
IL-33 Enhances ACE2 Expression on Epidermal Keratinocytes in Atopic Dermatitis: A Plausible Issue for SARS-CoV-2 Transmission in Inflamed Atopic Skin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Skin Samples for Immunohistochemistry
2.2. Culture for Primary Keratinocytes
2.3. Immunohistochemistry
2.4. Quantitative RT-PCR
2.5. Flow Cytometry
2.6. Immunofluorescent Exam
3. Results
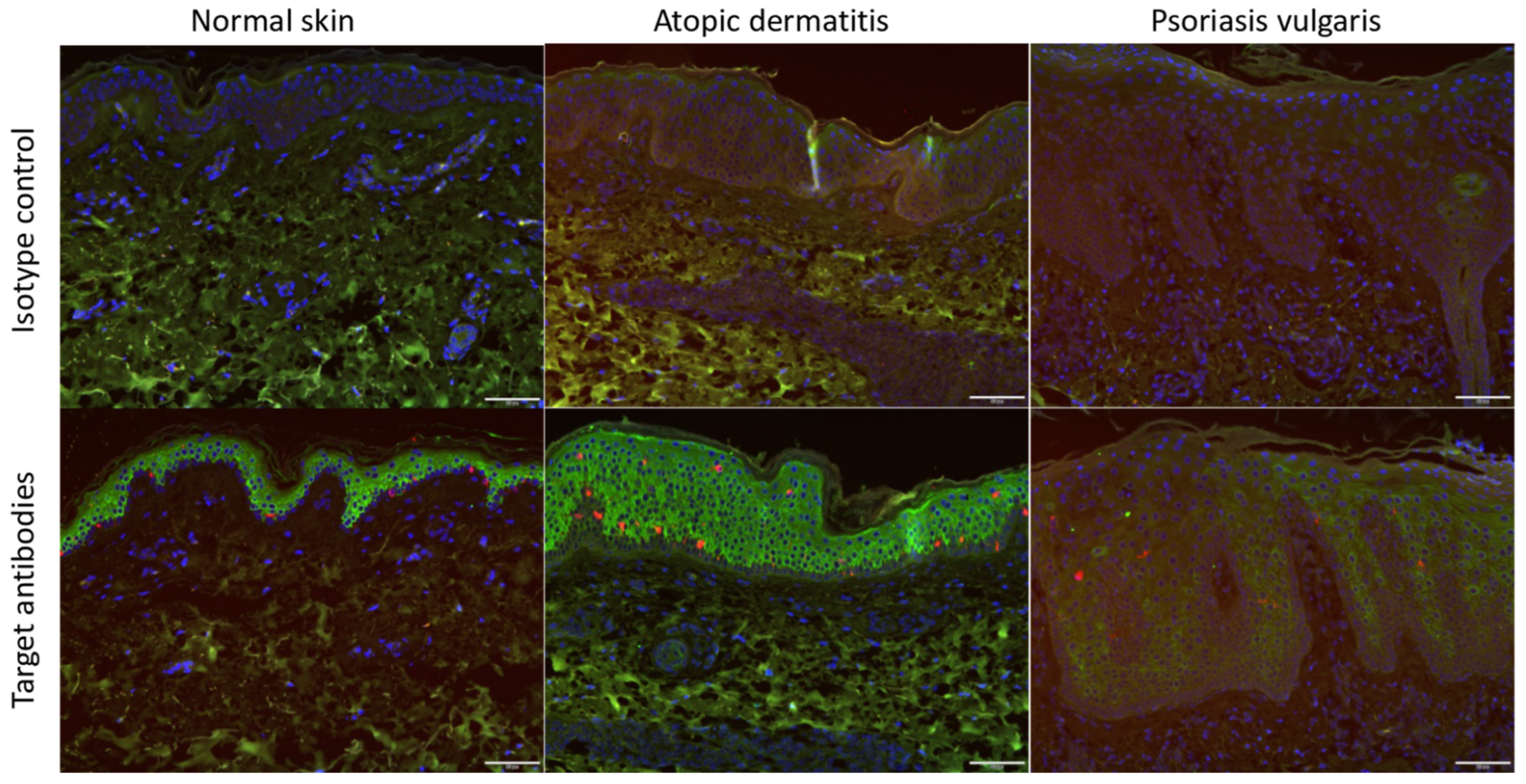
3.1. ACE2 Expression Was Increased in Atopic Dermatitis Compared with That in Normal Skin
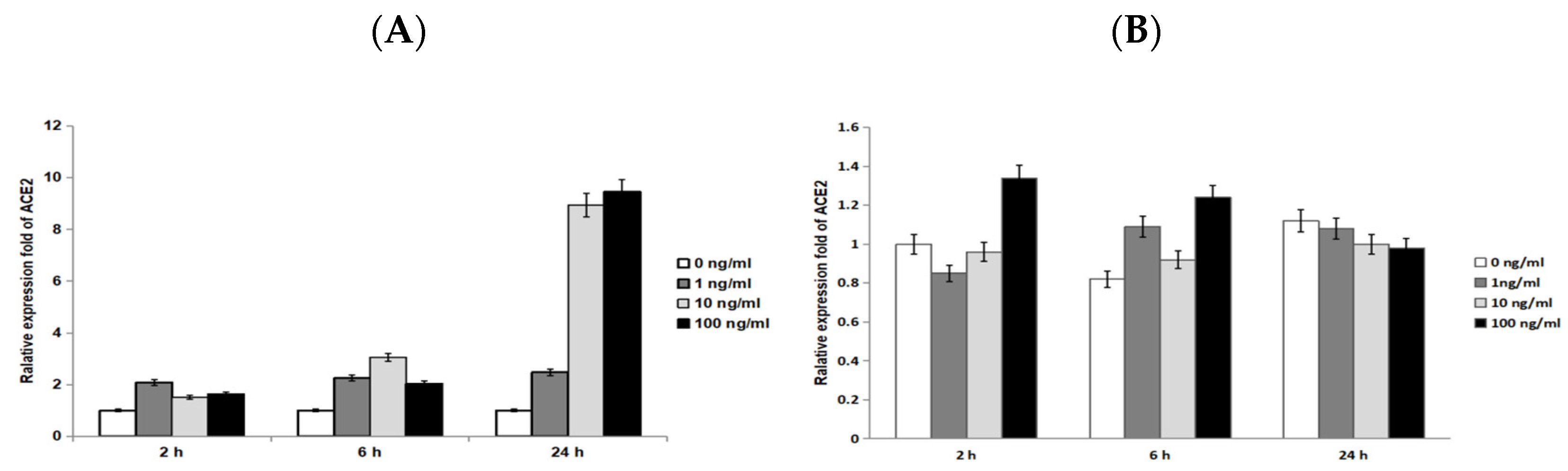
3.2. Through Quantitative RT-PCR, ACE2 mRNA Expression Increased in a Time-Dependent Manner under IL-33 Stimulation
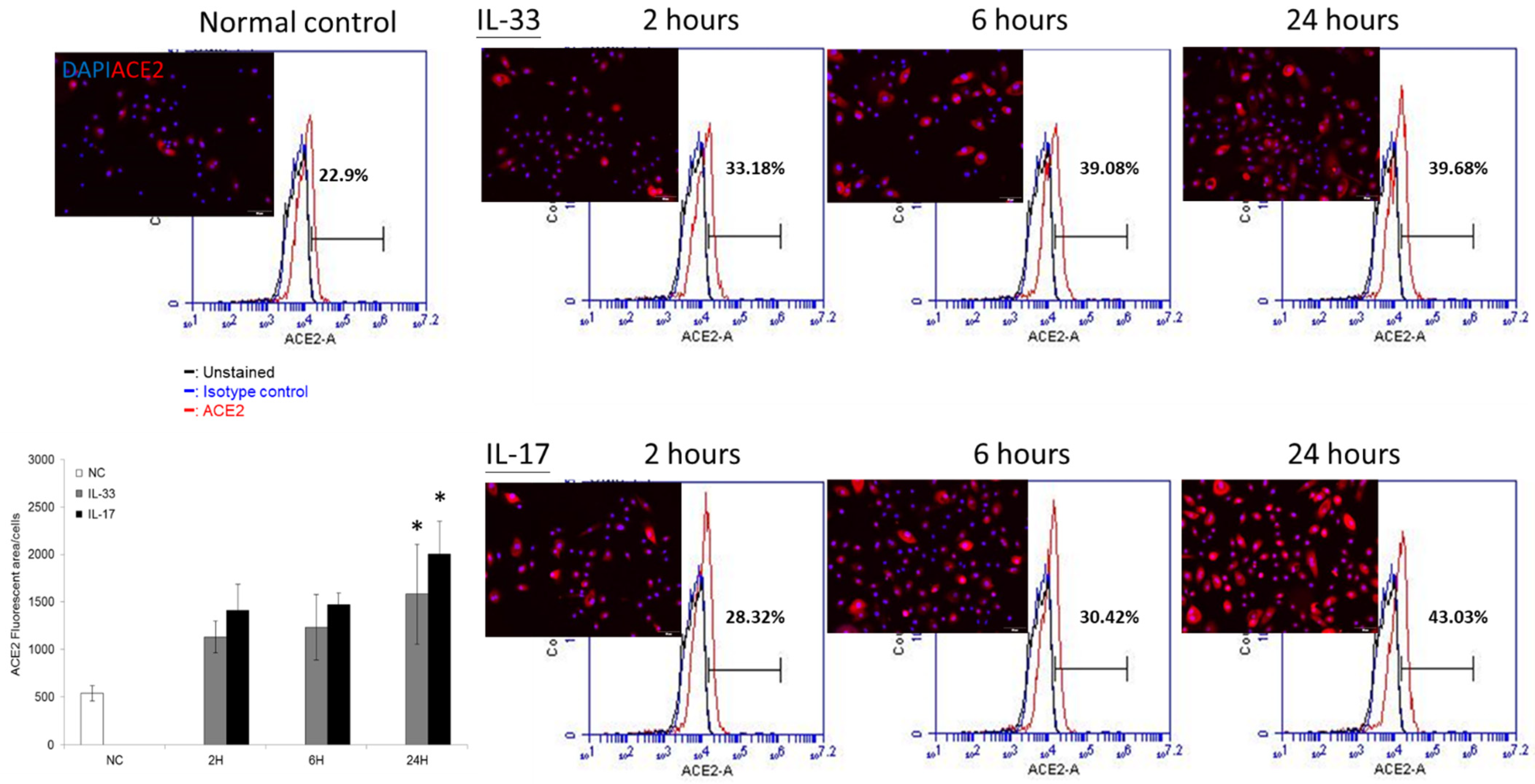
3.3. ACE2 Is Constitutively Expressed in the Cultured Keratinocytes — Both IL-33 and IL-17 Enhance the ACE2 Protein Expression in a Time-Dependent Manner
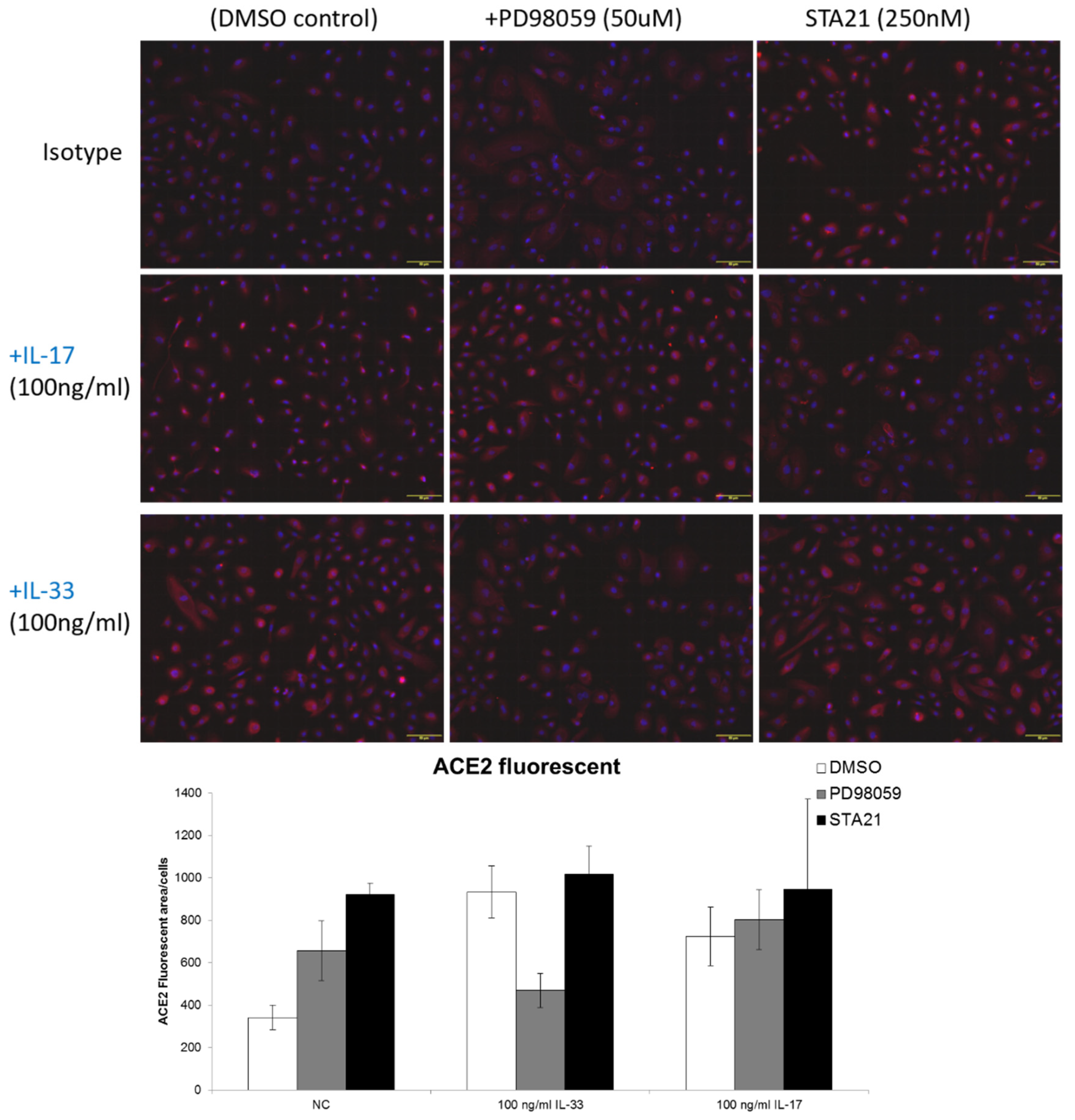
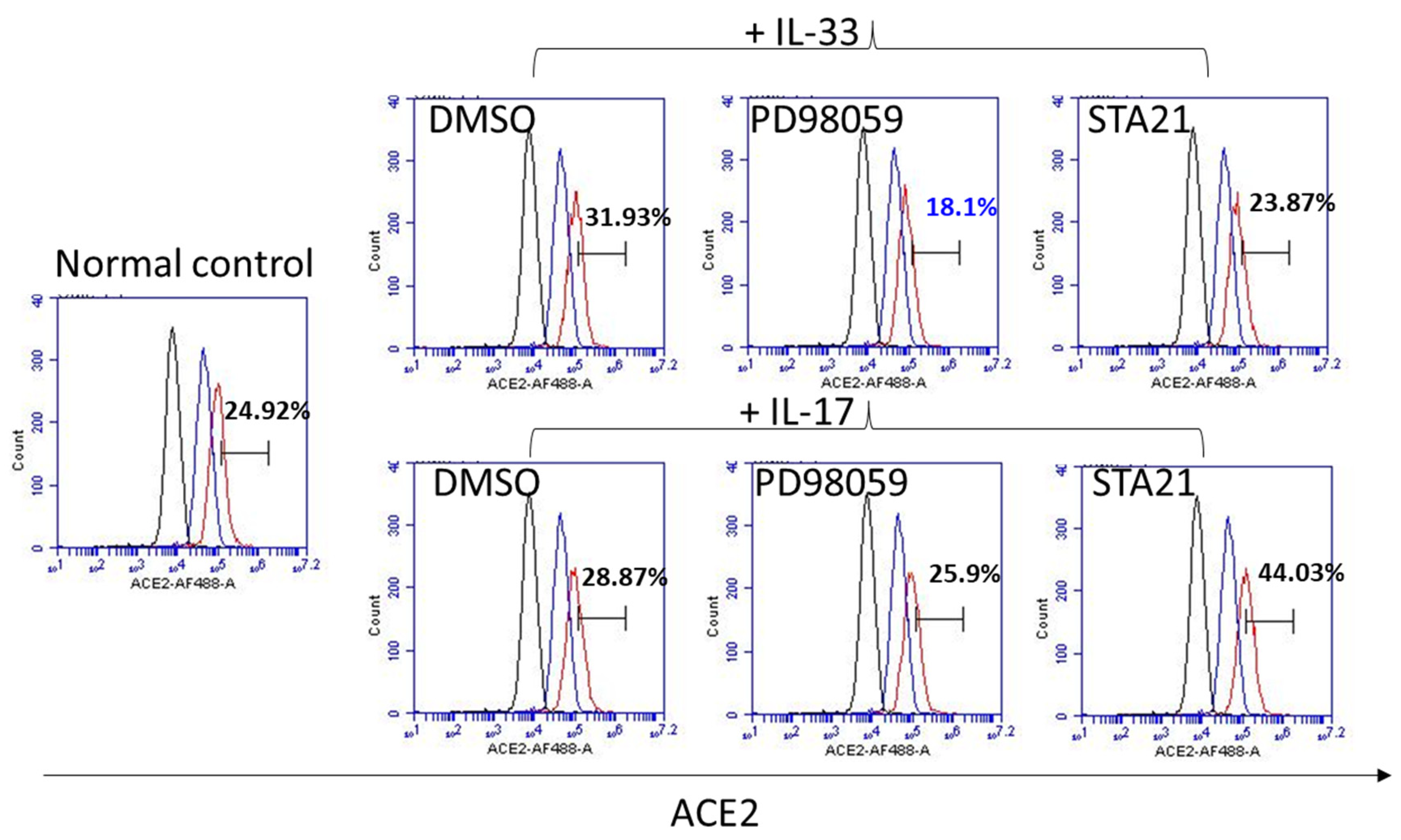
3.4. IL-33 Induces the ACE2 Expression, Which Is Abrogated by Pretreatment with PD98059
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nutten, S. Atopic Dermatitis: Global Epidemiology and Risk Factors. Ann. Nutr. Metab. 2015, 66 (Suppl. S1), 8–16. [Google Scholar] [CrossRef]
- Nakajima, S.; Nomura, T.; Common, J.; Kabashima, K. Insights into atopic dermatitis gained from genetically defined mouse models. J. Allergy Clin. Immunol. 2019, 143, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33–driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Stott, B.; Lavender, P.; Lehmann, S.; Pennino, D.; Durham, S.; Schmidt-Weber, C. Human IL-31 is induced by IL-4 and promotes TH2-driven inflammation. J. Allergy Clin. Immunol. 2013, 132, 446–454.e5. [Google Scholar] [CrossRef] [PubMed]
- Coyle, A.J.; Lloyd, C.; Tian, J.; Nguyen, T.; Erikkson, C.; Wang, L.; Ottoson, P.; Persson, P.; Delaney, T.; Lehar, S.; et al. Crucial Role of the Interleukin 1 Receptor Family Member T1/St2 in T Helper Cell Type 2–Mediated Lung Mucosal Immune Responses. J. Exp. Med. 1999, 190, 895–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savinko, T.; Matikainen, S.; Saarialho-Kere, U.; Lehto, M.; Wang, G.; Lehtimäki, S.; Karisola, P.; Reunala, T.; Wolff, H.; Lauerma, A.; et al. IL-33 and ST2 in Atopic Dermatitis: Expression Profiles and Modulation by Triggering Factors. J. Investig. Dermatol. 2012, 132, 1392–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J. The IL-1-Like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [Green Version]
- Petra, I.; Tsilioni, I.; Taracanova, A.; Katsarou-Katsari, A.; Theoharides, C. Interleukin 33 and interleukin 4 regulate interleukin 31 gene expression and secretion from human laboratory of allergic diseases 2 mast cells stimulated by substance P and/or immunoglobulin E. Allergy. Asthma. Proc. 2018, 39, 153–160. [Google Scholar] [CrossRef]
- Kaur, D.; Gomez, E.; Doe, C.; Berair, R.; Woodman, L.; Saunders, R.; Hollins, F.; Rose, F.R.; Amrani, Y.; May, R.; et al. IL-33 drives airway hyperresponsiveness through IL-13-mediated mast cell: Airay smooth muscle crosstalk. Allergy 2015, 70, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, C.; Otsuka, A.; Kabashima, K. Interleukin-31 and interleukin-31 receptor: New therapeutic targets for atopic dermatitis. Exp. Derm. 2018, 27, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Mai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13921–13926. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, H.; Nagao, K.; Kubo, A.; Hata, T.; Shimizu, A.; Mizuno, H.; Yamada, T.; Amagai, M. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J. Allergy Clin. Immunol. 2012, 129, 1538–1546.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Londei, M.; Kenney, B.; Los, G.; Marino, M.H. A phase 1 study of ANB020, an anti-IL-33 monoclonal antibody, in healthy volunteers. J. Am. Acad. Dermatol. 2017, 76, AB20. [Google Scholar] [CrossRef]
- Polak, S.; Van Gool, I.; Cohen, D.; von der Thüsen, J.; van Paassen, J. A systematic review of pathological findings in COVID-19: A pathophysiological timeline and possible mechanisms of disease progression. Mod. Pathol. 2020, 33, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kobayashi, T.; Iijima, K.; Hsu, F.; Kita, H. IL-33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J. Allergy Clin. Immunol. 2017, 140, 1351–1363.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Tang, J.; Yang, X.; Zanvit, P.; Cui, K.; Ku, W.L.; Jin, W.; Zhang, D.; Goldberg, N.; Cain, A.; et al. TGF-β induces ST2 and programs ILC2 development. Nat. Commun. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Guggino, G.; Ciccia, F.; Di Liberto, D.; Lo Pizzo, M.; Ruscitti, P.; Cipriani, P.; Ferrante, A.; Sireci, G.; Dieli, F.; Fourniè, J.J.; et al. Interleukin (IL)-9/IL-9R axis drives γδ T cells activation in psoriatic arthritis patients. Clin. Exp. Immunol. 2016, 186, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Häsler, R.; Wesch, D.; Kabelitz, D. Human Vδ2 T cells are a major source of interleukin-9. Proc. Natl. Acad. Sci. USA 2016, 113, 12520–12525. [Google Scholar] [CrossRef] [Green Version]
- Halim, T.; Krauß, R.; Sun, A.; Takei, F. Lung Natural Helper Cells Are a Critical Source of Th2 Cell-Type Cytokines in Protease Allergen-Induced Airway Inflammation. Immunity 2012, 36, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Yang, F.; Zhou, Y.; Yang, H.; Low, P.Y.; Kemeny, D.M.; Tan, P.; Moh, A.; Kaplan, M.H.; Zhang, Y.; et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014, 24, 1387–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papotto, P.; Reinhardt, A.; Prinz, I.; Silva-Santos, B. Innately versatile: γδ17 T cells in inflammatory and autoimmune diseases. J. Autoimmun. 2018, 87, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Willart, M.A.; Deswarte, K.; Pouliot, P.; Braun, H.; Beyaert, R.; Lambrecht, B.N.; Hammad, H. Interleukin-1α controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J. Exp. Med. 2012, 209, 1505–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llop-Guevara, A.; Chu, D.K.; Walker, T.D.; Goncharova, S.; Fattouh, R.; Silver, J.S.; Moore, C.L.; Xie, J.L.; O’Byrne, P.M.; Coyle, A.J.; et al. A GM-CSF/IL-33 Pathway Facilitates Allergic Airway Responses to Sub-Threshold House Dust Mite Exposure. PLoS ONE 2014, 9, e88714. [Google Scholar] [CrossRef] [Green Version]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef] [PubMed]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef]
- Marcu-Malina, V.; Balbir-Gurman, A.; Dardik, R.; Braun-Moscovici, Y.; Segel, M.; Bank, I. A Novel Prothrombotic Pathway in Systemic Sclerosis Patients: Possible Role of Bisphosphonate-Activated Î3Î’ T Cells. Front. Immunol. 2014, 5, 414. [Google Scholar] [CrossRef] [Green Version]
- George, P.; Wells, A.; Jenkins, R. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Cesare, A.; Meglio, P.; Nestle, F. A Role for Th17 Cells in the Immunopathogenesis of Atopic Dermatitis? J. Investig. Dermatol. 2008, 128, 2569–2571. [Google Scholar] [CrossRef] [Green Version]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Selvakumar, T.A.; McPherson, T.; Taylor, S.; Ogg, G.S. IL-17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp. Derm. 2012, 21, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, A.; Nograles, K.E.; Johnson-Huang, L.M.; Fuentes-Duculan, J.; Cardinale, I.; Bonifacio, K.M.; Gulati, N.; Mitsui, H.; Guttman-Yassky, E.; Suárez-Fariñas, M. IL-17 Induces an Expanded Range of Downstream Genes in Reconstituted Human Epidermis Model. PLoS ONE 2014, 9, e90284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crish, J.; Gopalakrishnan, R.; Bone, F.; Gilliam, A.; Eckert, R. The Distal and Proximal Regulatory Regions of the Involucrin Gene Promoter have Distinct Functions and Are Required for In Vivo Involucrin Expression. J. Investig. Dermatol. 2006, 126, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, T. The Angiotensin AT2 Receptor: A major constituent of the “Protective Arm” of the Renin-Angiotensin System. Hypertens. News 2018. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Shi, Y.; Tan, Y.; Xiao, R. ACE2 Expression on the Keratinocytes and SARS-CoV-2 Percutaneous Transmission: Are they Related? J. Investig. Dermatol. 2021, 141, 197–198. [Google Scholar] [CrossRef]
- Xue, X.; Mi, Z.; Wang, Z.; Pang, Z.; Liu, H.; Zhang, F. High Expression of ACE2 on Keratinocytes Reveals Skin as a Potential Target for SARS-CoV-2. J. Investig. Dermatol. 2021, 141, 206–209.e1. [Google Scholar] [CrossRef]
- Tembhre, M.K.; Parihar, A.S.; Sharma, V.K.; Imran, S.; Bhari, N.; Lakshmy, R.; Bhalla, A. Enhanced expression of angiotensin-converting enzyme 2 in psoriatic skin and its upregulation in keratinocytes by interferon-γ: Implication of inflammatory milieu in skin tropism of SARS-CoV-2. Br. J. Dermatol. 2020, 184, 577–579. [Google Scholar] [CrossRef]
- Lee, C.; Wu, S.; Hong, C.; Chen, G.; Wei, Y.; Yu, H. Involvement of mtDNA Damage Elicited by Oxidative Stress in the Arsenical Skin Cancers. J. Investig. Dermatol. 2013, 133, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Ryu, W.; Lee, H.; Bae, H.; Ryu, H.; Son, S. IL-33 down-regulates filaggrin expression by inducing STAT3 and ERK phosphorylation in human keratinocytes. J. Derm. Sci. 2016, 82, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Ryu, W.I.; Lee, H.; Bae, H.C.; Jeon, J.; Ryu, H.J.; Kim, J.; Kim, J.H.; Son, J.W.; Kim, J.; Imai, Y. IL-33 down-regulates CLDN1 expression through the ERK/STAT3 pathway in keratinocytes. J. Derm. Sci. 2018, 90, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Lei, T.; Guo, P.; Yu, J.; Xu, Q.; Luo, Y.; Ke, R.; Huang, D. Mechanisms shaping the role of ERK1/2 in cellular sene scence (Review). Mol. Med. Rep. 2018, 19, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Mohammad, I.; Liu, Z. Overview of the STAT-3 signaling pathway in cancer and the development of specific inhibitors (Review). Oncol. Lett. 2020, 19, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J., Jr. STATs and Gene Regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Haura, E.; Turkson, J.; Jove, R. Mechanisms of Disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat. Clin. Pract. Oncol. 2005, 2, 315–324. [Google Scholar] [CrossRef]
- Buettner, R.; Huang, M.; Gritsko, T.; Karras, J.; Enkemann, S.; Mesa, T.; Nam, S.; Yu, H.; Jove, R. Activated Signal Transducers and Activators of Transcription 3 Signaling Induces CD46 Expression and Protects Human Cancer Cells from Complement-Dependent Cytotoxicity. Mol. Cancer Res. 2007, 5, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Furue, M. Regulation of Skin Barrier Function via Competition between AHR Axis versus IL-13/IL-4–JAK–STAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis. J. Clin. Med. 2020, 9, 3741. [Google Scholar] [CrossRef]
- Tan, Q.; Yang, H.; Liu, E.; Wang, H. P38/ERK MAPK signaling pathways are involved in the regulation of filaggrin and involucrin by IL-17. Mol. Med. Rep. 2017, 16, 8863–8867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, J.G.; Murrell, D.F.; Garcet, S.; Navrazhina, K.; Lee, P.C.; Muscianisi, E.; Blauvelt, A. Secukinumab lowers expression of ACE2 in affected skin of patients with psoriasis. J. Allergy Clin. Immunol. 2021, 147, 1107–1109.e.2. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, E.-C.; Hong, C.-H. IL-33 Enhances ACE2 Expression on Epidermal Keratinocytes in Atopic Dermatitis: A Plausible Issue for SARS-CoV-2 Transmission in Inflamed Atopic Skin. Biomedicines 2022, 10, 1183. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051183
Lin E-C, Hong C-H. IL-33 Enhances ACE2 Expression on Epidermal Keratinocytes in Atopic Dermatitis: A Plausible Issue for SARS-CoV-2 Transmission in Inflamed Atopic Skin. Biomedicines. 2022; 10(5):1183. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051183
Chicago/Turabian StyleLin, En-Cheng, and Chien-Hui Hong. 2022. "IL-33 Enhances ACE2 Expression on Epidermal Keratinocytes in Atopic Dermatitis: A Plausible Issue for SARS-CoV-2 Transmission in Inflamed Atopic Skin" Biomedicines 10, no. 5: 1183. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051183