
Distinct Cellular Tools of Mild Hyperthermia-Induced Acquired Stress Tolerance in Chinese Hamster Ovary Cells

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blotting
2.3. Cell Survival Protocol
2.4. Determination of ATP Content
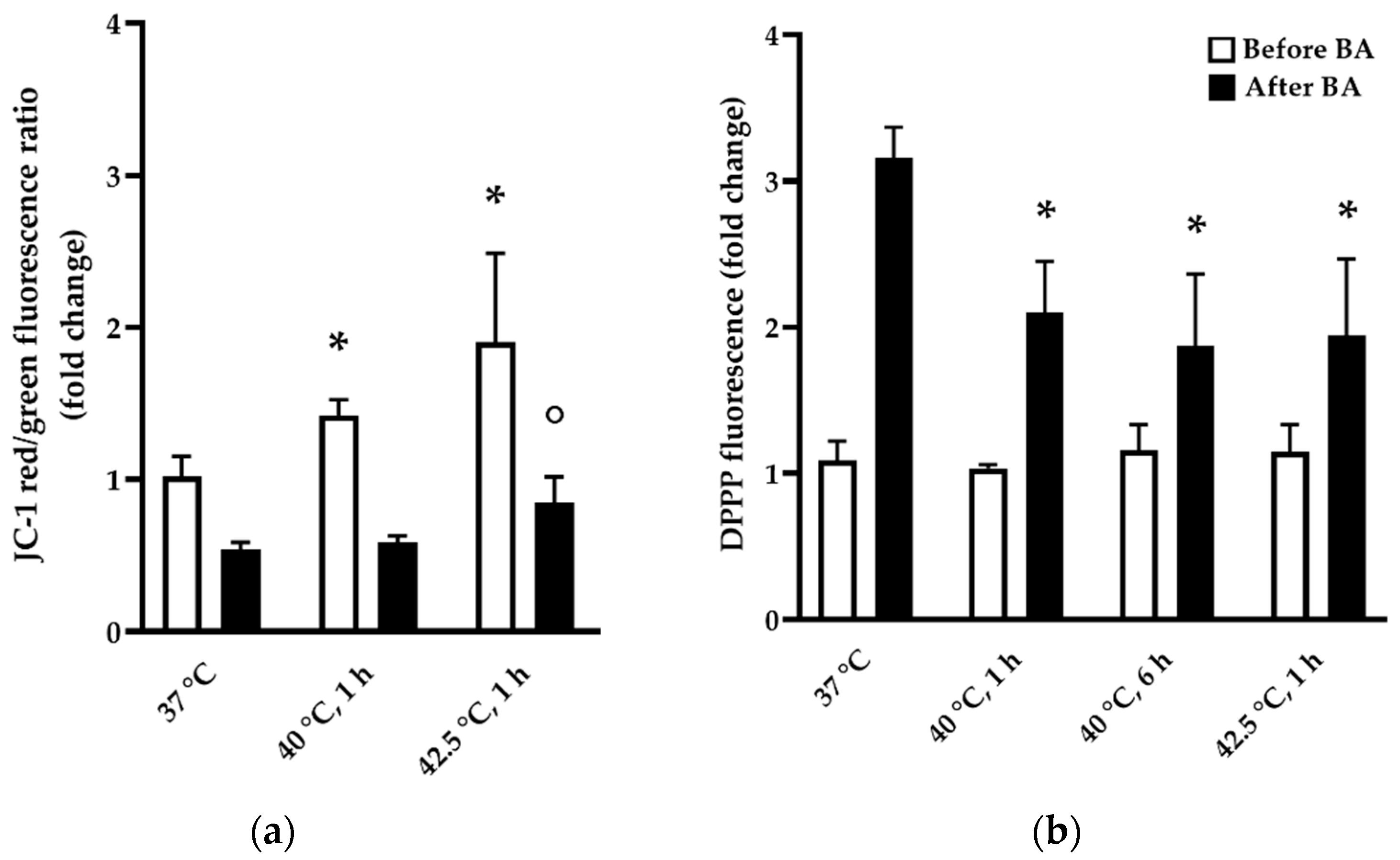
2.5. Mitochondrial Membrane Potential Assay
2.6. Lipid Peroxidation Assay
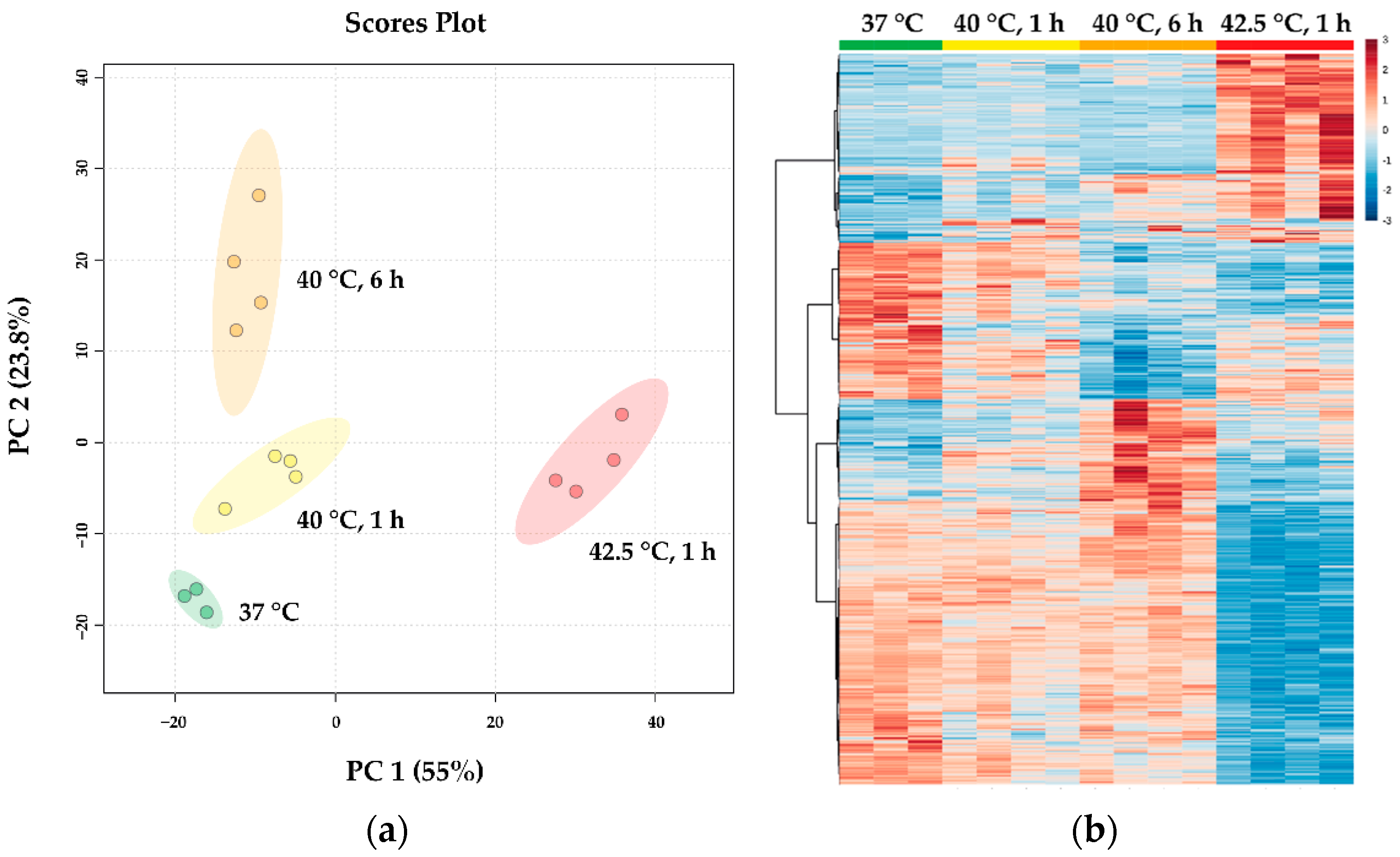
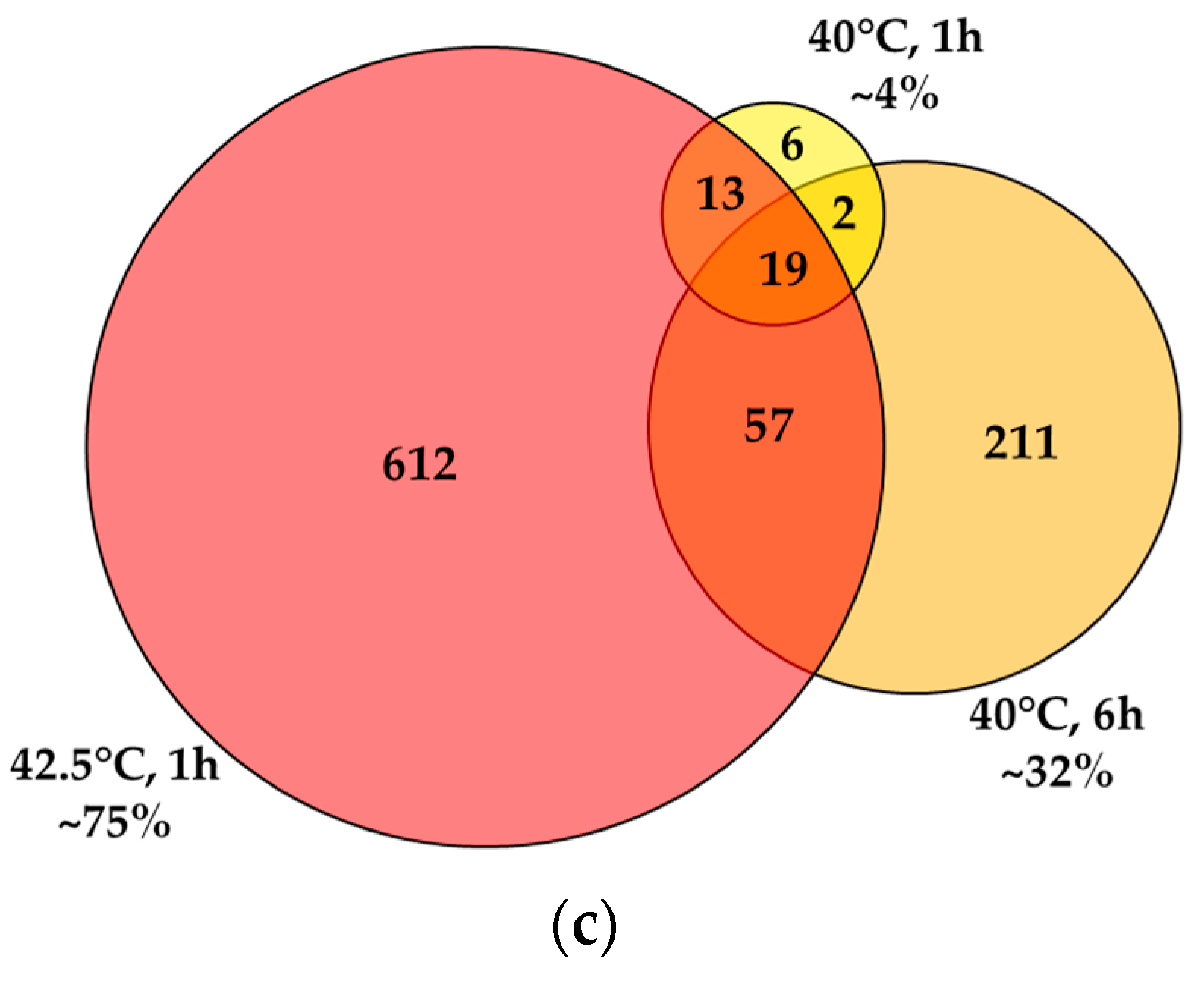
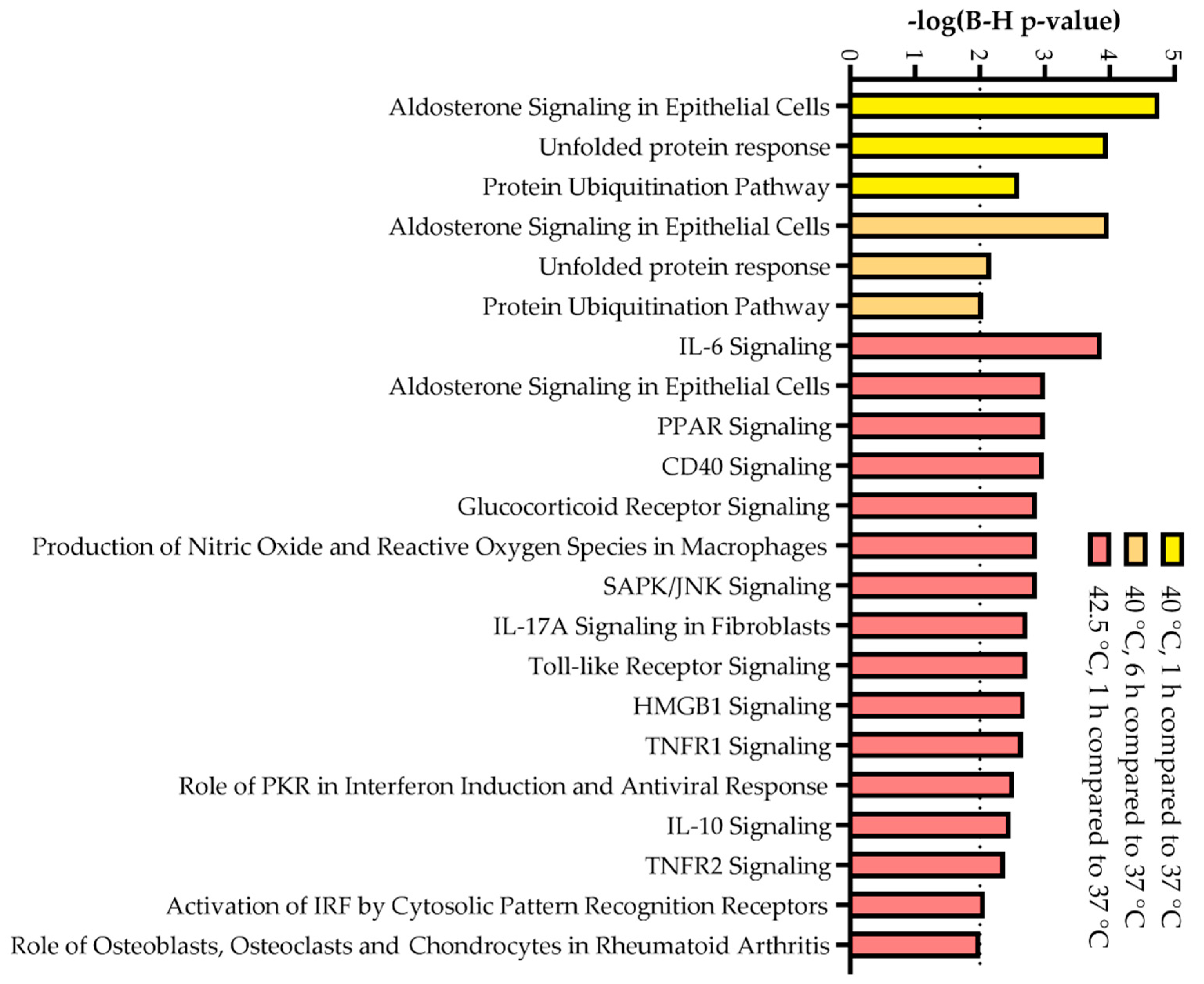
2.7. RNA Preparation, Sequencing and Bioinformatic Analysis
2.8. Lipidomics
2.9. Statistical Analyses
3. Results
3.1. Duration of Fever-like Hyperthermia Determines the Threshold of Hsp Induction
3.2. Mild Heat-Induced Acquired Stress Tolerance Does Not Require Hsp Induction
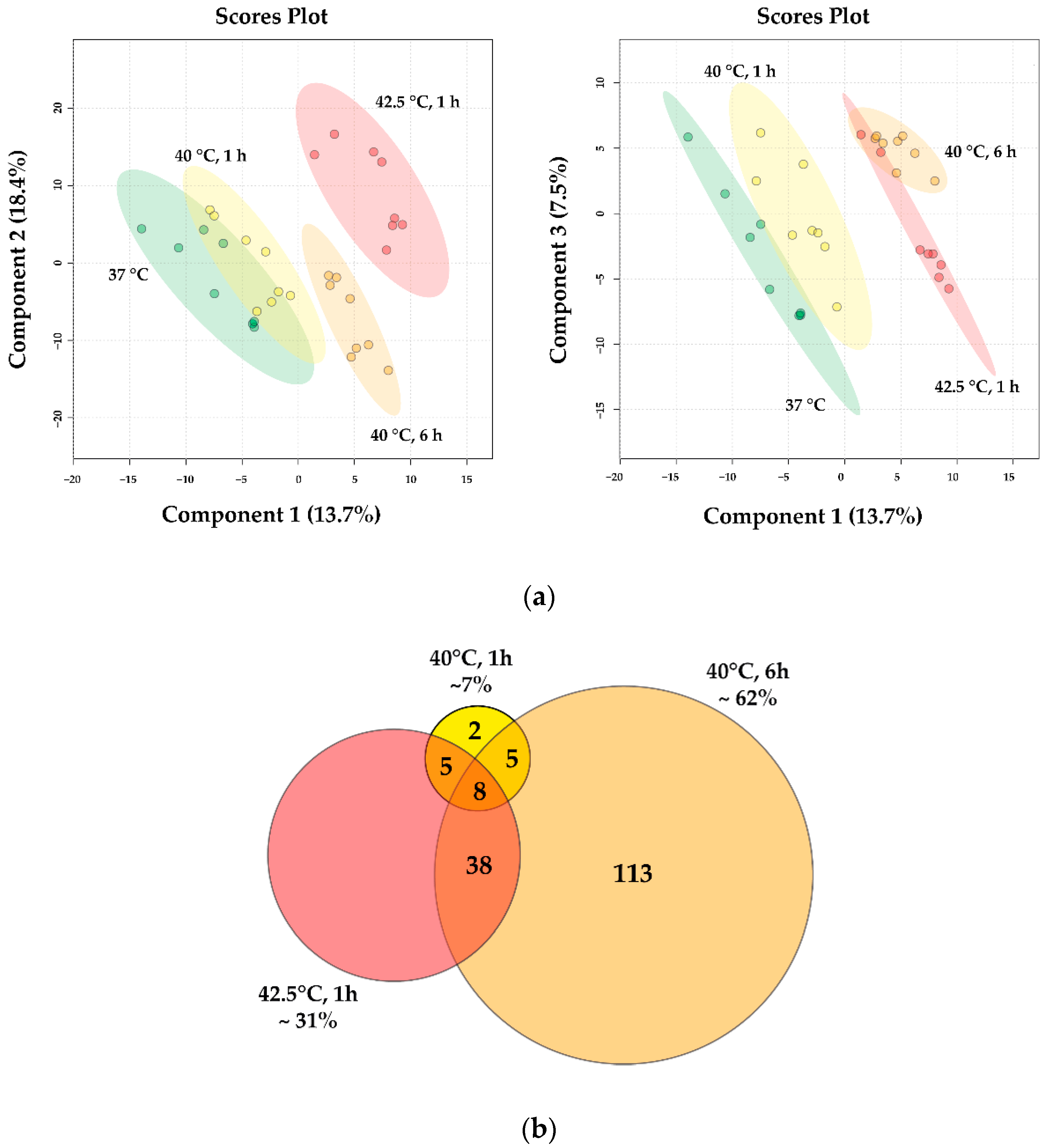
3.3. RNA Sequencing Data Reveal Distinct, Dose-Dependent Stress Transcriptomes
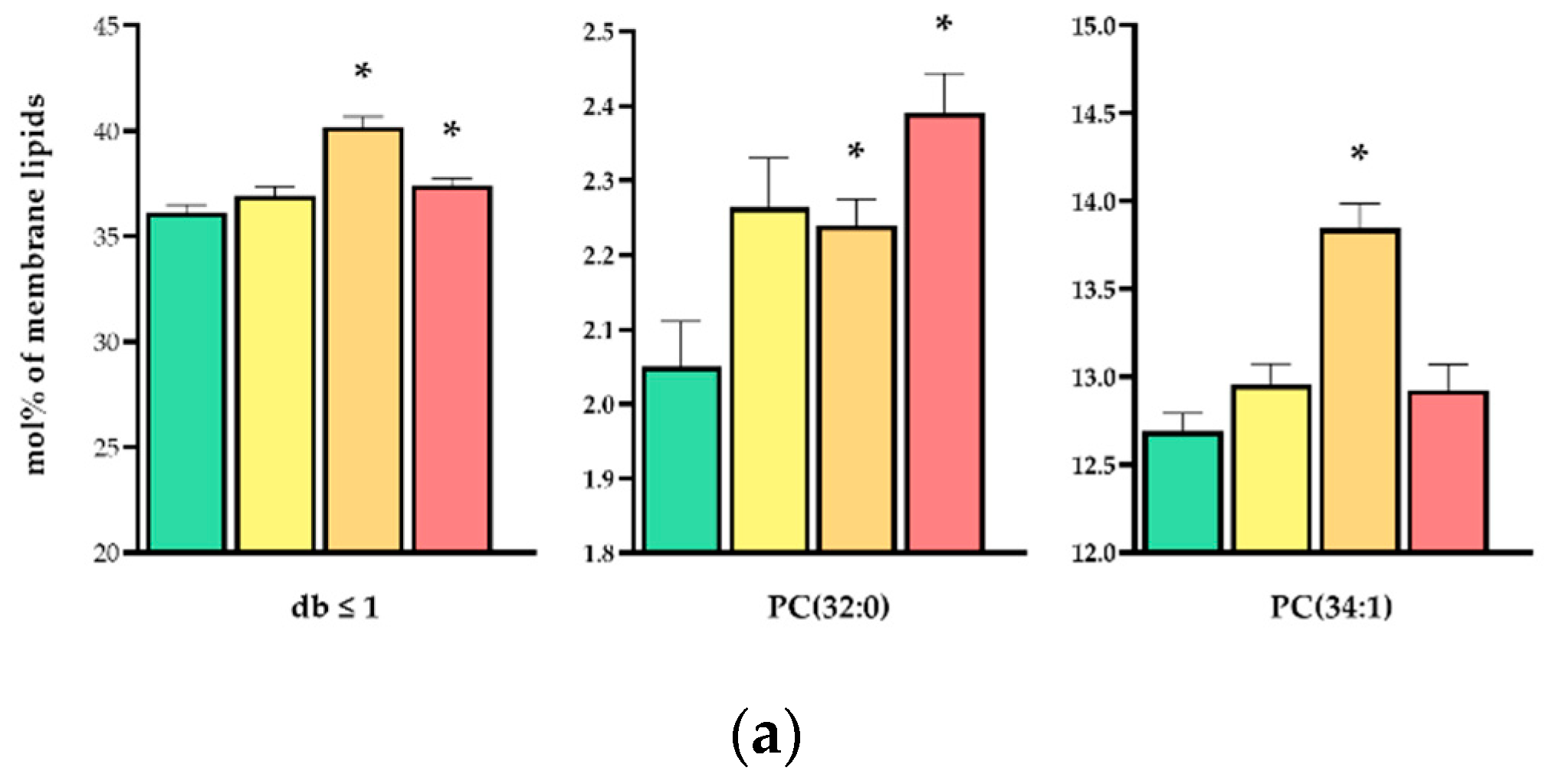
3.4. Lipidomic Data Reveal Distinct, Dose-Dependent Stress Lipidomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Habib, K.E.; Gold, P.W.; Chrousos, G.P. Neuroendocrinology of Stress. Endocrinol. Metab. Clin. N. Am. 2001, 30, 695–728. [Google Scholar] [CrossRef]
- Chrousos, G.P.; Gold, P.W. The Concepts of Stress and Stress System Disorders. Overview of Physical and Behavioral Homeostasis. JAMA 1992, 267, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Selye, H. Stress without Distress. In Psychopathology of Human Adaptation; Serban, G., Ed.; Springer: Boston, MA, USA, 1976; pp. 137–146. ISBN 978-1-4684-2240-5. [Google Scholar]
- Peksel, B.; Gombos, I.; Péter, M.; Vigh, L.; Tiszlavicz, Á.; Brameshuber, M.; Balogh, G.; Schütz, G.J.; Horváth, I.; Vigh, L.; et al. Mild Heat Induces a Distinct “Eustress” Response in Chinese Hamster Ovary Cells but Does Not Induce Heat Shock Protein Synthesis. Sci. Rep. 2017, 7, 15643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Yamakawa, N.; Mori, E.; Ohnishi, K.; Yokota, S.; Sugo, N.; Aratani, Y.; Koyama, H.; Ohnishi, T. Development of Thermotolerance Requires Interaction between Polymerase-β and Heat Shock Proteins. Cancer Sci. 2008, 99, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Q.; Zhang, X.; Qian, Q.; Xu, J.; Ni, P.; Qian, Y. Mild Endoplasmic Reticulum Stress Ameliorates Lipopolysaccharide-Induced Neuroinflammation and Cognitive Impairment via Regulation of Microglial Polarization. J. Neuroinflamm. 2017, 14, 233. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Ye, S.; Zhang, J.; He, M.; Dong, C.; Tu, W.; Liu, P.; Shao, C. Protective Effect of Mild Endoplasmic Reticulum Stress on Radiation-Induced Bystander Effects in Hepatocyte Cells. Sci. Rep. 2016, 6, 38832. [Google Scholar] [CrossRef] [Green Version]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.-J.; Sambrook, J. The Presence of Malfolded Proteins in the Endoplasmic Reticulum Signals the Induction of Glucose-Regulated Proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hänelt, I.; Hummer, G.; Ernst, R. Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol. Cell 2017, 67, 673–684.e8. [Google Scholar] [CrossRef]
- Xu, J.; Taubert, S. Beyond Proteostasis: Lipid Metabolism as a New Player in ER Homeostasis. Metabolites 2021, 11, 52. [Google Scholar] [CrossRef]
- Fun, X.H.; Thibault, G. Lipid Bilayer Stress and Proteotoxic Stress-Induced Unfolded Protein Response Deploy Divergent Transcriptional and Non-Transcriptional Programmes. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2020, 1865, 158449. [Google Scholar] [CrossRef]
- Gianfrancesco, M.A.; Paquot, N.; Piette, J.; Legrand-Poels, S. Lipid Bilayer Stress in Obesity-Linked Inflammatory and Metabolic Disorders. Biochem. Pharmacol. 2018, 153, 168–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, A.B.; Roberts, L.S.; Chandra, V.; Rivera, I.G.; Nomura, D.K.; Forbes, D.J.; Niwa, M. The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev. Cell 2018, 46, 327–343.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, R.; Ballweg, S.; Levental, I. Cellular Mechanisms of Physicochemical Membrane Homeostasis. Curr. Opin. Cell Biol. 2018, 53, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Diotallevi, A.; Magnani, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Infection by Intracellular Parasites. Future Sci. OA 2017, 3, FSO198. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A. Regulation of Cytokine Production by the Unfolded Protein Response; Implications for Infection and Autoimmunity. Front. Immunol. 2018, 9, 422. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Vigh, L.; Horváth, I.; Maresca, B.; Harwood, J.L. Can the Stress Protein Response Be Controlled by ‘Membrane-Lipid Therapy’? Trends Biochem. Sci. 2007, 32, 357–363. [Google Scholar] [CrossRef]
- Balogh, G.; Péter, M.; Glatz, A.; Gombos, I.; Török, Z.; Horváth, I.; Harwood, J.L.; Vígh, L. Key Role of Lipids in Heat Stress Management. FEBS Lett. 2013, 587, 11. [Google Scholar] [CrossRef] [Green Version]
- Török, Z.; Crul, T.; Maresca, B.; Schütz, G.J.; Viana, F.; Dindia, L.; Piotto, S.; Brameshuber, M.; Balogh, G.; Péter, M.; et al. Plasma Membranes as Heat Stress Sensors: From Lipid-Controlled Molecular Switches to Therapeutic Applications. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1594–1618. [Google Scholar] [CrossRef] [Green Version]
- Kluger, M.J.; Kozak, W.; Corm, C.A.; Leon, L.R.; Soszynski, D. The Adaptive Value of Fever. Infect. Dis. Clin. N. Am. 1996, 10, 1–20. [Google Scholar] [CrossRef]
- Kozak, W. Fever: A Possible Strategy for Membrane Homeostasis during Infection. Perspect. Biol. Med. 1993, 37, 14–34. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.-P.; Repasky, E.A.; Subjeck, J.R. Distribution of HSP70, Protein Kinase C, and Spectrin Is Altered in Lymphocytes during a Fever-like Hyperthermia Exposure. J. Cell. Physiol. 1997, 172, 44–54. [Google Scholar] [CrossRef]
- Mace, T.A.; Zhong, L.; Kokolus, K.M.; Repasky, E.A. Effector CD8+T Cell IFN-γ Production and Cytotoxicity Are Enhanced by Mild Hyperthermia. Int. J. Hyperth. 2012, 28, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabuchi, Y.; Takasaki, I.; Wada, S.; Zhao, Q.-L.; Hori, T.; Nomura, T.; Ohtsuka, K.; Kondo, T. Genes and Genetic Networks Responsive to Mild Hyperthermia in Human Lymphoma U937 Cells. Int. J. Hyperth. 2008, 24, 613–622. [Google Scholar] [CrossRef]
- Gordon, L.M.; Sauerheber, R.D.; Esgate, J.A.; Dipple, I.; Marchmont, R.J.; Houslay, M.D. The Increase in Bilayer Fluidity of Rat Liver Plasma Membranes Achieved by the Local Anesthetic Benzyl Alcohol Affects the Activity of Intrinsic Membrane Enzymes. J. Biol. Chem. 1980, 255, 4519–4527. [Google Scholar] [CrossRef]
- Horvath, I.; Glatz, A.; Varvasovszki, V.; Torok, Z.; Pali, T.; Balogh, G.; Kovacs, E.; Nadasdi, L.; Benko, S.; Joo, F.; et al. Membrane Physical State Controls the Signaling Mechanism of the Heat Shock Response in Synechocystis PCC 6803: Identification of Hsp17 as a “Fluidity Gene”. Proc. Natl. Acad. Sci. USA 1998, 95, 3513–3518. [Google Scholar] [CrossRef] [Green Version]
- Balogh, G.; Horváth, I.; Nagy, E.; Hoyk, Z.; Benkõ, S.; Bensaude, O.; Vígh, L. The Hyperfluidization of Mammalian Cell Membranes Acts as a Signal to Initiate the Heat Shock Protein Response: Membrane Fluidity and Heat Shock Response. FEBS J. 2005, 272, 6077–6086. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Tretter, L.; Adam-Vizi, V. Depolarization of In Situ Mitochondria Due to Hydrogen Peroxide-Induced Oxidative Stress in Nerve Terminals: Inhibition of α-Ketoglutarate Dehydrogenase. J. Neurochem. 2002, 73, 220–228. [Google Scholar] [CrossRef]
- Wang, Y.; Feinstein, S.I.; Fisher, A.B. Peroxiredoxin 6 as an Antioxidant Enzyme: Protection of Lung Alveolar Epithelial Type II Cells from H2O2-induced Oxidative Stress. J. Cell. Biochem. 2008, 104, 1274–1285. [Google Scholar] [CrossRef] [Green Version]
- Tóth, M.E.; Dukay, B.; Péter, M.; Balogh, G.; Szűcs, G.; Zvara, Á.; Szebeni, G.J.; Hajdu, P.; Sárközy, M.; Puskás, L.G.; et al. Male and Female Animals Respond Differently to High-Fat Diet and Regular Exercise Training in a Mouse Model of Hyperlipidemia. IJMS 2021, 22, 4198. [Google Scholar] [CrossRef]
- Péter, M.; Gudmann, P.; Kóta, Z.; Török, Z.; Vígh, L.; Glatz, A.; Balogh, G. Lipids and Trehalose Actively Cooperate in Heat Stress Management of Schizosaccharomyces Pombe. IJMS 2021, 22, 13272. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.; Schwudke, D.; Schuhmann, K.; Sampaio, J.L.; Bornstein, S.R.; Schroeder, M.; Shevchenko, A. A Novel Informatics Concept for High-Throughput Shotgun Lipidomics Based on the Molecular Fragmentation Query Language. Genome Biol. 2011, 12, R8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebisch, G.; Vizcaíno, J.A.; Köfeler, H.; Trötzmüller, M.; Griffiths, W.J.; Schmitz, G.; Spener, F.; Wakelam, M.J.O. Shorthand Notation for Lipid Structures Derived from Mass Spectrometry. J. Lipid Res. 2013, 54, 1523–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, J.D.; Tibshirani, R. Statistical Significance for Genomewide Studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Wishart, D.S. Web-Based Inference of Biological Patterns, Functions and Pathways from Metabolomic Data Using MetaboAnalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef]
- Duan, S.; Hájek, P.; Lin, C.; Shin, S.K.; Attardi, G.; Chomyn, A. Mitochondrial Outer Membrane Permeability Change and Hypersensitivity to Digitonin Early in Staurosporine-Induced Apoptosis. J. Biol. Chem. 2003, 278, 1346–1353. [Google Scholar] [CrossRef] [Green Version]
- Chazotte, B.; Vanderkooi, G. Multiple Sites of Inhibition of Mitochondrial Electron Transport by Local Anesthetics. Biochim. Biophys. Acta (BBA)-Bioenerg. 1981, 636, 153–161. [Google Scholar] [CrossRef]
- Kikusato, M.; Toyomizu, M. Crucial Role of Membrane Potential in Heat Stress-Induced Overproduction of Reactive Oxygen Species in Avian Skeletal Muscle Mitochondria. PLoS ONE 2013, 8, e64412. [Google Scholar] [CrossRef]
- Heise, K.; Puntarulo, S.; Nikinmaa, M.; Abele, D.; Pörtner, H.-O. Oxidative Stress during Stressful Heat Exposure and Recovery in the North Sea Eelpout Zoarces Viviparus L. J. Exp. Biol. 2006, 209, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Fedyaeva, A.V.; Stepanov, A.V.; Lyubushkina, I.V.; Pobezhimova, T.P.; Rikhvanov, E.G. Heat Shock Induces Production of Reactive Oxygen Species and Increases Inner Mitochondrial Membrane Potential in Winter Wheat Cells. Biochem. Mosc. 2014, 79, 1202–1210. [Google Scholar] [CrossRef]
- Kassis, S.; Grondin, M.; Averill-Bates, D.A. Heat Shock Increases Levels of Reactive Oxygen Species, Autophagy and Apoptosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 118924. [Google Scholar] [CrossRef] [PubMed]
- Eze, M.O. Membrane Fluidity, Reactive Oxygen Species, and Cell-Mediated Immunity: Implications in Nutrition and Disease. Med. Hypotheses 1992, 37, 220–224. [Google Scholar] [CrossRef]
- Lee, J.; Yu, B.; Herlihy, J. Modulation of Cardiac Mitochondrial Membrane Fluidity by Age and Calorie Intake. Free Radic. Biol. Med. 1999, 26, 260–265. [Google Scholar] [CrossRef]
- Sergent, O.; Pereira, M.; Belhomme, C.; Chevanne, M.; Huc, L.; Lagadic-Gossmann, D. Role for Membrane Fluidity in Ethanol-Induced Oxidative Stress of Primary Rat Hepatocytes. J. Pharm. Exp. Ther. 2005, 313, 104–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, M.E.; Vígh, L.; Sántha, M. Alcohol Stress, Membranes, and Chaperones. Cell Stress Chaperones 2014, 19, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Panniers, R. Translational Control during Heat Shock. Biochimie 1994, 76, 737–747. [Google Scholar] [CrossRef]
- McCormick, C.; Khaperskyy, D.A. Translation Inhibition and Stress Granules in the Antiviral Immune Response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef]
- Katz, A.; Orell, O. Protein Synthesis and the Stress Response. In Cell-Free Protein Synthesis; Biyani, M., Ed.; IntechOpen: London, UK, 2012; ISBN 978-953-51-0803-0. [Google Scholar]
- Shamovsky, I.; Nudler, E. New Insights into the Mechanism of Heat Shock Response Activation. Cell. Mol. Life Sci. 2008, 65, 855–861. [Google Scholar] [CrossRef]
- Larkindale, J.; Vierling, E. Core Genome Responses Involved in Acclimation to High Temperature. Plant Physiol. 2008, 146, 323–324. [Google Scholar] [CrossRef] [Green Version]
- Finka, A.; Sood, V.; Quadroni, M.; Rios, P.D.L.; Goloubinoff, P. Quantitative Proteomics of Heat-Treated Human Cells Show an across-the-Board Mild Depletion of Housekeeping Proteins to Massively Accumulate Few HSPs. Cell Stress Chaperones 2015, 20, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Li, G.C.; Werb, Z. Correlation between Synthesis of Heat Shock Proteins and Development of Thermotolerance in Chinese Hamster Fibroblasts. Proc. Natl. Acad. Sci. USA 1982, 79, 3218–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, R.; Fodor, E. Cell-Specific Expression of Heat Shock Proteins in Chicken Reticulocytes and Lymphocytes. J. Cell Biol. 1984, 99, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettaieb, A.; Averill-Bates, D.A. Thermotolerance Induced at a Mild Temperature of 40 °C Protects Cells against Heat Shock-Induced Apoptosis. J. Cell. Physiol. 2005, 205, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Averill-Bates, D.A. Thermotolerance Induced at a Fever Temperature of 40 °C Protects Cells against Hyperthermia-Induced Apoptosis Mediated by Death Receptor Signalling. Biochem. Cell Biol. 2008, 86, 521–538. [Google Scholar] [CrossRef]
- Shigapova, N.; Török, Z.; Balogh, G.; Goloubinoff, P.; Vígh, L.; Horváth, I. Membrane Fluidization Triggers Membrane Remodeling Which Affects the Thermotolerance in Escherichia Coli. Biochem. Biophys. Res. Commun. 2005, 328, 1216–1223. [Google Scholar] [CrossRef]
- González-Cruz, J.; Pastenes, C. Water-Stress-Induced Thermotolerance of Photosynthesis in Bean (Phaseolus vulgaris L.) Plants: The Possible Involvement of Lipid Composition and Xanthophyll Cycle Pigments. Environ. Exp. Bot. 2012, 77, 127–140. [Google Scholar] [CrossRef]
- Armston, A.E.; Halestrap, A.P.; Scott, R.D. The Nature of the Changes in Liver Mitochondrial Function Induced by Glucagon Treatment of Rats. The Effects of Intramitochondrial Volume, Aging and Benzyl Alcohol. Biochim. Biophys. Acta (BBA)-Bioenerg. 1982, 681, 429–439. [Google Scholar] [CrossRef]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an Intermediate in Metabolism, Signal Transduction, ROS, Hypoxia, and Tumorigenesis. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Zhu, W.; Yu, J.; Wang, Q.; Zhang, J.; Cui, Y.; Pan, X.; Gao, X.; Sun, H. Succinate Accumulation Induces Mitochondrial Reactive Oxygen Species Generation and Promotes Status Epilepticus in the Kainic Acid Rat Model. Redox Biol. 2020, 28, 101365. [Google Scholar] [CrossRef]
- Chang, Y.-S.; Lin, C.-F.; Wu, C.-L.; Kuo, P.-Y.; Wu, F.-S.; Shieh, C.-C.; Gean, P.-W.; Chen, S.-T.; Chen, M.-S.; Wu, W.-C.; et al. Mechanisms Underlying Benzyl Alcohol Cytotoxicity (Triamcinolone Acetonide Preservative) in Human Retinal Pigment Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4214. [Google Scholar] [CrossRef]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of Mitochondrial Lipid Oxidation: Role of 4-Hydroxynonenal (4-HNE) and Other Bioactive Lipids in Mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Kalmar, B.; Greensmith, L. Induction of Heat Shock Proteins for Protection against Oxidative Stress. Adv. Drug Deliv. Rev. 2009, 61, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Balogi, Z.; Khachatryan, W.; Gao, H.; Vígh, L.; Multhoff, G. Membrane-Associated Heat Shock Proteins in Oncology: From Basic Research to New Theranostic Targets. Cells 2020, 9, 1263. [Google Scholar] [CrossRef] [PubMed]
- Kariya, A.; Tabuchi, Y.; Yunoki, T.; Kondo, T. Identification of Common Gene Networks Responsive to Mild Hyperthermia in Human Cancer Cells. Int. J. Mol. Med. 2013, 32, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettaieb, A.; Averill-Bates, D.A. Thermotolerance Induced at a Mild Temperature of 40 °C Alleviates Heat Shock-Induced ER Stress and Apoptosis in HeLa Cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Gupta, S.; Hu, W.; McGrath, B.C.; Cavener, D.R. Hyperthermia Induces the ER Stress Pathway. PLoS ONE 2011, 6, e23740. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, J.; Mattes, C.; Väth, K.; Radanović, T.; Surma, M.A.; Klose, C.; Ernst, R. A Quantitative Analysis of Cellular Lipid Compositions During Acute Proteotoxic ER Stress Reveals Specificity in the Production of Asymmetric Lipids. Front. Cell Dev. Biol. 2020, 8, 756. [Google Scholar] [CrossRef]
- Balogh, G.; Péter, M.; Liebisch, G.; Horváth, I.; Török, Z.; Nagy, E.; Maslyanko, A.; Benkő, S.; Schmitz, G.; Harwood, J.L.; et al. Lipidomics Reveals Membrane Lipid Remodelling and Release of Potential Lipid Mediators during Early Stress Responses in a Murine Melanoma Cell Line. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2010, 1801, 1036–1047. [Google Scholar] [CrossRef]
- Ariyama, H.; Kono, N.; Matsuda, S.; Inoue, T.; Arai, H. Decrease in Membrane Phospholipid Unsaturation Induces Unfolded Protein Response. J. Biol. Chem. 2010, 285, 22027–22035. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro, R.; Falasca, M. Lysophosphatidylinositol Signalling: New Wine from an Old Bottle. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2012, 1821, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Vong, C.T.; Tseng, H.H.L.; Kwan, Y.W.; Lee, S.M.-Y.; Hoi, M.P.M. Novel Protective Effect of O-1602 and Abnormal Cannabidiol, GPR55 Agonists, on ER Stress-Induced Apoptosis in Pancreatic β-Cells. Biomed. Pharmacother. 2019, 111, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.M.; Citir, M.; Terzi, E.M.; Cimen, I.; Yildirim, Z.; Dogan, A.E.; Kocaturk, B.; Onat, U.I.; Arditi, M.; Weber, C.; et al. Inositol-requiring Enzyme-1 Regulates Phosphoinositide Signaling Lipids and Macrophage Growth. EMBO Rep. 2020, 21, e51462. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Balogi, Z.; Gombos, I.; Åkerfelt, M.; Björkbom, A.; Balogh, G.; Török, Z.; Maslyanko, A.; Fiszer-Kierzkowska, A.; Lisowska, K.; et al. Hyperfluidization-Coupled Membrane Microdomain Reorganization Is Linked to Activation of the Heat Shock Response in a Murine Melanoma Cell Line. Proc. Natl. Acad. Sci. USA 2007, 104, 7945–7950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, R.; Ejsing, C.S.; Antonny, B. Homeoviscous Adaptation and the Regulation of Membrane Lipids. J. Mol. Biol. 2016, 428, 4776–4791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buda, C.; Dey, I.; Balogh, N.; Horvath, L.I.; Maderspach, K.; Juhasz, M.; Yeo, Y.K.; Farkas, T. Structural Order of Membranes and Composition of Phospholipids in Fish Brain Cells during Thermal Acclimatization. Proc. Natl. Acad. Sci. USA 1994, 91, 8234–8238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, T.; Kitajka, K.; Fodor, E.; Csengeri, I.; Lahdes, E.; Yeo, Y.K.; Krasznai, Z.; Halver, J.E. Docosahexaenoic Acid-Containing Phospholipid Molecular Species in Brains of Vertebrates. Proc. Natl. Acad. Sci. USA 2000, 97, 6362–6366. [Google Scholar] [CrossRef] [Green Version]
- Sinensky, M. Homeoviscous Adaptation—A Homeostatic Process That Regulates the Viscosity of Membrane Lipids in Escherichia Coli. Proc. Natl. Acad. Sci. USA 1974, 71, 522–525. [Google Scholar] [CrossRef] [Green Version]
- Singer, S.J.; Nicolson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes: Cell Membranes Are Viewed as Two-Dimensional Solutions of Oriented Globular Proteins and Lipids. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Vereb, G.; Szöllősi, J.; Matkó, J.; Nagy, P.; Farkas, T.; Vígh, L.; Mátyus, L.; Waldmann, T.A.; Damjanovich, S. Dynamic, yet Structured: The Cell Membrane Three Decades after the Singer–Nicolson Model. Proc. Natl. Acad. Sci. USA 2003, 100, 8053–8058. [Google Scholar] [CrossRef] [Green Version]
- Horváth, I.; Multhoff, G.; Sonnleitner, A.; Vígh, L. Membrane-Associated Stress Proteins: More than Simply Chaperones. Biochim. Biophys. Acta (BBA)-Biomembr. 2008, 1778, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Escribá, P.V.; Busquets, X.; Inokuchi, J.; Balogh, G.; Török, Z.; Horváth, I.; Harwood, J.L.; Vígh, L. Membrane Lipid Therapy: Modulation of the Cell Membrane Composition and Structure as a Molecular Base for Drug Discovery and New Disease Treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vígh, L.; Literáti, P.N.; Horváth, I.; Török, Z.; Balogh, G.; Glatz, A.; Kovács, E.; Boros, I.; Ferdinándy, P.; Parkas, B.; et al. Bimoclomol: A Nontoxic, Hydroxylamine Derivative with Stress Protein-Inducing Activity and Cytoprotective Effects. Nat. Med. 1997, 3, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Rosselló, C.A.; Fernández-García, P.; Lladó, V.; Kakhlon, O.; Escribá, P.V. The Implications for Cells of the Lipid Switches Driven by Protein–Membrane Interactions and the Development of Membrane Lipid Therapy. IJMS 2020, 21, 2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csoboz, B.; Balogh, G.E.; Kusz, E.; Gombos, I.; Peter, M.; Crul, T.; Gungor, B.; Haracska, L.; Bogdanovics, G.; Torok, Z.; et al. Membrane Fluidity Matters: Hyperthermia from the Aspects of Lipids and Membranes. Int. J. Hyperth. 2013, 29, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapeinos, C.; Marino, A.; Battaglini, M.; Migliorin, S.; Brescia, R.; Scarpellini, A.; De Julián Fernández, C.; Prato, M.; Drago, F.; Ciofani, G. Stimuli-Responsive Lipid-Based Magnetic Nanovectors Increase Apoptosis in Glioblastoma Cells through Synergic Intracellular Hyperthermia and Chemotherapy. Nanoscale 2019, 11, 72–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, P.; Wang, T.; Guzman, M.L.; Peter, R.I.; Chiosis, G. Chaperome Networks–Redundancy and Implications for Cancer Treatment. In HSF1 and Molecular Chaperones in Biology and Cancer; Mendillo, M.L., Pincus, D., Scherz-Shouval, R., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Germany, 2020; Volume 1243, pp. 87–99. ISBN 978-3-030-40203-7. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) The strongest expression change for sample pair 40 °C, 1 h compared to 37 °C. | |||
| Symbol (Mouse Orthologs) | Entrez Gene Name | Fold Change | FDR |
| HSPA1A/HSPA1B | heat shock protein family A (Hsp70) member 1A | 9.82 | 0.00 × 100 |
| GLYATL3 | glycine-N-acyltransferase like 3 | 4.65 | 7.30 × 103 |
| ZFAND2A | zinc finger AN1-type containing 2A | 3.29 | 0.00 × 100 |
| DNAJB1 | DnaJ heat shock protein family (Hsp40) member B1 | 3.17 | 0.00 × 100 |
| IL11 | interleukin 11 | 2.41 | 9.05 × 10−9 |
| CLK1 | CDC like kinase 1 | 2.35 | 0.00 × 100 |
| HSPB1 | heat shock protein family B (small) member 1 | 2.35 | 9.02 × 10−12 |
| HSPH1 | heat shock protein family H (Hsp110) member 1 | 2.23 | 2.18 × 10−8 |
| JUN | Jun proto-oncogene, AP-1 transcription factor subunit | 2.09 | 0.00 × 100 |
| IER5L | immediate early response 5 like | 2.08 | 3.18 × 10−12 |
| RSRP1 | arginine and serine rich protein 1 | 2.03 | 0.00 × 100 |
| RBAK | RB associated KRAB zinc finger | 1.86 | 6.28 × 10−7 |
| BAG3 | BCL2 associated athanogene 3 | 1.84 | 4.00 × 10−8 |
| DEDD2 | death effector domain containing 2 | 1.83 | 9.10 × 10−7 |
| CEP295NL | CEP295 N-terminal like | 1.77 | 1.83 × 10−4 |
| HSPA8 | heat shock protein family A (Hsp70) member 8 | 1.65 | 6.05 × 10−10 |
| TIPARP | TCDD inducible poly(ADP-ribose) polymerase | 1.63 | 3.27 × 10−9 |
| DUSP8 | dual specificity phosphatase 8 | 1.61 | 1.10 × 10−7 |
| NUAK2 | NUAK family kinase 2 | 1.60 | 1.00 × 10−2 |
| CCDC117 | coiled-coil domain containing 117 | 1.58 | 1.73 × 10−5 |
| RIN2 | Ras and Rab interactor 2 | −309.83 | 4.55 × 10−9 |
| HOXB3 | homeobox B3 | −2.33 | 5.07 × 10−3 |
| LCMT2 | leucine carboxyl methyltransferase 2 | −2.30 | 1.39 × 10−4 |
| NAA20 | N(alpha)-acetyltransferase 20, NatB catalytic subunit | −2.13 | 8.40 × 10−3 |
| PLK2 | polo like kinase 2 | −1.90 | 0.00 × 100 |
| PTP4A1 | protein tyrosine phosphatase 4A1 | −1.73 | 4.00 × 10−2 |
| MARCHF7 | membrane associated ring-CH-type finger 7 | −1.66 | 2.68 × 10−9 |
| GAS8 | growth arrest specific 8 | −1.58 | 4.63 × 10−3 |
| FDXACB1 | ferredoxin-fold anticodon binding domain containing 1 | −1.52 | 9.58 × 10−3 |
| (b) The strongest expression change for sample pair 40 °C, 6 h compared to 37 °C. | |||
| Symbol (Mouse Orthologs) | Entrez Gene Name | Fold Change | FDR |
| CRNKL1 | crooked neck pre-mRNA splicing factor 1 | 187.36 | 2.00 × 10−2 |
| CEL | carboxyl ester lipase | 13.19 | 4.00 × 10−2 |
| DNAH10 | dynein axonemal heavy chain 10 | 8.81 | 2.00 × 10−2 |
| GLYATL3 | glycine-N-acyltransferase like 3 | 7.06 | 5.88 × 10−6 |
| CCDC154 | coiled-coil domain containing 154 | 7.04 | 1.04 × 10−3 |
| WDR66 | WD repeat domain 66 | 6.16 | 4.99 × 10−6 |
| HSPA1A/HSPA1B | heat shock protein family A (Hsp70) member 1A | 5.50 | 1.05 × 10−13 |
| ARHGEF25 | Rho guanine nucleotide exchange factor 25 | 4.85 | 2.00 × 10−2 |
| PTP4A1 | protein tyrosine phosphatase 4A1 | 4.81 | 5.93 × 10−3 |
| COL24A1 | collagen type XXIV alpha 1 chain | 4.69 | 5.00 × 10−2 |
| SPEF1 | sperm flagellar 1 | 4.50 | 9.98 × 10−3 |
| HHIPL2 | HHIP like 2 | 4.19 | 3.53 × 10−6 |
| TNFRSF25 | TNF receptor superfamily member 25 | 3.77 | 1.00 × 10−2 |
| MTMR7 | myotubularin related protein 7 | 3.24 | 3.76 × 10−4 |
| HSPB1 | heat shock protein family B (small) member 1 | 3.05 | 0.00 × 100 |
| RGL3 | ral guanine nucleotide dissociation stimulator like 3 | 3.04 | 1.00 × 10−2 |
| RHBDL1 | rhomboid like 1 | 2.94 | 1.00 × 10−2 |
| HDAC9 | histone deacetylase 9 | 2.92 | 8.08 × 10−5 |
| TMED6 | transmembrane p24 trafficking protein 6 | 2.89 | 3.00 × 10−2 |
| ANKRD13D | ankyrin repeat domain 13D | 2.87 | 3.01 × 10−3 |
| RIN2 | Ras and Rab interactor 2 | −42.68 | 6.65 × 10−6 |
| IYD | iodotyrosine deiodinase | −8.96 | 6.90 × 10−3 |
| NAA20 | N(alpha)-acetyltransferase 20, NatB catalytic subunit | −5.69 | 1.33 × 10−12 |
| THEMIS2 | thymocyte selection associated family member 2 | −5.08 | 3.00 × 10−2 |
| FNDC10 | fibronectin type III domain containing 10 | −4.07 | 8.76 × 10−4 |
| TENM3 | teneurin transmembrane protein 3 | −3.66 | 4.01 × 10−4 |
| SDCBP2 | syndecan binding protein 2 | −3.60 | 7.06 × 10−3 |
| Ccdc74a | coiled-coil domain containing 74A | −3.14 | 4.00 × 10−2 |
| CAPN8 | calpain 8 | −3.11 | 2.45 × 10−5 |
| Dnajb3 | DnaJ heat shock protein family (Hsp40) member B3 | −2.98 | 1.98 × 10−4 |
| Gm1110 | predicted gene 1110 | −2.98 | 2.35 × 10−4 |
| Ccl2 | chemokine (C-C motif) ligand 2 | −2.97 | 0.00 × 100 |
| Ccl7 | chemokine (C-C motif) ligand 7 | −2.95 | 5.68 × 10−11 |
| EGR1 | early growth response 1 | −2.88 | 0.00 × 100 |
| EGR3 | early growth response 3 | −2.82 | 0.00 × 100 |
| CST6 | cystatin E/M | −2.78 | 4.00 × 10−2 |
| CXCL10 | C-X-C motif chemokine ligand 10 | −2.74 | 3.00 × 10−2 |
| CCR7 | C-C motif chemokine receptor 7 | −2.67 | 0.00 × 100 |
| HNRNPDL | heterogeneous nuclear ribonucleoprotein D like | −2.63 | 0.00 × 100 |
| BCLAF1 | BCL2 associated transcription factor 1 | −2.53 | 5.19 × 10−3 |
| (c) The strongest expression change for sample pair 42.5 °C, 1 h compared to 37 °C. | |||
| Symbol (Mouse Orthologs) | Entrez Gene Name | Fold Change | FDR |
| HSPA1A/HSPA1B | heat shock protein family A (Hsp70) member 1A | 464.60 | 0.00 × 100 |
| CRNKL1 | crooked neck pre-mRNA splicing factor 1 | 182.50 | 1.00 × 10−2 |
| GAB2 | GRB2 associated binding protein 2 | 93.26 | 4.00 × 10−2 |
| KDF1 | keratinocyte differentiation factor 1 | 58.54 | 4.73 × 10−5 |
| AKAP5 | A-kinase anchoring protein 5 | 52.76 | 1.14 × 10−7 |
| ALDH8A1 | aldehyde dehydrogenase 8 family member A1 | 41.51 | 2.77 × 10−4 |
| FOS | Fos proto-oncogene, AP-1 transcription factor subunit | 30.14 | 0.00 × 100 |
| LRIT3 | leucine rich repeat, Ig-like and transmembrane domains 3 | 26.41 | 5.34 × 10−13 |
| MAB21L1 | mab-21 like 1 | 23.95 | 3.27 × 10−3 |
| KLB | klotho beta | 22.63 | 1.07 × 10−6 |
| TRIM72 | tripartite motif containing 72 | 22.12 | 4.56 × 10−3 |
| AREG | amphiregulin | 21.64 | 0.00 × 100 |
| HAS1 | hyaluronan synthase 1 | 20.11 | 6.67 × 10−3 |
| ZFAND2A | zinc finger AN1-type containing 2A | 16.36 | 0.00 × 100 |
| CEL | carboxyl ester lipase | 15.64 | 2.00 × 10−2 |
| PDE8A | phosphodiesterase 8A | 15.04 | 2.00 × 10−2 |
| DNAJB1 | DnaJ heat shock protein family (Hsp40) member B1 | 11.82 | 0.00 × 100 |
| LRRC4 | leucine rich repeat containing 4 | 10.84 | 5.73 × 10−3 |
| NLGN3 | neuroligin 3 | 10.25 | 8.92 × 10−4 |
| HTR2B | 5-hydroxytryptamine receptor 2B | 9.09 | 1.00 × 10−2 |
| RIN2 | Ras and Rab interactor 2 | −29.93 | 7.82 × 10−7 |
| CXCL2 | C-X-C motif chemokine ligand 2 | −8.46 | 0.00 × 100 |
| BCLAF1 | BCL2 associated transcription factor 1 | −7.79 | 2.97 × 10−5 |
| DDAH2 | dimethylarginine dimethylaminohydrolase 2 | −5.82 | 2.00 × 10−2 |
| TLR6 | toll like receptor 6 | −4.96 | 1.62 × 10−6 |
| Gm11214 | glyceraldehyde-3-phosphate dehydrogenase pseudogene | −4.70 | 3.00 × 10−2 |
| PTP4A1 | protein tyrosine phosphatase 4A1 | −4.37 | 1.19 × 10−14 |
| HPS6 | HPS6 biogenesis of lysosomal organelles complex 2 subunit 3 | −3.98 | 0.00 × 100 |
| DDX28 | DEAD-box helicase 28 | −3.94 | 0.00 × 100 |
| OSBPL7 | oxysterol binding protein like 7 | −3.85 | 0.00 × 100 |
| HOXD4 | homeobox D4 | −3.83 | 1.22 × 10−5 |
| HOXA1 | homeobox A1 | −3.55 | 7.93 × 10−3 |
| ABTB1 | ankyrin repeat and BTB domain containing 1 | −3.53 | 1.38 × 10−12 |
| LFNG | LFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase | −3.45 | 0.00 × 100 |
| NFKBIA | NFKB inhibitor alpha | −3.45 | 0.00 × 100 |
| CSF3 | colony stimulating factor 3 | −3.44 | 6.38 × 10−3 |
| AMT | aminomethyltransferase | −3.39 | 4.60 × 10−14 |
| Iigp1 | interferon inducible GTPase 1 | −3.38 | 6.35 × 10−3 |
| MYORG | myogenesis regulating glycosidase (putative) | −3.37 | 6.90 × 10−8 |
| EPHB3 | EPH receptor B3 | −3.32 | 2.74 × 10−7 |
| Lipid Species | 37 °C | 40 °C, 1 h | 40 °C, 6 h | 42.5 °C, 1 h |
|---|---|---|---|---|
| Glycerophospholipids | ||||
| PC(32:1) | 2.568 ± 0.023 | 2.564 ± 0.025 | 2.680 ± 0.016 * | 2.537 ± 0.040 |
| PC(32:0) | 2.050 ± 0.061 | 2.264 ± 0.066 | 2.239 ± 0.036 * | 2.391 ± 0.053 * |
| PC(34:2) | 2.949 ± 0.029 | 2.936 ± 0.030 | 3.115 ± 0.025 * | 2.842 ± 0.044 |
| PC(34:1) | 12.691 ± 0.102 | 12.957 ± 0.117 | 13.844 ± 0.144 * | 12.924 ± 0.145 |
| PC(36:4) | 2.894 ± 0.060 | 2.847 ± 0.075 | 2.487 ± 0.031 * | 2.664 ± 0.054 * |
| PC(36:1) | 1.942 ± 0.028 | 1.970 ± 0.023 | 2.172 ± 0.051 * | 1.892 ± 0.028 |
| PC(38:6) | 1.468 ± 0.017 | 1.460 ± 0.037 | 1.322 ± 0.035 * | 1.256 ± 0.024 * |
| PC(38:5) | 2.438 ± 0.046 | 2.434 ± 0.075 | 1.953 ± 0.050 * | 2.231 ± 0.052 * |
| PC(38:4) | 1.657 ± 0.042 | 1.685 ± 0.057 | 1.359 ± 0.042 * | 1.588 ± 0.045 |
| PC(40:6) | 0.778 ± 0.009 | 0.770 ± 0.018 | 0.688 ± 0.022 * | 0.651 ± 0.013 * |
| PC(O-32:0) | 0.188 ± 0.007 | 0.202 ± 0.012 | 0.288 ± 0.013 * | 0.212 ± 0.010 |
| PC(O-34:1) | 1.755 ± 0.062 | 1.819 ± 0.056 | 2.172 ± 0.058 * | 1.941 ± 0.053 |
| PC(O-36:1) | 0.229 ± 0.016 | 0.236 ± 0.017 | 0.329 ± 0.016 * | 0.231 ± 0.015 |
| PC(O-38:5) | 1.174 ± 0.023 | 1.219 ± 0.020 | 1.069 ± 0.026 * | 1.298 ± 0.020 * |
| PE(34:1) | 0.730 ± 0.011 | 0.728 ± 0.018 | 0.805 ± 0.019 * | 0.671 ± 0.016 * |
| PE(38:6) | 0.406 ± 0.014 | 0.380 ± 0.006 | 0.339 ± 0.006 * | 0.355 ± 0.009 * |
| PE(38:5) | 0.714 ± 0.028 | 0.659 ± 0.013 | 0.571 ± 0.013 * | 0.630 ± 0.017 * |
| PE(38:4) | 1.221 ± 0.051 | 1.159 ± 0.032 | 1.007 ± 0.023 * | 1.111 ± 0.036 |
| PE(P-38:5) | 0.988 ± 0.027 | 0.920 ± 0.014 | 0.879 ± 0.016 * | 0.985 ± 0.026 |
| PE(P-40:7) | 0.434 ± 0.019 | 0.408 ± 0.009 | 0.343 ± 0.009 * | 0.456 ± 0.008 |
| PE(P-40:6) | 0.698 ± 0.015 | 0.683 ± 0.009 | 0.612 ± 0.011 * | 0.728 ± 0.014 |
| PE(P-40:5) | 0.583 ± 0.020 | 0.568 ± 0.010 | 0.493 ± 0.013 * | 0.625 ± 0.014 |
| PI(34:1) | 0.313 ± 0.012 | 0.336 ± 0.019 | 0.520 ± 0.026 * | 0.317 ± 0.013 |
| PI(36:1) | 0.268 ± 0.008 | 0.282 ± 0.012 | 0.428 ± 0.015 * | 0.349 ± 0.010 * |
| PI(38:5) | 0.485 ± 0.012 | 0.476 ± 0.006 | 0.424 ± 0.007 * | 0.438 ± 0.007 * |
| PI(38:4) | 4.089 ± 0.120 | 4.040 ± 0.078 | 3.612 ± 0.076 * | 4.189 ± 0.088 |
| PS(38:4) | 0.196 ± 0.004 | 0.177 ± 0.007 | 0.138 ± 0.003 * | 0.218 ± 0.006 * |
| PG(40:7) | 0.164 ± 0.006 | 0.155 ± 0.009 | 0.129 ± 0.007 * | 0.156 ± 0.012 |
| PG(44:12) | 0.056 ± 0.003 | 0.050 ± 0.002 | 0.024 ± 0.002 * | 0.045 ± 0.004 |
| PG(44:11) | 0.020 ± 0.002 | 0.018 ± 0.001 | 0.005 ± 0.001 * | 0.018 ± 0.002 |
| CL(72:6) | 0.196 ± 0.008 | 0.181 ± 0.003 | 0.165 ± 0.003 * | 0.186 ± 0.004 |
| CL(72:5) | 0.133 ± 0.004 | 0.124 ± 0.004 | 0.113 ± 0.002 * | 0.129 ± 0.004 |
| Lysophospholipids | ||||
| LPC(16:0) | 0.085 ± 0.007 | 0.091 ± 0.010 | 0.118 ± 0.011 * | 0.083 ± 0.008 |
| LPC(18:0) | 0.029 ± 0.003 | 0.034 ± 0.004 | 0.042 ± 0.002 * | 0.029 ± 0.002 |
| LPI(16:0) | 0.010 ± 0.000 | 0.012 ± 0.001 * | 0.022 ± 0.001 * | 0.013 ± 0.001 |
| LPI(18:0) | 0.062 ± 0.002 | 0.074 ± 0.003 * | 0.123 ± 0.005 * | 0.101 ± 0.010 * |
| Sphingolipids | ||||
| SM(34:1:2) | 6.201 ± 0.069 | 6.260 ± 0.077 | 6.305 ± 0.079 | 6.759 ± 0.071 * |
| SM(34:0:2) | 0.082 ± 0.004 | 0.087 ± 0.003 | 0.074 ± 0.005 | 0.099 ± 0.004 * |
| Cer(34:1:2) | 0.097 ± 0.004 | 0.090 ± 0.002 | 0.105 ± 0.002 | 0.142 ± 0.003 * |
| Cer(42:2:2) | 0.065 ± 0.002 | 0.073 ± 0.002 * | 0.090 ± 0.002 * | 0.090 ± 0.002 * |
| HexCer(34:1:2) | 0.155 ± 0.006 | 0.156 ± 0.006 | 0.145 ± 0.004 | 0.203 ± 0.005 * |
| HexCer(42:2:2) | 0.049 ± 0.002 | 0.049 ± 0.002 | 0.045 ± 0.001 | 0.061 ± 0.002 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiszlavicz, Á.; Gombos, I.; Péter, M.; Hegedűs, Z.; Hunya, Á.; Dukic, B.; Nagy, I.; Peksel, B.; Balogh, G.; Horváth, I.; et al. Distinct Cellular Tools of Mild Hyperthermia-Induced Acquired Stress Tolerance in Chinese Hamster Ovary Cells. Biomedicines 2022, 10, 1172. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051172
Tiszlavicz Á, Gombos I, Péter M, Hegedűs Z, Hunya Á, Dukic B, Nagy I, Peksel B, Balogh G, Horváth I, et al. Distinct Cellular Tools of Mild Hyperthermia-Induced Acquired Stress Tolerance in Chinese Hamster Ovary Cells. Biomedicines. 2022; 10(5):1172. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051172
Chicago/Turabian StyleTiszlavicz, Ádám, Imre Gombos, Mária Péter, Zoltán Hegedűs, Ákos Hunya, Barbara Dukic, István Nagy, Begüm Peksel, Gábor Balogh, Ibolya Horváth, and et al. 2022. "Distinct Cellular Tools of Mild Hyperthermia-Induced Acquired Stress Tolerance in Chinese Hamster Ovary Cells" Biomedicines 10, no. 5: 1172. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051172