
Synergy of Human Platelet-Derived Extracellular Vesicles with Secretome Proteins Promotes Regenerative Functions

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. HPL Preparation
2.2. Isolation of HPL EVs
2.3. EV Quantification by Tunable Resistive Pulse Sensing (TRPS)
2.4. Protein Quantification and Western Blot
2.5. Tandem Mass-Tag (TMT) Proteomics
2.5.1. Chemicals
2.5.2. Sample Preparation
2.5.3. High-Performance Liquid Chromatography Coupled to Mass Spectrometry
2.5.4. Data Evaluation
2.6. Skin-Organoids, Fibrosphere Formation and Immunohistochemistry
2.7. Immunomodulation Assay
2.8. Zeta Potential and Size Measurements by Nanoparticle Tracking Analysis (NTA)
2.9. Super-Resolution Microscopy
2.10. Angiogenesis Assay
2.11. Wound Healing Assay
2.12. Statistical Analysis
3. Results
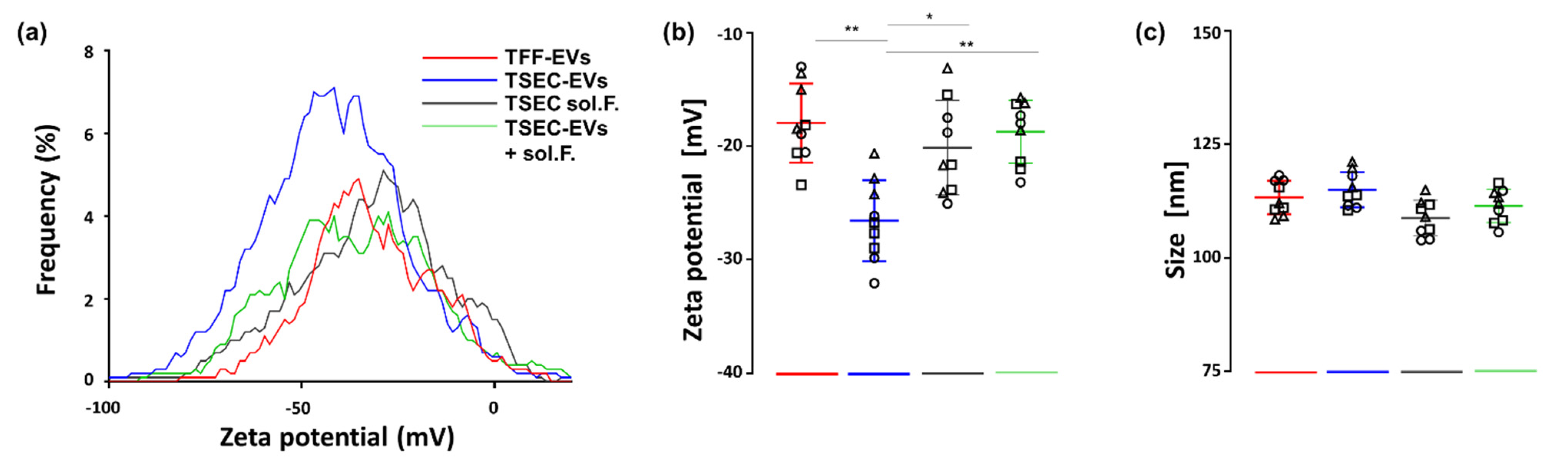
3.1. Purification and Characterization of HPL-Derived EVs
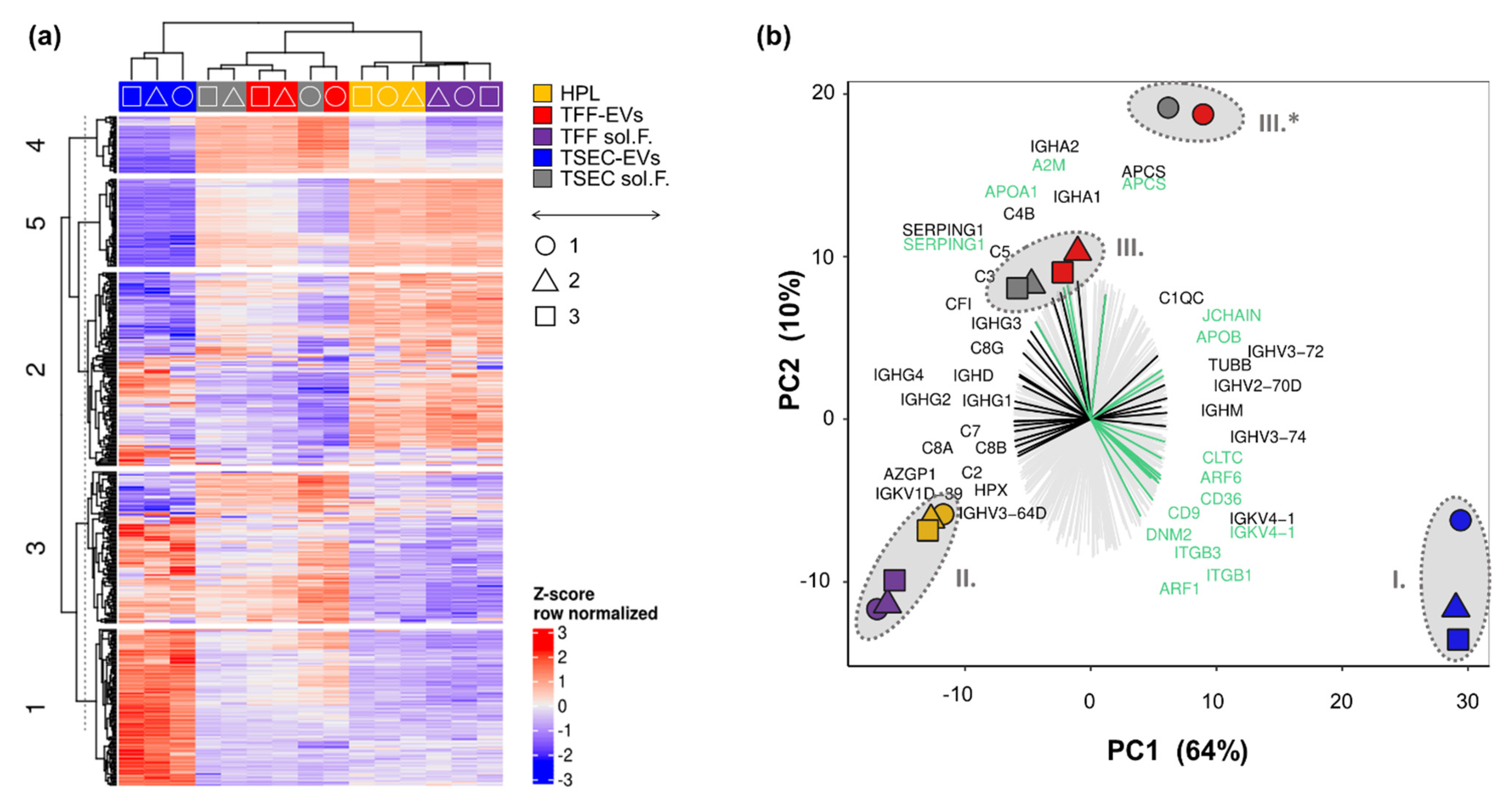
3.2. Proteomic Profiling of HPL-Derived Samples
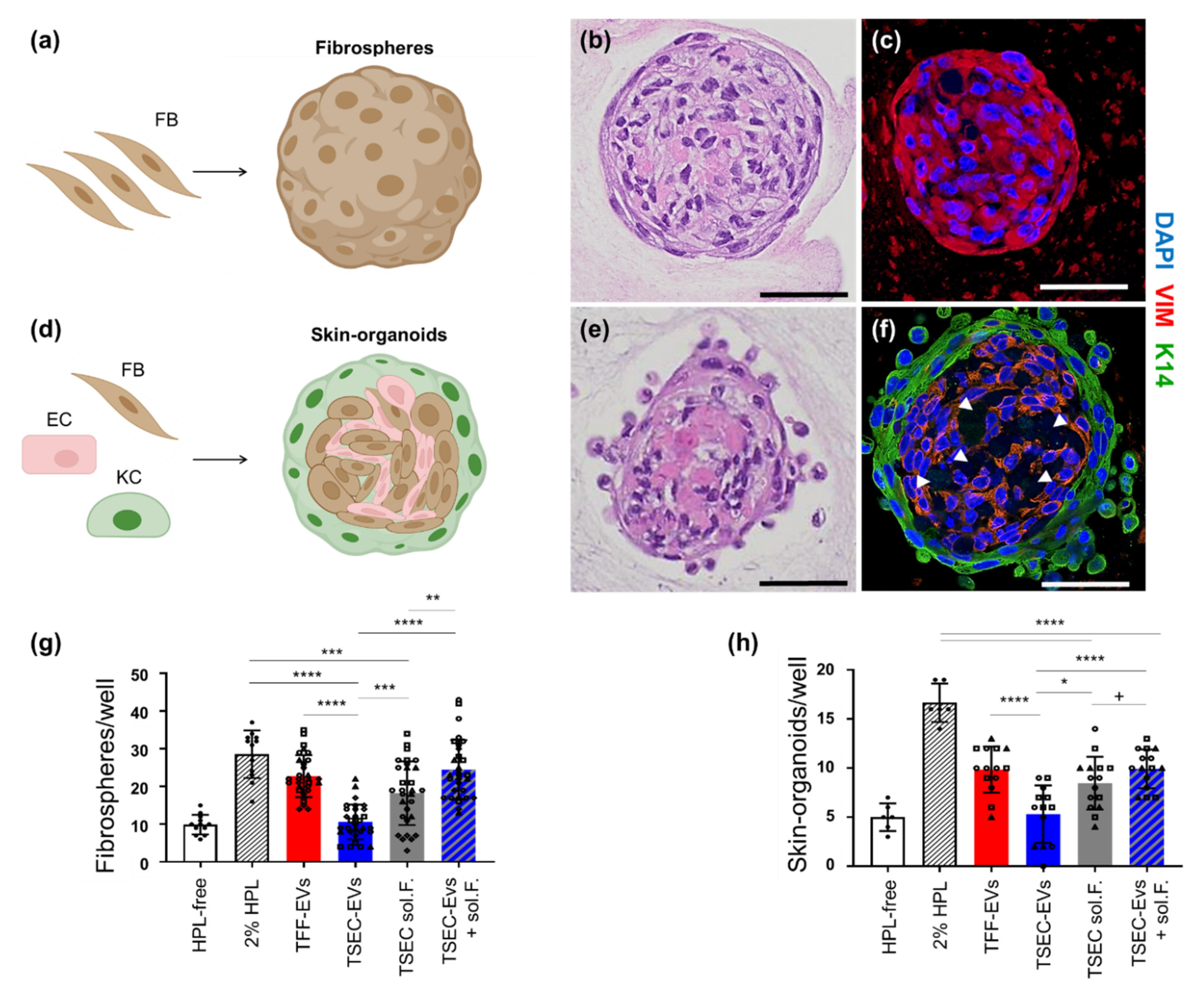
3.3. HPL EVs Support Fibrosphere and Skin-Organoid Formation in the Presence of Platelet Proteins
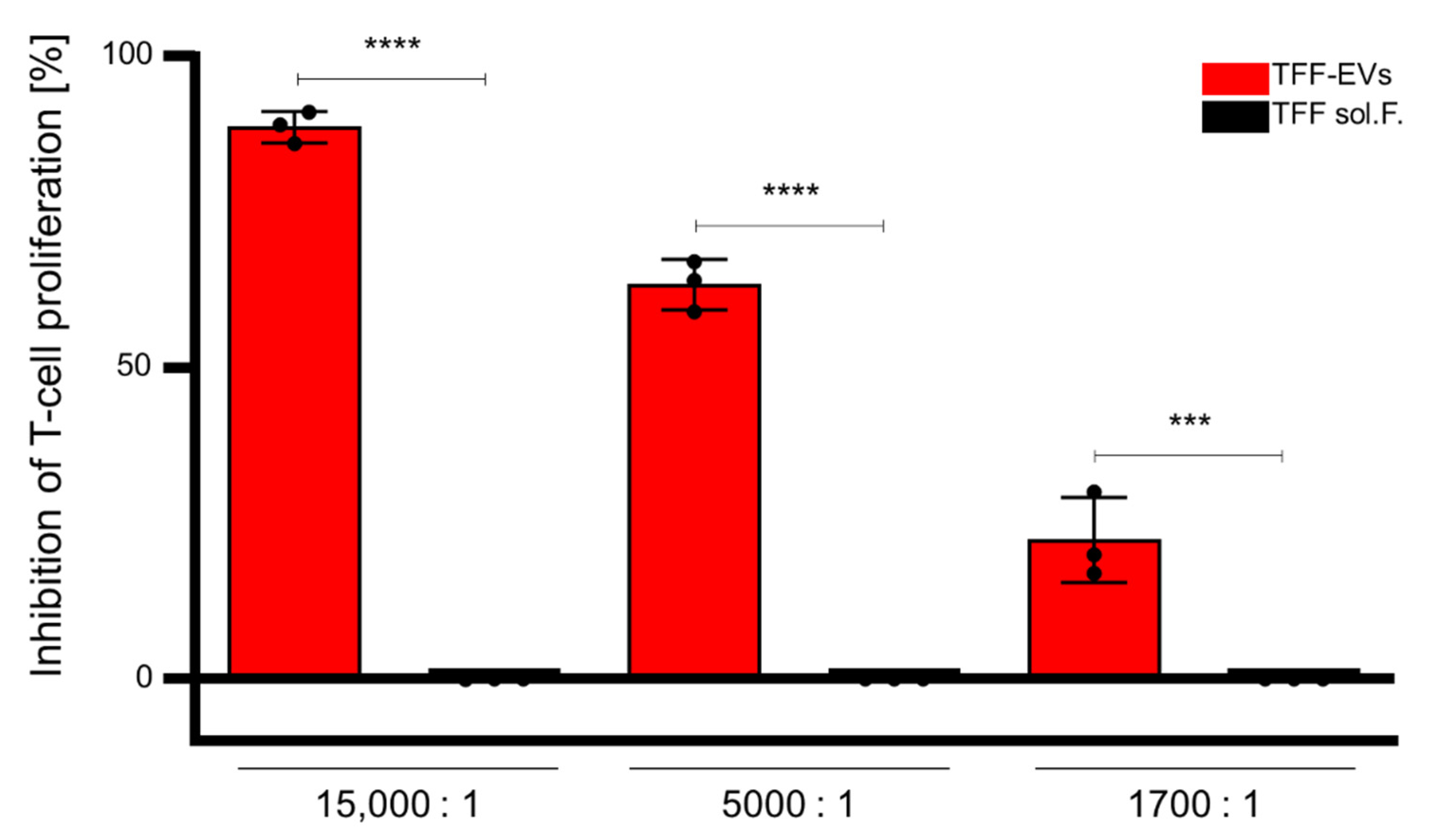
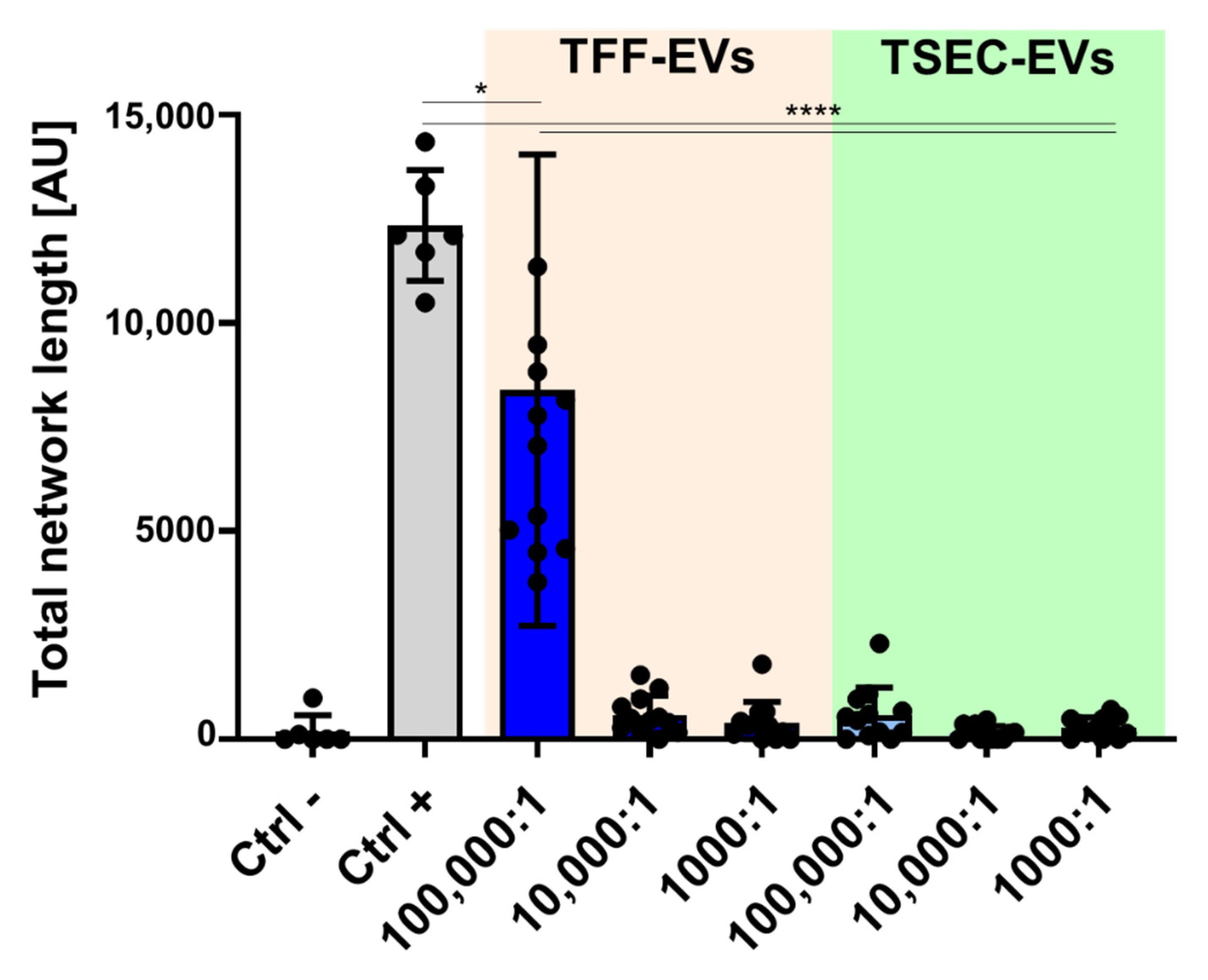
3.4. HPL-EVs Inhibit T-Cell Proliferation and Ameliorate Scratch Wound Healing In Vitro in the Presence of Platelet Proteins
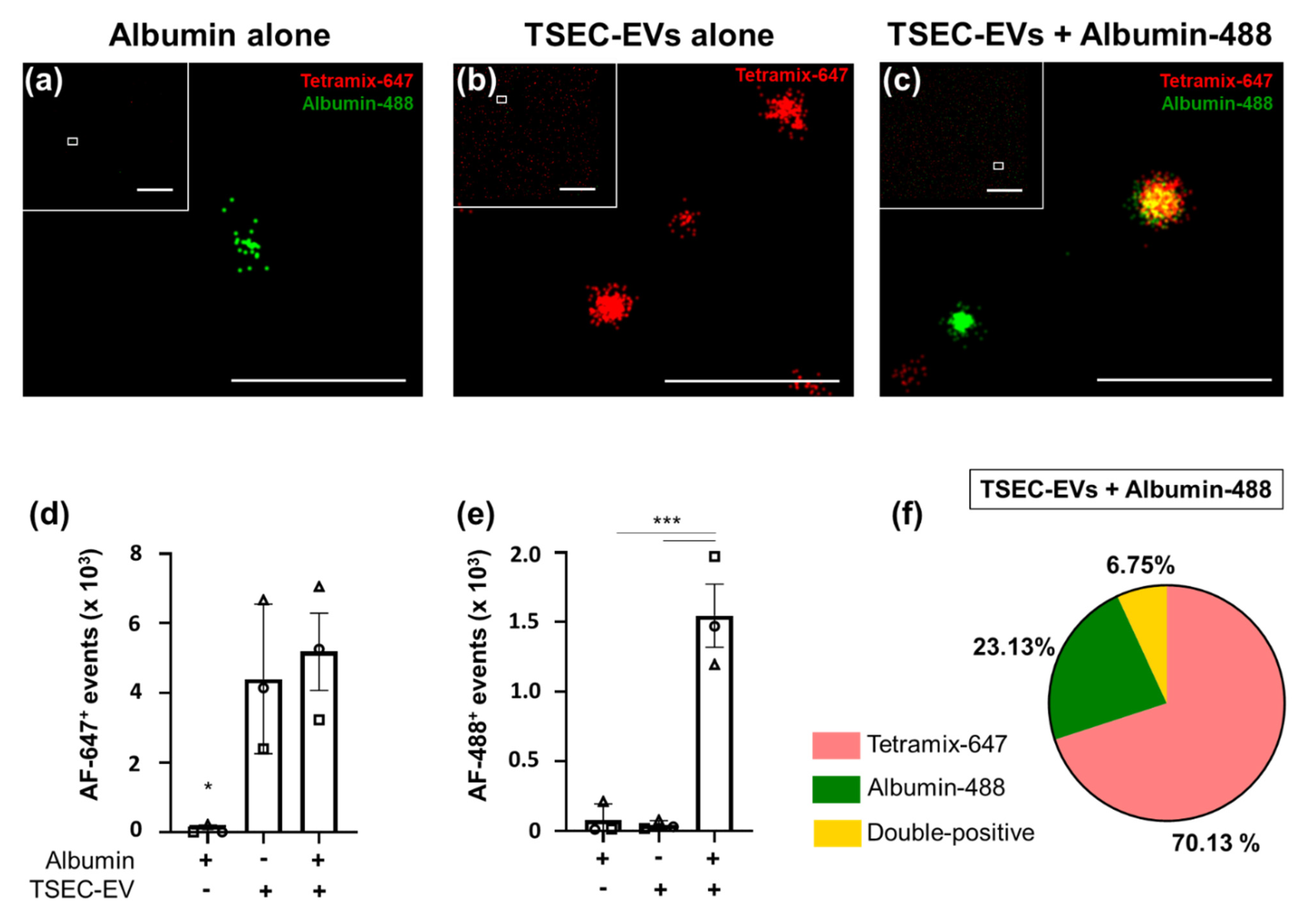
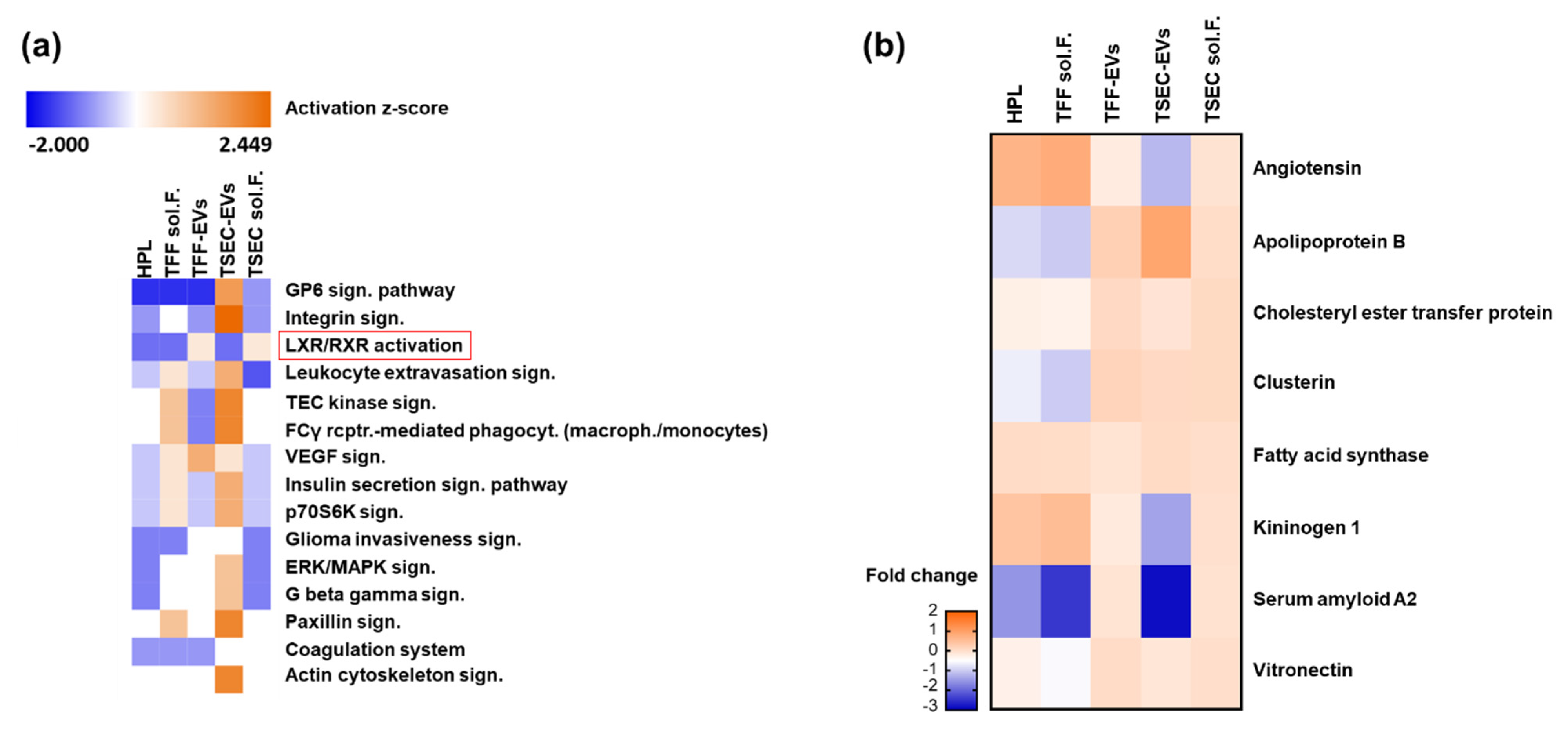
3.5. HPL-EVs Acquire a Protein Corona in a Protein-Rich Environment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nomenclature | ||||
| Use of generic term EV for HPL-EVs | ||||
| Collection and storage | ||||
| Sample information | cell type and origin | 10 expired buffy-coat derived platelet concentrates | ||
| preparation of HPL | platelet concentrate bags, freeze at −30 °C overnight, thawed a 37 °C, pooled in one bag and frozen second time at −30 °C | |||
| HPL preparations | Lysates thawed and centrifuged 4000× g, 16 min aliquoted in 50 mL tubes and frozen at −20 °C | |||
| processed material | 1000 mL (Batch 1); 1000 mL (Batch 2); 1000 mL (Batch 3) | |||
| Aliquots used | 20 × 50 mL tubes per batch | |||
| Processing conditions | dilution medium | α-MEM or RPMI with final concentration of 200 mg/L CaCl2 (clotting) | ||
| time of incubation | 37 °C for 1 h, kept overnight at 4 °C | |||
| harvesting medium | clotted media diluted 1:2 in α-MEM without supplementation | |||
| EV pellets | storage in 0.9% saline + 10 mM HEPES | |||
| EV preparations | storage temperature: −80 °C | |||
| Isolation | ||||
| Differential centrifugation | 3000× g for 10 min, SX4750, Allegra X-15R, Beckman Coulter Diluted 1:2 in α-MEM without supplementation, further filtration through 0.22 µm pore size filter | |||
| Ultrafiltration (TFF) | Tangential flow filtration followed concentration of clotted media, column surface area: 1600 cm2, pore size: 500 kDa | |||
| Size-exclusion chromatography (TSEC) | TFF-EVs were loaded in a qEV2 size-exclusion chromatography column and separated into 25 fractions using 0.22 µm-filtered 0.9% saline + 10mM HEPES as buffer, fractions 7–9 (TSEC-EVs) and 19–21 (TSEC-soluble fractions) | |||
| Characterization | ||||
| Parameter | Unit | Method | ||
| Quantification | Size & concentration | particle number | TFF-EVs—2.17 ± 0.76 x 1012/mL TSEV-EVs—7.20 ± 3.90 x 1011/mL | TRPS |
| particle size (mode) | TFF-EVs—113.3 ± 3.7 nm TSEV-EVs—115.0 ± 3.9 nm | TRPS | ||
| Composition | protein content | TFF-EVs—132.30 ± 16.08 mg/mL TSEC-EVs—0.56 ± 0.50 mg/mL | DC-Protein | |
| protein identification | Enrichment of CD9 and ITGB3 in TSEC-EV EVs fractions | Proteomic | ||
| Identity | trans membrane proteins | CD9 and CD63 | WB | |
| contaminants | HSA and ApoA1 | WB | ||
| Super-resolution microscopy (tetramix—CD9/63/81-AF647 and BSA-AF488) | ||||
| Functional studies | ||||
| Immunomodulation assay (IMA) | Effect of EVs on PBMC proliferation induced by PHA or CD3/28 | |||
| Angiogenesis assays | Effect of EVs on network formation of endothelial cells in matrigel | |||
| Organoid formation and fibrosphere assays | Effect of EVs on fibroblast, keratinocyte and endothelial organoid/spheroid formation | |||
| Wound-healing assay | Effect of EVs on fibroblast migration | |||
| Sample | Protein | Fold-Change | Description | Function |
|---|---|---|---|---|
| HPL | ||||
| CXCL7 | 2.10 | Platelet basic protein | DNA synthesis, mitosis, glycolysis, intracellular cAMP accumulation, prostaglandin E2 secretion, and synthesis of hyaluronic acid and sulfated glycosaminoglycan. | |
| G6PI | 1.22 | Glucose-6-phosphate isomerase | Conversion of glucose-6-phosphate to fructose-6-phosphate. Also acts as a secreted cytokine: angiogenic factor (AMF) that stimulates endothelial cell motility. | |
| PGS1 | 1.15 | Biglycan | May be involved in collagen fiber assembly. | |
| PROF1 | 1.14 | Profilin-1 | Binds to actin and affects the structure of the cytoskeleton. | |
| PEDF | 1.11 | Pigment epithelium-derived factor | Neurotrophic protein; induces extensive neuronal differentiation in retinoblastoma cells. Potent inhibitor of angiogenesis. | |
| FETUA | 1.07 | Alpha-2-HS-glycoprotein | Promotes endocytosis, possesses opsonic properties and influences the mineral phase of bone. | |
| TAGL2 | 1.06 | Transgelin-2 | Regulates Mlc, Ras homolog, F Actin, Actin. Binds Cadherin. | |
| A1AG1 | 1.00 | Alpha-1-acid glycoprotein 1 | Functions as transport protein in the blood stream. Appears to function in modulating the activity of the immune system during the acute-phase reaction. | |
| TRFE | 0.97 | Serotransferrin | Iron binding transport proteins. May also have a further role in stimulating cell proliferation. | |
| HEMO | 0.95 | Hemopexin | Binds heme and transports it to the liver for breakdown and iron recovery. | |
| ALBU | 0.93 | Albumin | Binds water, Ca2+, Na+, K+, fatty acids, hormones and bilirubin. | |
| A1AT | 0.92 | Alpha-1-antitrypsin | Inhibitor of serine proteases. Irreversibly inhibits trypsin, chymotrypsin and plasminogen activator. | |
| PGK1 | 0.90 | Phosphoglycerate kinase 1 | Catalyzes the conversion of 1,3-diphosphoglycerate to 3-phosphoglycerate. Polymerase alpha cofactor protein. | |
| CAH2 | 0.89 | Carbonic anhydrase 2 | Bone resorption and osteoclast differentiation. Reversible hydration of carbon dioxide. Involved in the regulation of fluid secretion into the anterior chamber of the eye. | |
| ZA2G | 0.88 | Zinc-alpha-2-glycoprotein | Stimulates lipid degradation in adipocytes. | |
| LUM | 0.83 | Lumican | Regulates TNF, KERA, MMP14, DNA endogenous promoter, DNA promoter, FAS, Mmp, CXCL2, CDKN1A, IL6, IL1B, RNA polymerase II, TGFB1, COL1A2, COL1A1. Binds Collagen. | |
| KAIN | 0.78 | Kallistatin | Inhibits human amidolytic and kininogenase activities of tissue kallikrein. | |
| CFAB | 0.78 | Complement factor B | Is cleaved by factor D into 2 fragments: Ba and Bb. Bb then combines with complement factor 3b to generate the C3 or C5 convertase. Ba inhibits the proliferation of preactivated B-lymphocytes. | |
| RET4 | 0.77 | Retinol-binding protein 4 | Mediates retinol transport in blood plasma. | |
| TTHY | 0.75 | Transthyretin | Thyroid hormone-binding protein. | |
| TFF-EVs | ||||
| C1QC | 0.55 | Complement C1q subcomponent subunit C | C1q associates with the proenzymes C1r and C1s to yield C1, the first component of the serum complement system. | |
| C1QA | 0.53 | Complement C1q subcomponent subunit A | C1q associates with the proenzymes C1r and C1s to yield C1, the first component of the serum complement system. | |
| C4BPB | 0.47 | C4b-binding protein beta chain | Controls the classical pathway of complement activation. | |
| C4BPA | 0.44 | C4b-binding protein alpha chain | Controls the classical pathway of complement activation. | |
| HV146 | 0.43 | Immunoglobulin heavy variable 1-46 | V region of the variable domain of immunoglobulin heavy chains that participates in the antigen recognition. | |
| C1QB | 0.40 | Complement C1q subcomponent subunit B | C1q associates with the proenzymes C1r and C1s to yield C1, the first component of the serum complement system. | |
| TSP1 | 0.36 | Thrombospondin-1 | Adhesive glycoprotein that mediates cell-to-cell and cell-to-matrix interactions. Binds heparin. | |
| C1S | 0.36 | Complement C1s subcomponent | C1s B chain combines with C1q and C1r to form C1, component of the classical pathway of the complement system. | |
| C1R | 0.36 | Complement C1r subcomponent | C1r activates C1s so that it can, in turn, activate C2 and C4. | |
| DUS29 | 0.35 | Dual specificity phosphatase 29 | Able to dephosphorylate phosphotyrosine, phosphoserine and phosphothreonine residues within the same substrate. Involved in the modulation of intracellular signaling cascades. | |
| PROS | 0.34 | Vitamin K-dependent protein S | Anticoagulant plasma protein; cofactor to activated protein C. Prevents coagulation and stimulates fibrinolysis. | |
| IGHM | 0.33 | Immunoglobulin heavy constant mu | Constant region of immunoglobulin heavy chains. | |
| LG3BP | 0.30 | Galectin-3-binding protein | Promotes integrin-mediated cell adhesion. May stimulate host defense against viruses and tumor cells. | |
| GRP2 | 0.30 | RAS guanyl-releasing protein 2 | Functions in aggregation of platelets and adhesion of T-lymphocytes and neutrophils. | |
| PLF4 | 0.29 | Platelet factor 4 | Released during platelet aggregation. Chemotactic for neutrophils and monocytes. | |
| PRG4 | 0.28 | Proteoglycan 4 | Plays a role in boundary lubrication within articulating joints. | |
| FCN2 | 0.28 | Ficolin-2 | May function in innate immunity through activation of the lectin complement pathway. | |
| CD5L | 0.27 | CD5 antigen-like | Secreted protein that acts as a key regulator of lipid synthesis. | |
| FA5 | 0.27 | Coagulation factor V | Central regulator of hemostasis. Critical cofactor for conversion of prothrombin to thrombin. | |
| HV374 | 0.26 | Immunoglobulin heavy variable 3-74 | V region of the variable domain of immunoglobulin heavy chains that participates in the antigen recognition. | |
| TFF sol.F. | ||||
| CXCL7 | 2.41 | Platelet basic protein | DNA synthesis, mitosis, glycolysis, intracellular cAMP accumulation, prostaglandin E2 secretion, and synthesis of hyaluronic acid and sulfated glycosaminoglycan. | |
| G6PI | 1.38 | Glucose-6-phosphate isomerase | Conversion of glucose-6-phosphate to fructose-6-phosphate. Also acts as a secreted cytokine: angiogenic factor (AMF) that stimulates endothelial cell motility. | |
| PGS1 | 1.38 | Biglycan | May be involved in collagen fiber assembly. | |
| PEDF | 1.32 | Pigment epithelium-derived factor | Neurotrophic protein; induces extensive neuronal differentiation in retinoblastoma cells. Potent inhibitor of angiogenesis. | |
| PROF1 | 1.31 | Profilin-1 | Binds to actin and affects the structure of the cytoskeleton. | |
| TAGL2 | 1.25 | Transgelin-2 | Regulates Mlc, Ras homolog, F Actin, Actin. Binds Cadherin. | |
| FETUA | 1.25 | Alpha-2-HS-glycoprotein | Promotes endocytosis, possesses opsonic properties and influences the mineral phase of bone. | |
| HEMO | 1.17 | Hemopexin | Binds heme and transports it to the liver for breakdown and iron recovery. | |
| A1AG1 | 1.16 | Alpha-1-acid glycoprotein 1 | Functions as transport protein in the blood stream. Appears to function in modulating the activity of the immune system during the acute-phase reaction. | |
| TRFE | 1.13 | Serotransferrin | Iron binding transport proteins. May also have a further role in stimulating cell proliferation. | |
| ALBU | 1.11 | Albumin | Binds water, Ca2+, Na+, K+, fatty acids, hormones and bilirubin. | |
| A1AT | 1.10 | Alpha-1-antitrypsin | Inhibitor of serine proteases. Irreversibly inhibits trypsin, chymotrypsin and plasminogen activator. | |
| CAH2 | 1.08 | Carbonic anhydrase 2 | Bone resorption and osteoclast differentiation. Reversible hydration of carbon dioxide. Involved in the regulation of fluid secretion into the anterior chamber of the eye. | |
| ZA2G | 1.04 | Zinc-alpha-2-glycoprotein | Stimulates lipid degradation in adipocytes. | |
| PGK1 | 1.01 | Phosphoglycerate kinase 1 | Catalyzes the conversion of 1,3-diphosphoglycerate to 3-phosphoglycerate. Polymerase alpha cofactor protein. | |
| TTHY | 0.99 | Transthyretin | Thyroid hormone-binding protein. | |
| HV333 | 0.99 | Immunoglobulin heavy variable 3-33 | V region of the variable domain of immunoglobulin heavy chains that participates in the antigen recognition. | |
| ICAM2 | 0.97 | Intercellular adhesion molecule 2 | May play a role in lymphocyte recirculation by blocking LFA-1-dependent cell adhesion. | |
| LUM | 0.96 | Lumican | Regulates TNF, KERA, MMP14, DNA endogenous promoter, DNA promoter, FAS, Mmp, CXCL2, CDKN1A, IL6, IL1B, RNA polymerase II, TGFB1, COL1A2, COL1A1. Binds Collagen. | |
| GSTP1 | 0.95 | Glutathione S-transferase P | Conjugation of reduced glutathione to a wide number of exogenous and endogenous hydrophobic electrophiles. | |
| TSEC-EVs | ||||
| MYH14 | 2.89 | Myosin-14 | Cellular myosin that appears to play a role in cytokinesis, cell shape, and specialized functions such as secretion and capping. | |
| GIT1 | 2.53 | ARF GTPase-activating protein GIT1 | Multidomain scaffold protein that interacts with numerous proteins, participating in receptor internalization, focal adhesion remodeling, and signaling by both G protein-coupled receptors and tyrosine kinase receptors. | |
| CD9 | 2.14 | CD9 antigen | Integrin-associated tetraspanin, which regulates platelet activation and aggregation, and cell adhesion. | |
| GTR3 | 2.10 | Glucose transporter type 3 | Facilitative glucose transporter that can also mediate the uptake of various other monosaccharides. | |
| MYH9 | 1.90 | Myosin-9 | Cellular myosin that plays a role in cytokinesis, cell shape, and specialized functions such as secretion and capping. | |
| ITB6 | 1.81 | Integrin beta-6 | Integrin alpha-V:beta-6 (ITGAV:ITGB6) is a receptor for fibronectin and cytotactin. | |
| FCN2 | 1.62 | Ficolin-2 | May function in innate immunity through activation of the lectin complement pathway. | |
| DUS29 | 1.59 | Dual specificity phosphatase 29 | Able to dephosphorylate phosphotyrosine, phosphoserine and phosphothreonine residues within the same substrate. Involved in the modulation of intracellular signaling cascades. | |
| MYH11 | 1.59 | Myosin-11 | Muscle contraction. | |
| ANO6 | 1.57 | Anoctamin-6 | Small-conductance channel which acts as a regulator of phospholipid scrambling in platelets and osteoblasts. | |
| FRIH | 1.56 | Ferritin heavy chain | Stores iron in a soluble, non-toxic, readily available form. Important for iron homeostasis. | |
| ITA2B | 1.50 | Integrin alpha-IIb | Integrin alpha-IIb/beta-3 is a receptor for fibronectin, fibrinogen, plasminogen, prothrombin, thrombospondin and vitronectin. | |
| CD36 | 1.47 | Platelet glycoprotein 4 | Multifunctional glycoprotein that acts as receptor for a broad range of ligands. | |
| KATL2 | 1.47 | Katanin p60 ATPase-containing subunit A-like 2 | Severs microtubules in vitro in an ATP-dependent manner. | |
| CD47 | 1.45 | Leukocyte surface antigen CD47 | Acts as an adhesion receptor on platelets, and in the modulation of integrins. | |
| ML12A | 1.44 | Myosin regulatory light chain 12A | Plays a role in regulation of both smooth muscle and nonmuscle cell contractile activity via its phosphorylation. | |
| VWF | 1.44 | von Willebrand factor | Promotes adhesion of platelets to the sites of vascular injury. | |
| CTL1 | 1.42 | Choline transporter-like protein 1 | Choline transporter. Involved in membrane synthesis and myelin production. | |
| IGHM | 1.42 | Immunoglobulin heavy constant mu | Constant region of immunoglobulin heavy chains. | |
| SPTN1 | 1.39 | Spectrin alpha chain | Calcium-dependent movement of the cytoskeleton at the membrane. | |
| TSEC sol.F. | ||||
| EF1D | 0.38 | Elongation factor 1-delta | Regulates induction of heat-shock-responsive genes. | |
| HV102 | 0.33 | Immunoglobulin heavy variable 1-2 | V region of the variable domain of immunoglobulin heavy chains that participates in the antigen recognition. | |
| GRP2 | 0.26 | RAS guanyl-releasing protein 2 | Functions in aggregation of platelets and adhesion of T-lymphocytes and neutrophils. | |
| FILA2 | 0.25 | Filaggrin-2 | Essential for normal cell-cell adhesion in the cornified cell layers. | |
| K1C10 | 0.24 | Keratin. type I cytoskeletal 10 | Plays a role in the establishment of the epidermal barrier on plantar skin. | |
| HV146 | 0.22 | Immunoglobulin heavy variable 1-46 | V region of the variable domain of immunoglobulin heavy chains that participates in the antigen recognition. | |
| K2C5 | 0.22 | Keratin. type II cytoskeletal 5 | Regulates SERPINF1, VEGFA, ICAM1, MAPK3, MAPK1, KRT5, HIF1A. | |
| K22E | 0.21 | Keratin. type II cytoskeletal 2 epidermal | Associated with keratinocyte activation, proliferation and keratinization. | |
| HOOK3 | 0.19 | Protein Hook homolog 3 | Promote vesicle trafficking and/or fusion via the homotypic vesicular protein sorting complex (the HOPS complex). | |
| TYPH | 0.19 | Thymidine phosphorylase | May have a role in maintaining the integrity of the blood vessels. | |
| THIO | 0.18 | Thioredoxin | Participates in various redox reactions. | |
| GNA13 | 0.18 | Guanine nucleotide-binding protein subunit alpha-13 | Guanine nucleotide-binding proteins (G proteins) are involved in various transmembrane signaling systems. | |
| PSA3 | 0.17 | Proteasome subunit alpha type-3 | Involved in the proteolytic degradation of most intracellular proteins. | |
| K1C9 | 0.17 | Keratin. type I cytoskeletal 9 | Plays a role in keratin filament assembly. | |
| K2C6B | 0.17 | Keratin. type II cytoskeletal 6B | Regulates KRT6B, CEBPB, F Actin. | |
| TRI23 | 0.17 | E3 ubiquitin-protein ligase | Acts as an E3 ubiquitin-protein ligase. Plays an essential role in autophagy activation during viral infection. | |
| K1C16 | 0.17 | Keratin. type I cytoskeletal 16 | Acts as a regulator of innate immunity in response to skin barrier breach. | |
| K2C1 | 0.16 | Keratin. type II cytoskeletal 1 | May regulate the activity of kinases such as PKC and SRC via binding to ITB1 and RACK1. | |
| NEXN | 0.16 | Nexilin | Involved in regulating cell migration through association with the actin cytoskeleton. | |
| MASP2 | 0.16 | Mannan-binding lectin serine protease 2 | Serum protease that plays an important role in the activation of the complement system. |
References
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between Platelets and Coagulation. Blood Rev. 2021, 46, 100733. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Freedman, J. Platelets and Innate Immunity. Cell. Mol. Life Sci. CMLS 2010, 67, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.F.; Mustoe, T.A.; Altrock, B.W.; Deuel, T.F.; Thomason, A. Role of Platelet-derived Growth Factor in Wound Healing. J. Cell. Biochem. 1991, 45, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Jansen, E.E.; Braun, A.; Jansen, P.; Hartmann, M. Platelet-Therapeutics to Improve Tissue Regeneration Andwound Healing-Physiological Background Andmethods of Preparation. Biomedicines 2021, 9, 869. [Google Scholar] [CrossRef] [PubMed]
- Burnouf, T.; Strunk, D.; Koh, M.B.C.; Schallmoser, K. Human Platelet Lysate: Replacing Fetal Bovine Serum as a Gold Standard for Human Cell Propagation? Biomaterials 2016, 76, 371–387. [Google Scholar] [CrossRef]
- Harrison, P.; Alsousou, J.; Andia, I.; Burnouf, T.; Dohan Ehrenfest, D.; Everts, P.; Langer, H.; Magalon, J.; Marck, R.; Gresele, P. The Use of Platelets in Regenerative Medicine and Proposal for a New Classification System: Guidance from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1895–1900. [Google Scholar] [CrossRef]
- Wu, J.; Piao, Y.; Liu, Q.; Yang, X. Platelet-Rich Plasma-Derived Extracellular Vesicles: A Superior Alternative in Regenerative Medicine? Cell Prolif. 2021, 54, e13123. [Google Scholar] [CrossRef]
- Moraes, V.Y.; Lenza, M.; Tamaoki, M.J.; Faloppa, F.; Belloti, J.C. Platelet-Rich Therapies for Musculoskeletal Soft Tissue Injuries. Cochrane Database Syst. Rev. 2014, 2014, CD010071. [Google Scholar] [CrossRef]
- Etulain, J. Platelets in Wound Healing and Regenerative Medicine. Platelets 2018, 29, 556–568. [Google Scholar] [CrossRef]
- Schallmoser, K.; Henschler, R.; Gabriel, C.; Koh, M.B.C.; Burnouf, T. Production and Quality Requirements of Human Platelet Lysate: A Position Statement from the Working Party on Cellular Therapies of the International Society of Blood Transfusion. Trends Biotechnol. 2020, 38, 13–23. [Google Scholar] [CrossRef]
- Schallmoser, K.; Bartmann, C.; Rohde, E.; Reinisch, A.; Kashofer, K.; Stadelmeyer, E.; Drexler, C.; Lanzer, G.; Linkesch, W.; Strunk, D. Human Platelet Lysate Can Replace Fetal Bovine Serum for Clinical-Scale Expansion of Functional Mesenchymal Stromal Cells. Transfusion 2007, 47, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Oeller, M.; Laner-Plamberger, S.; Krisch, L.; Rohde, E.; Strunk, D.; Schallmoser, K. Human Platelet Lysate for Good Manufacturing Practice-compliant Cell Production. Int. J. Mol. Sci. 2021, 22, 5178. [Google Scholar] [CrossRef] [PubMed]
- Ebner-Peking, P.; Krisch, L.; Wolf, M.; Hochmann, S.; Hoog, A.; Vári, B.; Muigg, K.; Poupardin, R.; Scharler, C.; Schmidhuber, S.; et al. Self-Assembly of Differentiated Progenitor Cells Facilitates Spheroid Human Skin Organoid Formation and Planar Skin Regeneration. Theranostics 2021, 11, 8430–8447. [Google Scholar] [CrossRef] [PubMed]
- Spakova, T.; Janockova, J.; Rosocha, J. Characterization and Therapeutic Use of Extracellular Vesicles Derived from Platelets. Int. J. Mol. Sci. 2021, 22, 9701. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P. The Nature and Significance of Platelet Products in Human Plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Brisson, A.R.; Tan, S.; Linares, R.; Gounou, C.; Arraud, N. Extracellular Vesicles from Activated Platelets: A Semiquantitative Cryo-Electron Microscopy and Immuno-Gold Labeling Study. Platelets 2017, 28, 263–271. [Google Scholar] [CrossRef]
- Aatonen, M.T.; Öhman, T.; Nyman, T.A.; Laitinen, S.; Grönholm, M.; Siljander, P.R.M. Isolation and Characterization of Platelet-Derived Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 24692. [Google Scholar] [CrossRef]
- Puhm, F.; Boilard, E.; MacHlus, K.R. Platelet Extracellular Vesicles; beyond the Blood. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 87–96. [Google Scholar] [CrossRef]
- Poupardin, R.; Wolf, M.; Strunk, D. Adherence to Minimal Experimental Requirements for Defining Extracellular Vesicles and Their Functions. Adv. Drug Deliv. Rev. 2021, 176, 113872. [Google Scholar] [CrossRef]
- Simonsen, J.B.; Münter, R. Pay Attention to Biological Nanoparticles When Studying the Protein Corona on Nanomedicines. Angew. Chem. 2020, 132, 12684–12688. [Google Scholar] [CrossRef]
- Murphy, D.E.; de Jong, O.G.; Evers, M.J.W.; Nurazizah, M.; Schiffelers, R.M.; Vader, P. Natural or Synthetic RNA Delivery: A Stoichiometric Comparison of Extracellular Vesicles and Synthetic Nanoparticles. Nano Lett. 2021, 21, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J. Protein–Nanoparticle Interaction: Corona Formation and Conformational Changes in Proteins on Nanoparticles. Int. J. Nanomed. 2020, 15, 5783–5802. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Poupardin, R.W.; Ebner-Peking, P.; Andrade, A.C.; Blöchl, C.; Obermayer, A.; Gomes, F.G.; Vari, B.; Eminger, E.; Binder, H.-M.; et al. A Functional Corona around Extracellular Vesicles Enhances Angiogenesis during Skin Regeneration and Signals in Immune Cells. bioRxiv 2021, 808808. [Google Scholar]
- Tóth, E.; Turiák, L.; Visnovitz, T.; Cserép, C.; Mázló, A.; Sódar, B.W.; Försönits, A.I.; Petővári, G.; Sebestyén, A.; Komlósi, Z.; et al. Formation of a Protein Corona on the Surface of Extracellular Vesicles in Blood Plasma. J. Extracell. Vesicles 2021, 10, e12140. [Google Scholar] [CrossRef]
- Laner-Plamberger, S.; Lener, T.; Schmid, D.; Streif, D.A.; Salzer, T.; Öller, M.; Hauser-Kronberger, C.; Fischer, T.; Jacobs, V.R.; Schallmoser, K.; et al. Mechanical Fibrinogen-Depletion Supports Heparin-Free Mesenchymal Stem Cell Propagation in Human Platelet Lysate. J. Transl. Med. 2015, 13, 354. [Google Scholar] [CrossRef] [Green Version]
- Binder, H.; Maeding, N.; Wolf, M.; Andrade, A.C.; Vari, B.; Krisch, L.; Gomes, F.G.; Blöchl, C.; Muigg, K.; Poupardin, R.; et al. Scalable Enrichment of Immunomodulatory Human Acute Myeloid Leukemia Cell Line-Derived Extracellular Vesicles. Cells 2021, 10, 3321. [Google Scholar] [CrossRef]
- Andrade, A.C.; Wolf, M.; Binder, H.-M.M.; Gomes, F.G.; Manstein, F.; Ebner-Peking, P.; Poupardin, R.; Zweigerdt, R.; Schallmoser, K.; Strunk, D. Hypoxic Conditions Promote the Angiogenic Potential of Human Induced Pluripotent Stem Cell-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 3890. [Google Scholar] [CrossRef]
- Cleland, W.W. Dithiothreitol, a New Protective Reagent for SH Groups. Biochemistry 1964, 3, 480–482. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, A.; Hofmann, N.A.; Obenauf, A.C.; Kashofer, K.; Rohde, E.; Schallmoser, K.; Flicker, K.; Lanzer, G.; Linkesch, W.; Speicher, M.R.; et al. Humanized Large-Scale Expanded Endothelial Colony-Forming Cells Function in Vitro and in Vivo. Blood 2009, 113, 6716–6725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laner-Plamberger, S.; Oeller, M.; Rohde, E.; Schallmoser, K.; Strunk, D. Heparin and Derivatives for Advanced Cell Therapies. Int. J. Mol. Sci. 2021, 22, 12041. [Google Scholar] [CrossRef] [PubMed]
- Pachler, K.; Ketterl, N.; Desgeorges, A.; Dunai, Z.A.; Laner-Plamberger, S.; Streif, D.; Strunk, D.; Rohde, E.; Gimona, M. An In Vitro Potency Assay for Monitoring the Immunomodulatory Potential of Stromal Cell-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2017, 18, 1413. [Google Scholar] [CrossRef]
- Ketterl, N.; Brachtl, G.; Schuh, C.; Bieback, K.; Schallmoser, K.; Reinisch, A.; Strunk, D. A Robust Potency Assay Highlights Significant Donor Variation of Human Mesenchymal Stem/Progenitor Cell Immune Modulatory Capacity and Extended Radio-Resistance. Stem Cell Res. Ther. 2015, 6, 236. [Google Scholar] [CrossRef] [Green Version]
- Priglinger, E.; Strasser, J.; Buchroithner, B.; Weber, F.; Wolbank, S.; Auer, D.; Grasmann, E.; Arzt, C.; Sivun, D.; Grillari, J.; et al. Label-Free Characterization of an Extracellular Vesicle-Based Therapeutic. J. Extracell. Vesicles 2021, 10, e12156. [Google Scholar] [CrossRef]
- Dassati, S.; Waldner, A.; Schweigreiter, R. Apolipoprotein D Takes Center Stage in the Stress Response of the Aging and Degenerative Brain. Neurobiol. Aging 2014, 35, 1632–1642. [Google Scholar] [CrossRef] [Green Version]
- Olm, F.; Lim, H.C.; Schallmoser, K.; Strunk, D.; Laurell, T.; Scheding, S. Acoustophoresis Enables the Label-Free Separation of Functionally Different Subsets of Cultured Bone Marrow Stromal Cells. Cytom. Part A 2021, 99, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Midekessa, G.; Godakumara, K.; Ord, J.; Viil, J.; Lättekivi, F.; Dissanayake, K.; Kopanchuk, S.; Rinken, A.; Andronowska, A.; Bhattacharjee, S.; et al. Zeta Potential of Extracellular Vesicles: Toward Understanding the Attributes That Determine Colloidal Stability. ACS Omega 2020, 5, 16701–16710. [Google Scholar] [CrossRef] [PubMed]
- Laner-Plamberger, S.; Oeller, M.; Mrazek, C.; Hartl, A.; Sonderegger, A.; Rohde, E.; Strunk, D.; Schallmoser, K. Upregulation of Mitotic Bookmarking Factors during Enhanced Proliferation of Human Stromal Cells in Human Platelet Lysate. J. Transl. Med. 2019, 17, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deppermann, C.; Kubes, P. Start a Fire, Kill the Bug: The Role of Platelets in Inflammation and Infection. Innate Immun. 2018, 24, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Peerschke, E.I.B.; Yin, W.; Ghebrehiwet, B. Platelet Mediated Complement Activation. Adv. Exp. Med. Biol. 2008, 632, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Schallmoser, K.; Rohde, E.; Reinisch, A.; Bartmann, C.; Thaler, D.; Drexler, C.; Obenauf, A.C.; Lanzer, G.; Linkesch, W.; Strunk, D. Rapid Large-Scale Expansion of Functional Mesenchymal Stem Cells from Unmanipulated Bone Marrow without Animal Serum. Tissue Eng. Part C Methods 2008, 14, 185–196. [Google Scholar] [CrossRef]
- Li, Y.; Lin, F. Mesenchymal Stem Cells Are Injured by Complement after Their Contact with Serum. Blood 2012, 120, 3436–3443. [Google Scholar] [CrossRef] [Green Version]
- Patel, G.K.; Khan, M.A.; Zubair, H.; Srivastava, S.K.; Khushman, M.; Singh, S.; Singh, A.P. Comparative Analysis of Exosome Isolation Methods Using Culture Supernatant for Optimum Yield, Purity and Downstream Applications. Sci. Rep. 2019, 9, 5335. [Google Scholar] [CrossRef] [Green Version]
- Blundell, E.L.C.J.; Healey, M.J.; Holton, E.; Sivakumaran, M.; Manstana, S.; Platt, M. Characterisation of the Protein Corona Using Tunable Resistive Pulse Sensing: Determining the Change and Distribution of a Particle’s Surface Charge. Anal. Bioanal. Chem. 2016, 408, 5757–5768. [Google Scholar] [CrossRef] [Green Version]
- Röcker, C.; Pötzl, M.; Zhang, F.; Parak, W.J.; Nienhaus, G.U. A Quantitative Fluorescence Study of Protein Monolayer Formation on Colloidal Nanoparticles. Nat. Nanotechnol. 2009, 4, 577–580. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Göppert, T.M.; Müller, R.H. Adsorption Kinetics of Plasma Proteins on Solid Lipid Nanoparticles for Drug Targeting. Int. J. Pharm. 2005, 302, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Aurora, A.B.; Olson, E.N. Immune Modulation of Stem Cells and Regeneration. Cell Stem Cell 2014, 15, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zayed, M.; Iohara, K. Immunomodulation and Regeneration Properties of Dental Pulp Stem Cells: A Potential Therapy to Treat Coronavirus Disease 2019. Cell Transplant. 2020, 29, 0963689720952089. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, F.G.; Andrade, A.C.; Wolf, M.; Hochmann, S.; Krisch, L.; Maeding, N.; Regl, C.; Poupardin, R.; Ebner-Peking, P.; Huber, C.G.; et al. Synergy of Human Platelet-Derived Extracellular Vesicles with Secretome Proteins Promotes Regenerative Functions. Biomedicines 2022, 10, 238. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020238
Gomes FG, Andrade AC, Wolf M, Hochmann S, Krisch L, Maeding N, Regl C, Poupardin R, Ebner-Peking P, Huber CG, et al. Synergy of Human Platelet-Derived Extracellular Vesicles with Secretome Proteins Promotes Regenerative Functions. Biomedicines. 2022; 10(2):238. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020238
Chicago/Turabian StyleGomes, Fausto Gueths, André Cronemberger Andrade, Martin Wolf, Sarah Hochmann, Linda Krisch, Nicole Maeding, Christof Regl, Rodolphe Poupardin, Patricia Ebner-Peking, Christian G. Huber, and et al. 2022. "Synergy of Human Platelet-Derived Extracellular Vesicles with Secretome Proteins Promotes Regenerative Functions" Biomedicines 10, no. 2: 238. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10020238