) | Cd2+: 0.5–5 µM Pb2+: 0.075–5 µM Cu2+: 0.075–5 µM Hg2+: 0.1–5 µM | [37] |
| rGO/SnO2/PPy modified GCE | – | −0.78 | −0.58 | −0.11 | 0.20 | Well-resolved peaks | Cd2+: 0.5–3 µM Pb2+: 0.5–3 µM Cu2+: 0.5–3 µM Hg2+: 0.5–3 µM | [38] |
| P1,2-DAAQ modified GCE | – | −0.81 | −0.59 | −0.23 | 0.07 | Epeak shifts with increasing concentration. Peak shoulder for Pb(II) | Cd2+: 0–1.07 µM Pb2+: 0–0.58 µM Cu2+: 0–1.89 µM Hg2+: 0–0.60 µM | [39] |
| Metallophthalocyanine modified GCE | – | −0.75 | −0.47 | −0.16 | 0.06 | Epeak shifts with increasing concentration | Cu2+: 0–0.1 mM Cd2+: 0–0.1 mM Pb2+: 0–0.1 mM Hg2+: 0–0.1 mM | [40] |
| FGO modified GCE | – | −0.7 | −0.5 | 0.0 | 0.3 | Peak shoulder for Pb(II). Epeak shifts with increasing concentration | Cd2+: 1.0–6.0 µM Pb2+: 1.0–6.0 µM Cu2+: 1.0–6.0 µM Hg2+: 1.0–6.0 µM | [41] |
| AgNps/rGO modified magnetic GCE | – | −0.74 | −0.56 | −0.05 | 0.32 | Peak shoulder for Pb(II) | Cd2+: 0.05–1.5 µM Pb2+: 0.05–1.5 µM Cu2+: 0.05–1.5 µM Hg2+: 0.05–1.5 µM | [28] |
| rGO/NiWO4 modified CPE | – | −0.77 | −0.58 | −0.11 | 0.24 | Epeak shifts with increasing concentration | Cd2+: 0.2–1.0 µM Pb2+: 0.2–1.0 µM Cu2+: 0.2–1.0 µM Hg2+: 0.2–1.0 µM | [42] |
| HAP-Nafion modified GCE | – | −0.70 | −0.50 | −0.13 | 0.12 | Epeak shifts with increasing concentration | Cd2+: 3.0–10 µM Pb2+: 3.0–10 µM Cu2+: 3.0–10 µM Hg2+: 0.1–1.0 µM | [43] |
| GO/CeO2 modified GCE | – | −0.76 | −0.54 | −0.06 | 0.31 | Double peaks | Cd2+: 0.2–2.5 µM Pb2+: 0.2–2.5 µM Cu2+: 0.2–2.5 µM Hg2+: 0.2–2.5 µM | [44] |
| Cys/rGO modified GCE | – | −0.77 | −0.52 | −0.05 | 0.29 | Epeak shifts with increasing concentration. Peak shoulder for Pb(II) | Cu2+: 0.4–2.0 µM Cd2+: 0.4–2.0 µM Pb2+: 0.4–1.2 µM Hg2+: 0.4–2.0 µM | [45] |
| ASV | ICP-AES (ppm) | Ext-Int (%) | ICP-Ext (%) | ICP-Add (%) | ||
|---|---|---|---|---|---|---|
| Ext. Calibration (ppm) | Standard Addition (ppm) | |||||
| Zn2+ | 8.8 ± 0.3 | 13.3 ± 0.5 | 12.7 ± 0.2 | 33.8 | 30.7 | 4.7 |
| Pb2+ | 11.3 ± 0.3 | 18.3 ± 0.2 | 17.2 ± 0.1 | 38.3 | 34.3 | 6.4 |
| Cu2+ | 15.1 ± 0.1 | 16.1 ± 0.5 | 14.6 ± 0.2 | 6.2 | 3.4 | 10.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, K.; Pérez-Ràfols, C.; Marchoud, A.; Cuartero, M.; Crespo, G.A. Anodic Stripping Voltammetry with the Hanging Mercury Drop Electrode for Trace Metal Detection in Soil Samples. Chemosensors 2021, 9, 107. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9050107
Xu K, Pérez-Ràfols C, Marchoud A, Cuartero M, Crespo GA. Anodic Stripping Voltammetry with the Hanging Mercury Drop Electrode for Trace Metal Detection in Soil Samples. Chemosensors. 2021; 9(5):107. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9050107
Chicago/Turabian StyleXu, Kequan, Clara Pérez-Ràfols, Amine Marchoud, María Cuartero, and Gastón A. Crespo. 2021. "Anodic Stripping Voltammetry with the Hanging Mercury Drop Electrode for Trace Metal Detection in Soil Samples" Chemosensors 9, no. 5: 107. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9050107