
Small Hero with Great Powers: Vaccinia Virus E3 Protein and Evasion of the Type I IFN Response

Abstract
:1. Introduction
2. Early Studies on Poxviruses and the Interferon System
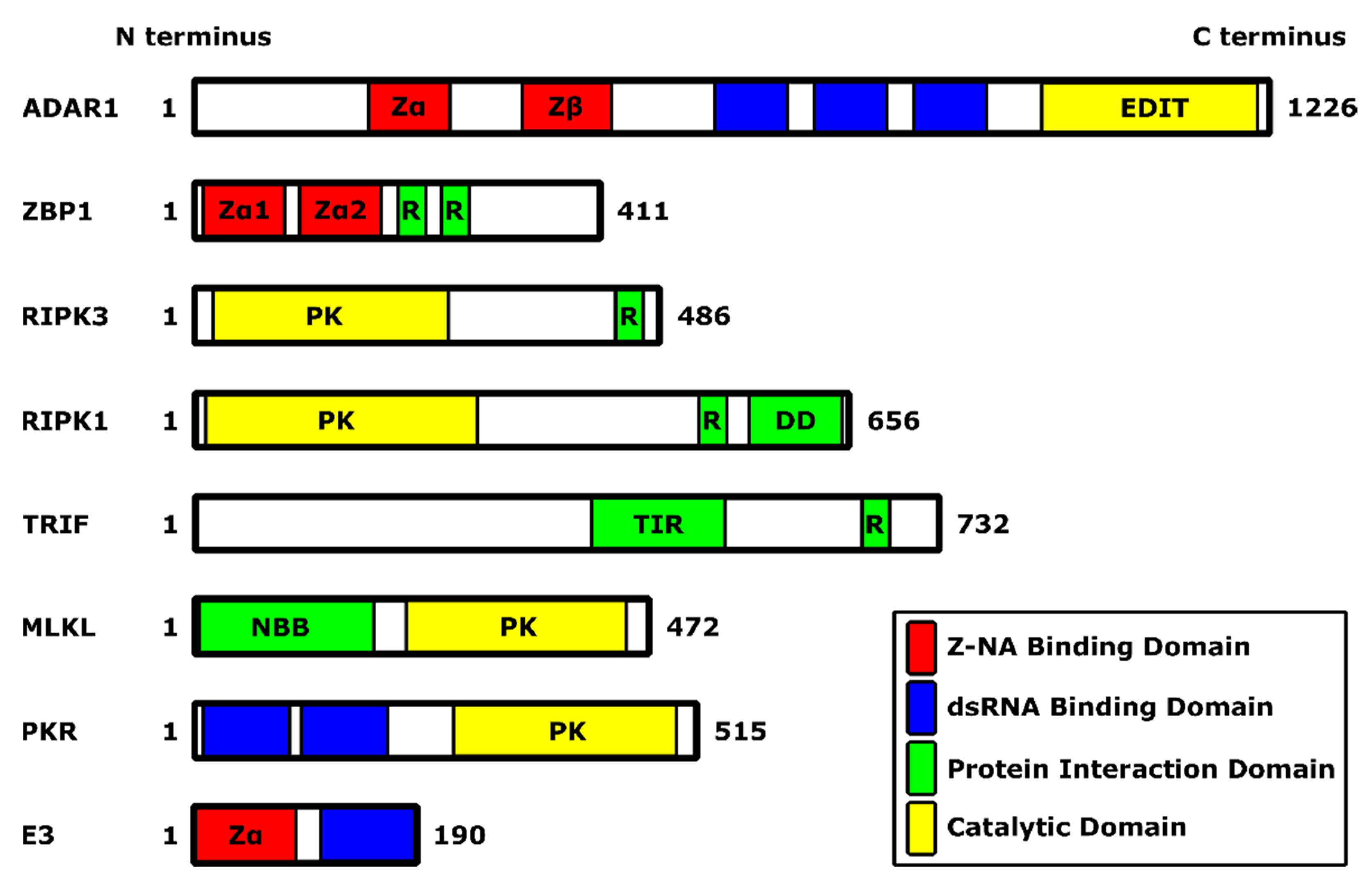
2.1. Discovery of E3 Protein
2.2. C Terminus of E3 Contains a dsRNA-Binding Domain
3. E3 Protein Function in Viral Pathogenicity: Studies In Vivo
3.1. E3 Protein Is VACV Virulence Factor
3.2. E3 Protein Is a Type I IFN Antagonist
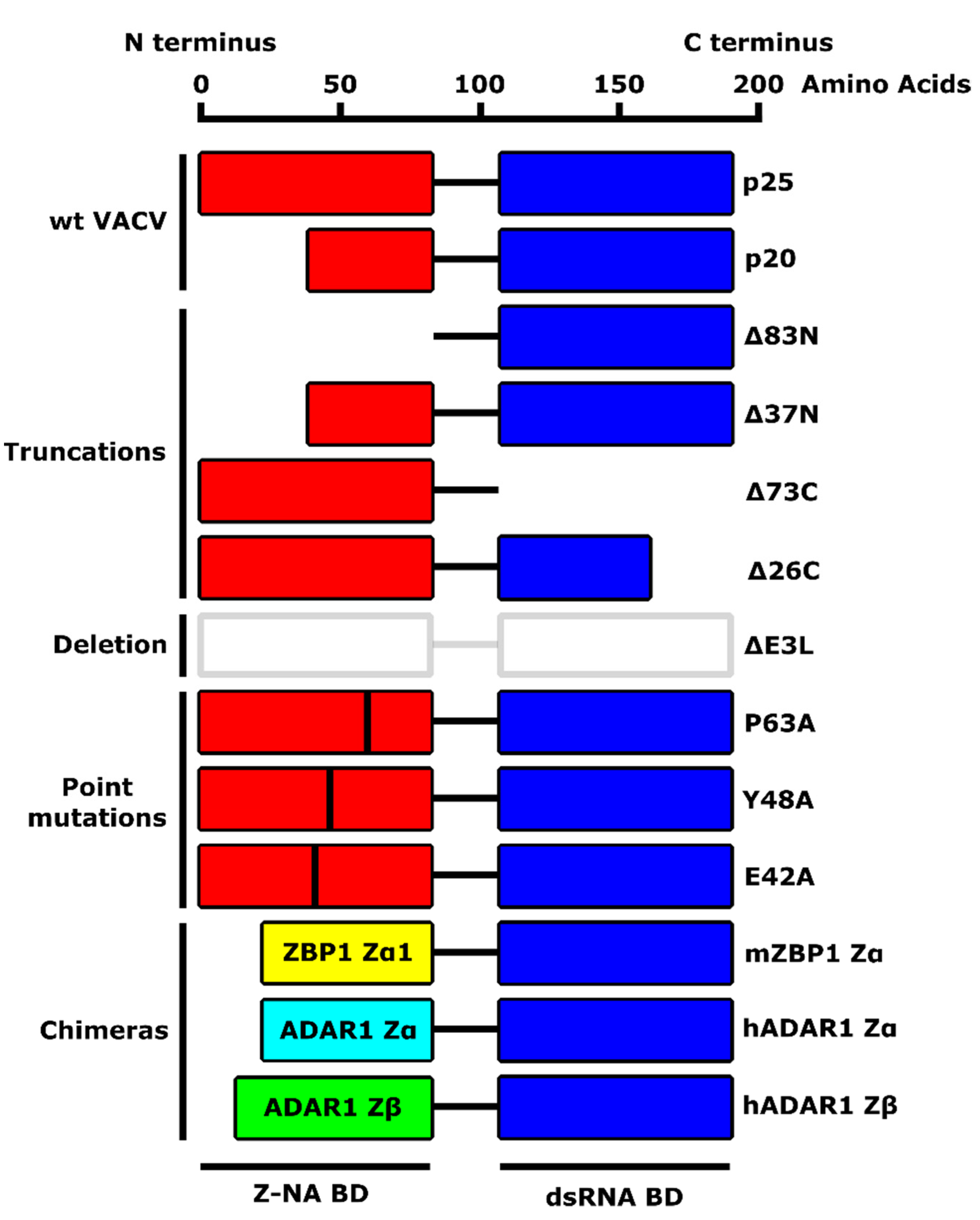
4. E3 Protein Binds to Z-Form Nucleic Acids
4.1. Discovery of Z-DNA
4.2. The N Terminus of E3 Is a ZBD and Binding to a Z-NA Is Required for VACV Pathogenesis
5. Zα Domain of E3 Plays a Role in Inhibition of Necroptosis
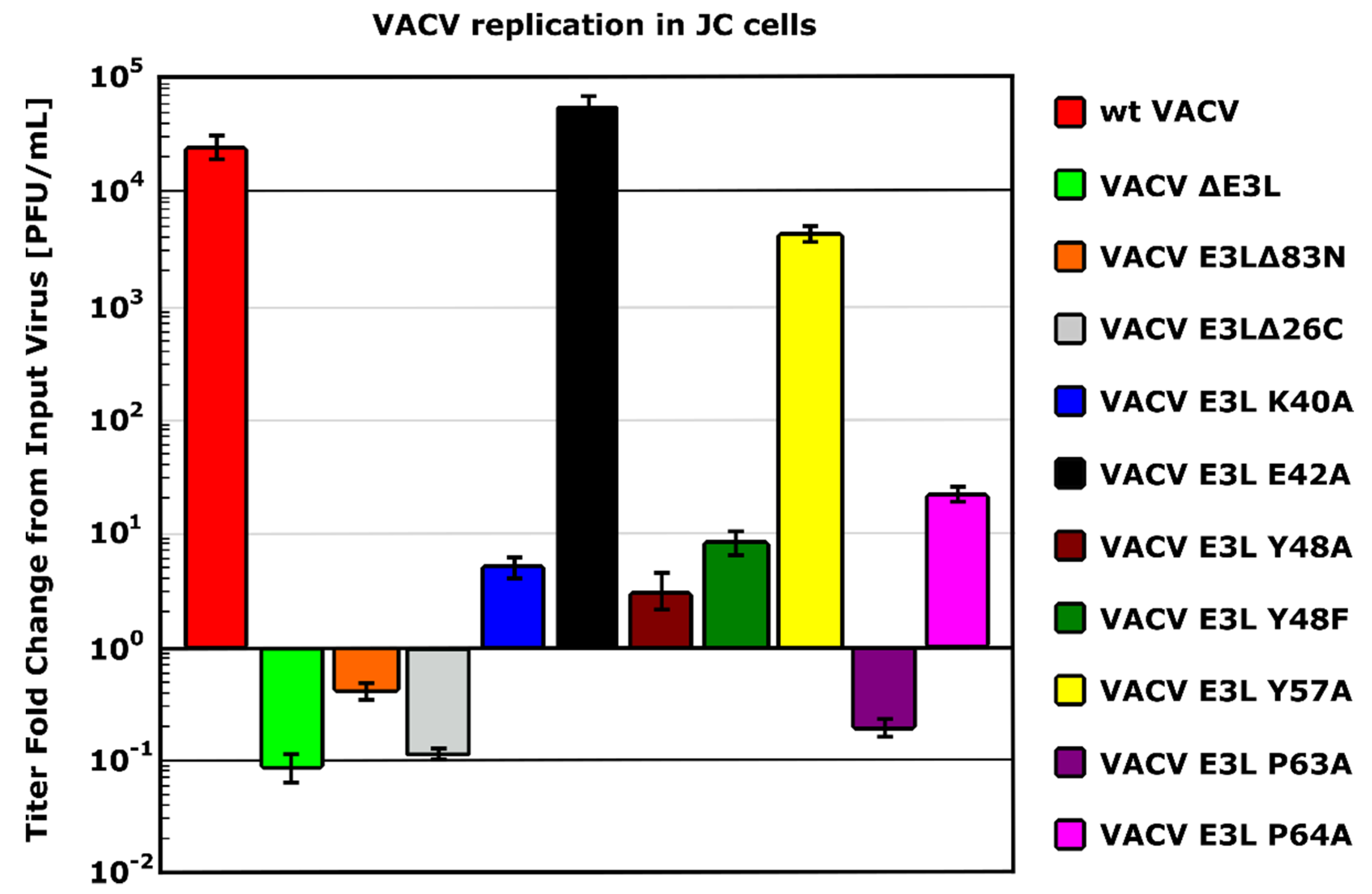
5.1. N Terminus of E3 Is Required for Full PKR Inhibition in 129 MEFs
5.2. Binding to Z-NAs by E3 Is Essential for VACV IFN Resistance
5.3. Necroptosis Inhibition Is Conferred by N Terminus of E3
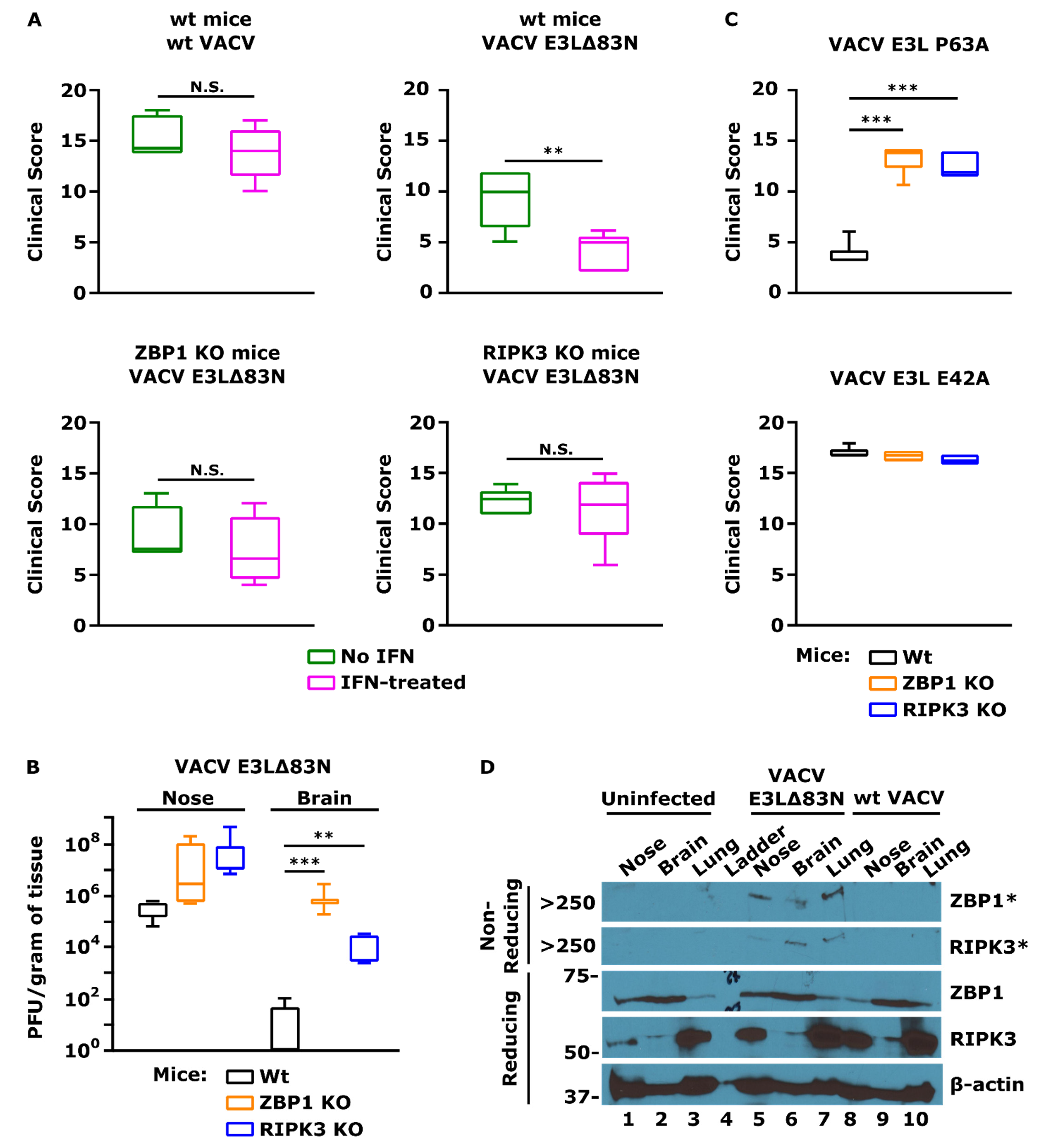
5.4. ZBP1 Mediates Necroptos Is Triggered by a VACV N-Terminal Mutant of E3
5.5. The ZBD of E3 Interacts with Z-RNA to Inhibit Necroptosis
6. Role of the ZBP1/RIPK3/MLKL Axis in Pathogenesis In Vivo
7. ZBP1 Binding of Z-Form NAs Plays a Role in Pathogenesis in Other Viruses
7.1. Cytomegaloviruses
7.2. Herpes Simplex Virus 1
7.3. Influenza A Virus
8. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haller, S.L.; Peng, C.; McFadden, G.; Rothenburg, S. Poxviruses and the evolution of host range and virulence. Infect. Genet. Evol. 2013, 21, 15–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFadden, G. Poxvirus tropism. Nat. Rev. Genet. 2005, 3, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.I.O.; De Oliveira, J.S.; Kroon, E.G.; Trindade, G.D.S.; Drumond, B.P. Here, There, and Everywhere: The Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses. Viruses 2020, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxvirus DNA Replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010199. [Google Scholar] [CrossRef] [Green Version]
- Gubser, C.; Hue, S.; Kellam, P.; Smith, G.L. Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 2004, 85, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Benfield, C.; de Motes, C.M.; Mazzon, M.; Ember, S.W.J.; Ferguson, B.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S. The Smallpox Eradication Game. Public Choice 2006, 130, 179–207. [Google Scholar] [CrossRef]
- Durski, K.N.; Mccollum, A.M.; Nakazawa, Y.; Petersen, B.W.; Reynolds, M.; Briand, S.; Djingarey, M.H.; Olson, V.; Damon, I.K.; Khalakdina, A. Emergence of Monkeypox—West and Central Africa, 1970–2017. MMWR. Morb. Mortal. Wkly. Rep. 2018, 67, 306–310. [Google Scholar] [CrossRef]
- Whitaker-Dowling, P.; Youngner, J.S. Vaccinia-mediated rescue of encephalomyocarditis virus from the inhibitory effects of interferon. Virology 1986, 152, 50–57. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.-Y.; He, X.-B.; Jia, H.-J.; Chen, G.-H.; Jin, Q.-W.; Long, Z.-L.; Jing, Z.-Z. The cGas–Sting Signaling Pathway Is Required for the Innate Immune Response Against Ectromelia Virus. Front. Immunol. 2018, 9, 1297. [Google Scholar] [CrossRef] [Green Version]
- Delaloye, J.; Roger, T.; Steiner-Tardivel, Q.-G.; LE Roy, D.; Reymond, M.K.; Akira, S.; Petrilli, V.; Gomez, C.E.; Perdiguero, B.; Tschopp, J.; et al. Innate Immune Sensing of Modified Vaccinia Virus Ankara (MVA) Is Mediated by TLR2-TLR6, MDA-5 and the NALP3 Inflammasome. PLOS Pathog. 2009, 5, e1000480. [Google Scholar] [CrossRef]
- Dempsey, A.; Keating, S.E.; Carty, M.; Bowie, A.G. Poxviral protein E3–altered cytokine production reveals that DExD/H-box helicase 9 controls Toll-like receptor–stimulated immune responses. J. Biol. Chem. 2018, 293, 14989–15001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Jesr, M.; Teir, M.; De Motes, C.M. Vaccinia Virus Activation and Antagonism of Cytosolic DNA Sensing. Front. Immunol. 2020, 11, 568412. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler, H.; Cotsmire, S.; Zhang, T.; Balachandran, S.; Upton, J.W.; Langland, J.; Kalman, D.; Jacobs, B.L.; Mocarski, E.S. Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe 2021, 29, 1266–1276.e5. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, B.; Benfield, C.; Ren, H.; Lee, V.; Frazer, G.L.; Strnadova, P.; Sumner, R.P.; Smith, G.L. Vaccinia virus protein N2 is a nuclear IRF3 inhibitor that promotes virulence. J. Gen. Virol. 2013, 94, 2070–2081. [Google Scholar] [CrossRef] [PubMed]
- Neidel, S.; Ren, H.; Torres, A.A.; Smith, G.L. NF-κB activation is a turn on for vaccinia virus phosphoprotein A49 to turn off NF-κB activation. Proc. Natl. Acad. Sci. USA 2019, 116, 5699–5704. [Google Scholar] [CrossRef] [Green Version]
- Stuart, J.H.; Sumner, R.P.; Lu, Y.; Snowden, J.S.; Smith, G.L. Vaccinia Virus Protein C6 Inhibits Type I IFN Signalling in the Nucleus and Binds to the Transactivation Domain of STAT2. PLOS Pathog. 2016, 12, e1005955. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.; McCaughan, C.; Wise, L.M.; Mercer, A.A.; Fleming, S.B. Orf virus inhibits interferon stimulated gene expression and modulates the JAK/STAT signalling pathway. Virus Res. 2015, 208, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFκB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef] [Green Version]
- Adomavicius, T.; Guaita, M.; Zhou, Y.; Jennings, M.D.; Latif, Z.; Roseman, A.M.; Pavitt, G.D. The structural basis of translational control by eIF2 phosphorylation. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kerr, I.M.; Brown, R.E.; Hovanessian, A.G. Nature of inhibitor of cell-free protein synthesis formed in response to interferon and double-stranded RNA. Nature 1977, 268, 540–542. [Google Scholar] [CrossRef]
- Langland, J.O.; Jacobs, B.L. Inhibition of PKR by vaccinia virus: Role of the N- and C-terminal domains of E3L. Virology 2004, 324, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, W.K.; Hovanessian, A.; Brown, R.E.; Clemens, M.J.; Kerr, I.M. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature 1976, 264, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G.; Brown, R.E.; Kerr, I.M. Synthesis of low molecular weight inhibitor of protein synthesis with enzyme from interferon-treated cells. Nature 1977, 268, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G.; Justesen, J. The human 2′-5′oligoadenylate synthetase family: Unique interferon-inducible enzymes catalyzing 2′-5′ instead of 3′-5′ phosphodiester bond formation. Biochimie 2007, 89, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.; Kerr, S.; Roberts, W.K.; Brown, R.E.; Kerr, I.M. Novel 2′,5′-oligoadenylates synthesized in interferon-treated, vaccinia virus-infected cells. J. Virol. 1985, 56, 1041–1044. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Choi, M.K.; Ban, T.; Yanai, H.; Negishi, H.; Lu, Y.; Tamura, T.; Takaoka, A.; Nishikura, K.; Taniguchi, T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. USA 2008, 105, 5477–5482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, W.J.; Upton, J.; Mocarski, E.S. Receptor-Interacting Protein Homotypic Interaction Motif-Dependent Control of NF-κB Activation via the DNA-Dependent Activator of IFN Regulatory Factors. J. Immunol. 2008, 181, 6427–6434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, T.; Behlke, J.; Lowenhaupt, K.; Heinemann, U.; Rich, A. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat. Genet. 2001, 8, 761–765. [Google Scholar] [CrossRef]
- Shin, S.-I.; Ham, S.; Park, J.; Seo, S.H.; Lim, C.H.; Jeon, H.; Huh, J.; Roh, T.-Y. Z-DNA-forming sites identified by ChIP-Seq are associated with actively transcribed regions in the human genome. DNA Res. 2016, 23, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Wittig, B.; Dorbic, T.; Rich, A. Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei. Proc. Natl. Acad. Sci. USA 1991, 88, 2259–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deigendesch, N.; Koch-Nolte, F.; Rothenburg, S. ZBP1 subcellular localization and association with stress granules is controlled by its Z-DNA binding domains. Nucleic Acids Res. 2006, 34, 5007–5020. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.K.; Weissbach, R.; Ronson, G.E.; Scadden, A.D.J. Proteins that contain a functional Z-DNA-binding domain localize to cytoplasmic stress granules. Nucleic Acids Res. 2013, 41, 9786–9799. [Google Scholar] [CrossRef] [Green Version]
- Weissbach, R.; Scadden, A. Tudor-SN and ADAR1 are components of cytoplasmic stress granules. RNA 2012, 18, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Koehler, H.; Cotsmire, S.; Langland, J.; Kibler, K.V.; Kalman, D.; Upton, J.W.; Mocarski, E.S.; Jacobs, B.L. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc. Natl. Acad. Sci. USA 2017, 114, 11506–11511. [Google Scholar] [CrossRef] [Green Version]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.S.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.-D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef] [Green Version]
- Maelfait, J.; Liverpool, L.; Bridgeman, A.; Ragan, K.B.; Upton, J.W.; Rehwinkel, J. Sensing of viral and endogenous RNA by ZBP 1/ DAI induces necroptosis. EMBO J. 2017, 36, 2529–2543. [Google Scholar] [CrossRef]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.; Benitez, A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 Complexes with RIP3 to Mediate Virus-Induced Programmed Necrosis that Is Targeted by Murine Cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Williams, C.M. Programmed cell death—III. Neural control of the breakdown of the intersegmental muscles of silkmoths. J. Insect Physiol. 1965, 11, 601–610. [Google Scholar] [CrossRef]
- Peter, M.E.; Krammer, P.H. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003, 10, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies Within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Bergsbaken, T.; Fink, S.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Genet. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal. Transduct. Target. Ther. 2021, 6, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Aguileta, M.A.; Goossens, V.J.; Dubuisson, C.; Grootjans, S.; Dejardin, E.; Vandenabeele, P.; Bertrand, M.J.M. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013, 20, 1381–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, D.; Weinlich, R.; Brown, S.L.; Guy, C.T.; Fitzgerald, P.J.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2015, 23, 76–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, Y.; Liu, S.; Yu, X.; Li, L.; Shi, C.; He, W.; Li, J.; Xu, L.; Hu, Z.; et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. USA 2014, 111, 15438–15443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.-N.; Yang, Z.-H.; Wang, X.-K.; Zhang, Y.; Wan, H.; Song, Y.; Chen, X.; Shao, J.; Han, J. Distinct roles of RIP1–RIP3 hetero- and RIP3–RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014, 21, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Najafov, A.; Mookhtiar, A.K.; Luu, H.S.; Ordureau, A.; Pan, H.; Amin, P.; Li, Y.; Lu, Q.; Yuan, J. TAM Kinases Promote Necroptosis by Regulating Oligomerization of MLKL. Mol. Cell 2019, 75, 457–468.e4. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Quade, B.; Wang, H.; Sun, L.; Wang, X.; Rizo, J. A Plug Release Mechanism for Membrane Permeation by MLKL. Structure 2014, 22, 1489–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Bruneau, R.; Brennan, G.; Rothenburg, S. Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion. Biomedicines 2021, 9, 765. [Google Scholar] [CrossRef]
- Thacore, H.R.; Youngner, J.S. Rescue of vesicular stomatitis virus from interferon-induced resistance by superinfection with vaccinia virus. Virology 1973, 56, 505–511. [Google Scholar] [CrossRef]
- Paez, E.; Esteban, M. Resistance of vaccinia virus to interferon is related to an interference phenomenon between the virus and the interferon system. Virology 1984, 134, 12–28. [Google Scholar] [CrossRef]
- Akkaraju, G.R.; Whitaker-Dowling, P.; Youngner, J.S.; Jagus, R. Vaccinia specific kinase inhibitory factor prevents trans-lational inhibition by double-stranded RNA in rabbit reticulocyte lysate. J. Biol. Chem. 1989, 264, 10321–10325. [Google Scholar] [CrossRef]
- Watson, J.C.; Chang, H.-W.; Jacobs, B.L. Characterization of a vaccinia virus-encoded double-stranded RNA-binding protein that may be involved in inhibition of the double-stranded rna-dependent protein kinase. Virology 1991, 185, 206–216. [Google Scholar] [CrossRef]
- Chang, H.W.; Watson, J.C.; Jacobs, B.L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 4825–4829. [Google Scholar] [CrossRef] [Green Version]
- Beattie, E.; Kauffman, E.B.; Martinez, H.; Perkus, M.E.; Jacobs, B.L.; Paoletti, E.; Tartaglia, J. Host-range restriction of vaccinia virus E3L-specific deletion mutants. Virus Genes 1996, 12, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.; Paoletti, E.; Tartaglia, J. Distinct Patterns of IFN Sensitivity Observed in Cells Infected with Vaccinia K3L- and E3L- Mutant Viruses. Virology 1995, 210, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Shors, S.T.; Beattie, E.; Paoletti, E.; Tartaglia, J.; Jacobs, B.L. Role of the Vaccinia Virus E3L and K3L Gene Products in Rescue of VSV and EMCV from the Effects of IFN-α. J. Interf. Cytokine Res. 1998, 18, 721–729. [Google Scholar] [CrossRef]
- Carroll, K.; Elroy-Stein, O.; Moss, B.; Jagus, R. Recombinant vaccinia virus K3L gene product prevents activation of dou-ble-stranded RNA-dependent, initiation factor 2 alpha-specific protein kinase. J. Biol. Chem. 1993, 268, 12837–12842. [Google Scholar] [CrossRef]
- Davies, M.V.; Elroy-Stein, O.; Jagus, R.; Moss, B.; Kaufman, R.J. The vaccinia virus K3L gene product potentiates translation by inhibiting double-stranded-RNA-activated protein kinase and phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J. Virol. 1992, 66, 1943–1950. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-W.; Jacobs, B.L. Identification of a Conserved Motif That Is Necessary for Binding of the Vaccinia Virus E3L Gene Products to Double-Stranded RNA. Virology 1993, 194, 537–547. [Google Scholar] [CrossRef]
- Chang, H.W.; Uribe, L.H.; Jacobs, B.L. Rescue of vaccinia virus lacking the E3L gene by mutants of E3L. J. Virol. 1995, 69, 6605–6608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shors, T.; Kibler, K.V.; Perkins, K.B.; Seidler-Wulff, R.; Banaszak, M.P.; Jacobs, B.L. Complementation of Vaccinia Virus Deleted of the E3L Gene by Mutants of E3L. Virology 1997, 239, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-G.; Muralinath, M.; Brandt, T.; Pearcy, M.; Hauns, K.; Lowenhaupt, K.; Jacobs, B.L.; Rich, A. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 6974–6979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, T.A.; Jacobs, B.L. Both Carboxy- and Amino-Terminal Domains of the Vaccinia Virus Interferon Resistance Gene, E3L, Are Required for Pathogenesis in a Mouse Model. J. Virol. 2001, 75, 850–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.D.; Jacobs, B.L. The Amino Terminus of the Vaccinia Virus E3 Protein Is Necessary To Inhibit the Interferon Response. J. Virol. 2012, 86, 5895–5904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Dai, P.; Wang, W.; Santagostino, S.; Serna-Tamayo, C.; Meyer, C.; Wang, Y.; Li, T.; Chen, Z.J.; Colonna, M.; et al. The cytosolic DNA- and RNA-sensing pathways play important and non-redundant roles in host defense against vac-cinia infection. J. Immunol. 2017, 198, 148.116. [Google Scholar]
- Herbert, A.; Lowenhaupt, K.; Spitzner, J.; Rich, A. Chicken double-stranded RNA adenosine deaminase has apparent specificity for Z-DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 7550–7554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, B.L.; Weintraub, H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef]
- Chung, H.; Calis, J.J.; Wu, X.; Sun, T.; Yu, Y.; Sarbanes, S.L.; Thi, V.L.D.; Shilvock, A.; Hoffmann, H.-H.; Rosenberg, B.; et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 2018, 172, 811–824. [Google Scholar] [CrossRef] [Green Version]
- George, C.X.; Samuel, C.E. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl. Acad. Sci. USA 1999, 96, 4621–4626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, A.; Alfken, J.; Kim, Y.-G.; Mian, I.S.; Nishikura, K.; Rich, A. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc. Natl. Acad. Sci. USA 1997, 94, 8421–8426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, T.; Lowenhaupt, K.; Kim, Y.-G.; Li, L.; Brown, B.A.; Herbert, A.; Rich, A. Proteolytic Dissection of Zab, the Z-DNA-binding Domain of Human ADAR1. J. Biol. Chem. 1999, 274, 2899–2906. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Comella, N.; Tognazzi, K.; Brown, L.F.; Dvorak, H.F.; Kocher, O. Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Gene 1999, 240, 157–163. [Google Scholar] [CrossRef]
- Ha, S.C.; Van Quyen, D.; Hwang, H.-Y.; Oh, D.-B.; Brown, B.A.; Lee, S.M.; Park, H.-J.; Ahn, J.-H.; Kim, K.K.; Kim, Y.-G. Biochemical characterization and preliminary X-ray crystallographic study of the domains of human ZBP1 bound to left-handed Z-DNA. Biochim. et Biophys. Acta (BBA)-Proteins Proteom. 2006, 1764, 320–323. [Google Scholar] [CrossRef]
- Schwartz, T.; Rould, M.A.; Lowenhaupt, K.; Herbert, A.; Rich, A. Crystal Structure of the Zα Domain of the Human Editing Enzyme ADAR1 Bound to Left-Handed Z-DNA. Science 1999, 284, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Cotsmire, S. Vaccinia virus’ E3 Protein Inhibits Cellular Recognition of Canonical dsRNA and ZRNA. Doctor of Philoso-phy Dissertation, Arizona State University, Tempe, AZ, USA, 2021. [Google Scholar]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.-L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2013, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.; Szczerba, M.; Bonner, J.; Jacobs, B.L. Monkeypox Virus Inhibits Necroptosis by Blocking TAM Kinase-Mediated MLKL PhosphorylationTempe; Biodesign Center for Immunotherapy, Vaccines and Virotherapy, Arizona State University: Tempe, AZ, USA, 2022; Manuscript in preparation, to be submitted. [Google Scholar]
- Hsu, H.; Huang, J.; Shu, H.-B.; Baichwal, V.; Goeddel, D.V. TNF-Dependent Recruitment of the Protein Kinase RIP to the TNF Receptor-1 Signaling Complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like Receptor 3-mediated Necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Liang, Y.; Zhao, S.; Ding, Y.; Zhuang, Q.; Shi, Q.; Ai, T.; Wu, S.-Q.; Han, J. ZBP1 mediates interferon-induced necroptosis. Cell. Mol. Immunol. 2019, 17, 356–368. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotsmire, S.M.; Szczerba, M.; Jacobs, B.L. Detecting Necroptosis in Virus-Infected Cells; Humana: New York, NY, USA, 2020; pp. 199–216. [Google Scholar] [CrossRef]
- Upton, J.; Kaiser, W.J.; Mocarski, E.S. Cytomegalovirus M45 Cell Death Suppression Requires Receptor-interacting Protein (RIP) Homotypic Interaction Motif (RHIM)-dependent Interaction with RIP1. J. Biol. Chem. 2008, 283, 16966–16970. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.; Kaiser, W.J.; Mocarski, E.S. Virus Inhibition of RIP3-Dependent Necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, H.; Ragan, K.B.; Guo, H.; Gilley, R.P.; Landsteiner, V.J.; Kaiser, W.J.; Upton, J.W. Murine cytomegalovirus IE 3-dependent transcription is required for DAI / ZBP 1-mediated necroptosis. EMBO Rep. 2017, 18, 1429–1441. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, S.-Q.; Liang, Y.; Zhou, X.; Chen, W.; Li, L.; Wu, J.; Chen, C.; Li, J.; Zhong, C.-Q.; et al. RIP1/RIP3 Binding to HSV-1 ICP6 Initiates Necroptosis to Restrict Virus Propagation in Mice. Cell Host Microbe 2015, 17, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes Simplex Virus Suppresses Necroptosis in Human Cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Wu, G.; Shu, Z.; Yu, D.; Nan, N.; Yuan, F.; Liu, X.; Wang, H. ICP6 Prevents RIP1 Activation to Hinder Necroptosis Signaling. Front. Cell Dev. Biol. 2020, 8, 595253. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, Y.; Chen, Q.; Su, C.; Zhang, Z.; Yang, C.; Hu, Z.; Hou, J.; Zhou, J.; Gong, L.; et al. Herpes Simplex Virus 1 (HSV-1) and HSV-2 Mediate Species-Specific Modulations of Programmed Necrosis through the Viral Ribonucleotide Reductase Large Subunit R1. J. Virol. 2016, 90, 1088–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Gilley, R.P.; Fisher, A.; Lane, R.; Landsteiner, V.J.; Ragan, K.B.; Dovey, C.M.; Carette, J.; Upton, J.; Mocarski, E.S.; et al. Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H.; Ingram, J.P.; Rodriguez, D.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Kesavardhana, S.; Kuriakose, T.; Guy, C.S.; Samir, P.; Malireddi, R.; Mishra, A.; Kanneganti, T.-D. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 2017, 214, 2217–2229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Lin, C.-L.G. Oxidative damage to RNA: Mechanisms, consequences, and diseases. Exp. 2010, 67, 1817–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teoh, M.L.T.; Turner, P.V.; Evans, D.H. Tumorigenic Poxviruses Up-Regulate Intracellular Superoxide To Inhibit Apoptosis and Promote Cell Proliferation. J. Virol. 2005, 79, 5799–5811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.; Dhaliwal, J.; Adjibade, P.; Uniacke, J.; Mazroui, R.; Zerges, W. Localized control of oxidized RNA. J. Cell Sci. 2015, 128, 4210–4219. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E3 Protein | dsRNA Binding | PKR Inhibition | Z-NA Binding | Necroptosis Inhibition | Pathogenicity | Reference |
|---|---|---|---|---|---|---|
| wt E3 | yes | yes | yes | yes | high | [16,26,40,72] |
| Δ83N | yes | no | no | no | low | [16,26,40,72] |
| Δ37N | yes | no | no | no | low | [16,26,40,72] |
| Δ73C | no | no | (yes) | yes | (low) | [16] |
| Δ26C | no | no | (yes) | yes | low | [16,72,76] |
| ΔE3L | no | no | (no) | yes | low | [16,26,72,76] |
| P63A | (yes) | yes | no | no | low | [16,75] |
| Y48A | (yes) | yes | no | no | low | [16,75] |
| E42A | (yes) | yes | (yes) | yes | high | [16] |
| mZBP1 Zα | (yes) | (yes) | yes | yes | high | [16,75] |
| hADAR1 Zα | (yes) | (yes) | yes | yes | high | [16,75] |
| hADAR1 Zβ | (yes) | (yes) | no | no | low | [16,75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczerba, M.; Subramanian, S.; Trainor, K.; McCaughan, M.; Kibler, K.V.; Jacobs, B.L. Small Hero with Great Powers: Vaccinia Virus E3 Protein and Evasion of the Type I IFN Response. Biomedicines 2022, 10, 235. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020235
Szczerba M, Subramanian S, Trainor K, McCaughan M, Kibler KV, Jacobs BL. Small Hero with Great Powers: Vaccinia Virus E3 Protein and Evasion of the Type I IFN Response. Biomedicines. 2022; 10(2):235. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020235
Chicago/Turabian StyleSzczerba, Mateusz, Sambhavi Subramanian, Kelly Trainor, Megan McCaughan, Karen V. Kibler, and Bertram L. Jacobs. 2022. "Small Hero with Great Powers: Vaccinia Virus E3 Protein and Evasion of the Type I IFN Response" Biomedicines 10, no. 2: 235. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020235