
A Single Oral Dose of Diclofenac Causes Transition of Experimental Subclinical Acute Kidney Injury to Chronic Kidney Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Diclofenac Treatment
2.3. Ischemia Reperfusion Injury (IRI)
2.4. Determination of Blood Urea Nitrogen (BUN) Clearance
2.5. Organ Preservation
2.6. Renal Morphology and Immunofluorescence
2.7. Cytokine Expression
2.8. Statistical Analysis
3. Results
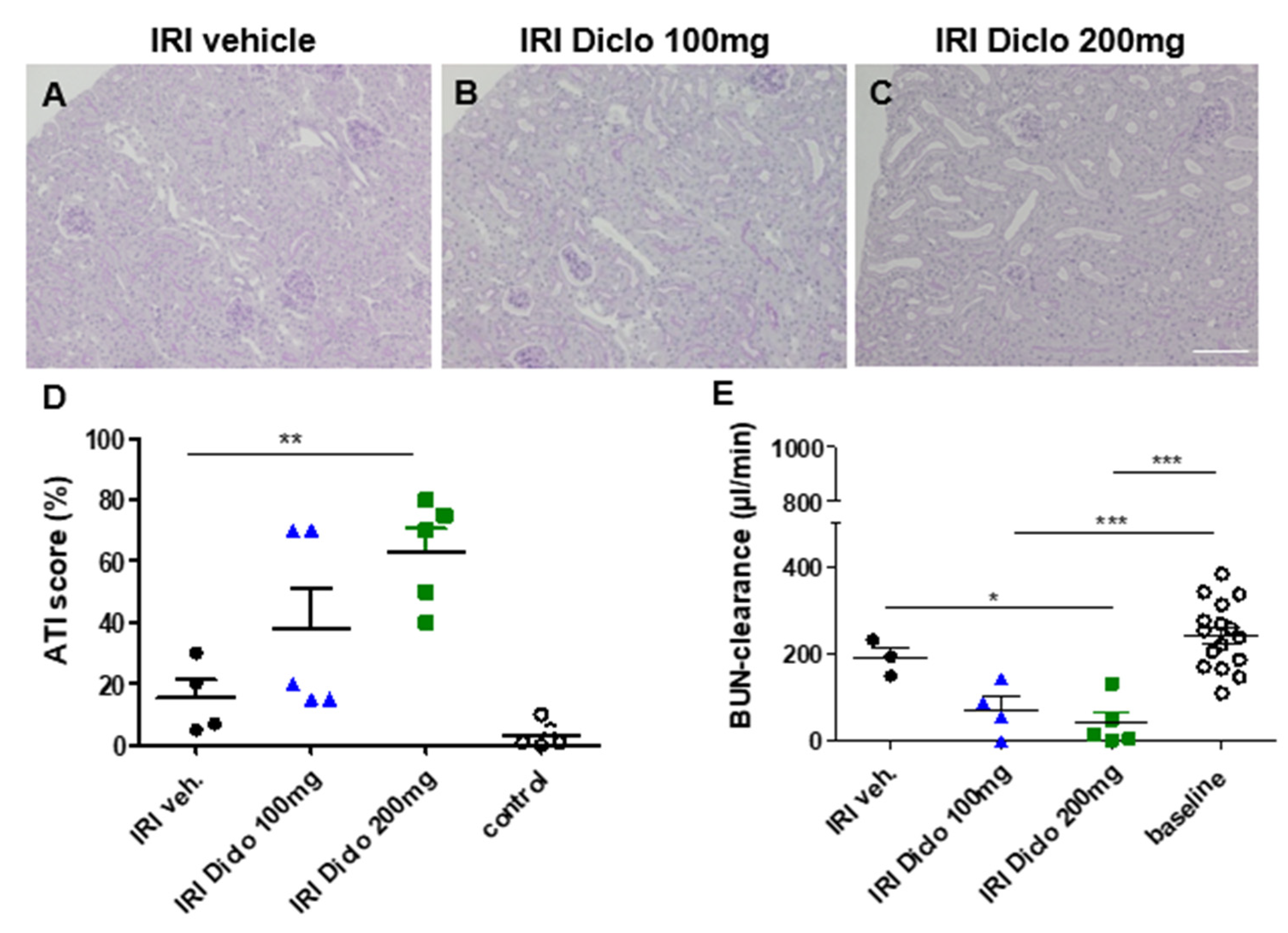
3.1. Diclofenac Causes Aggravation of Renal Damage and Renal Function Impairment 24 h after IRI in a Dose-Dependent Manner
3.1.1. Renal Morphology 24 h after Unilateral IRI and a Single Oral Dose of Diclofenac
3.1.2. Renal Function 24 h after Bilateral Renal IRI and a Single Oral Dose of Diclofenac
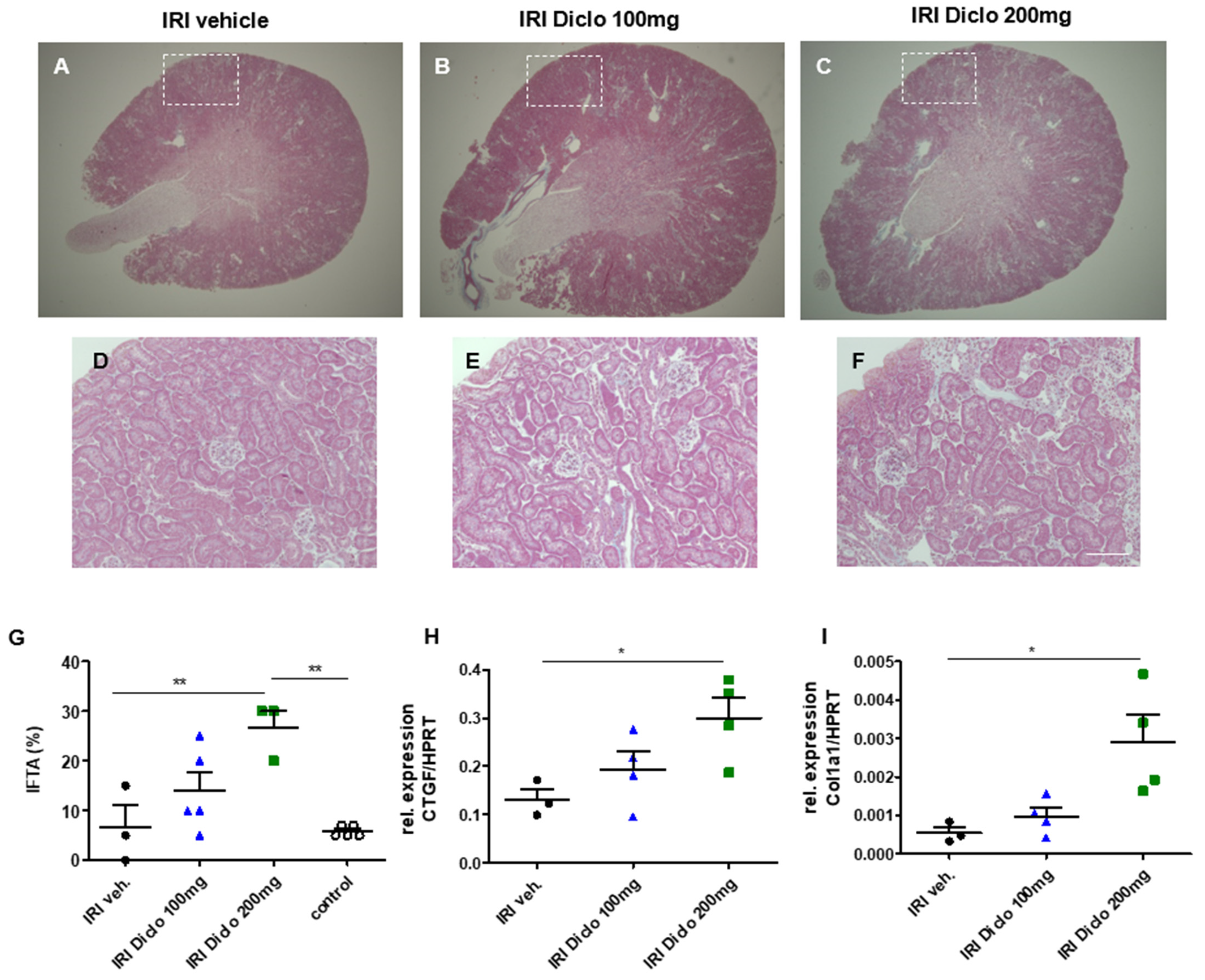
3.2. A Single Oral Dose of Diclofenac Causes Chronic Renal Damage in the Setting of Subclinical AKI
3.2.1. Interstitial Fibrosis and Tubular Atrophy (IFTA) Two Weeks after Unilateral IRI and a Single Dose of Diclofenac
3.2.2. Pro-Fibrotic Cytokines in the Renal Tissue 24 h after IRI
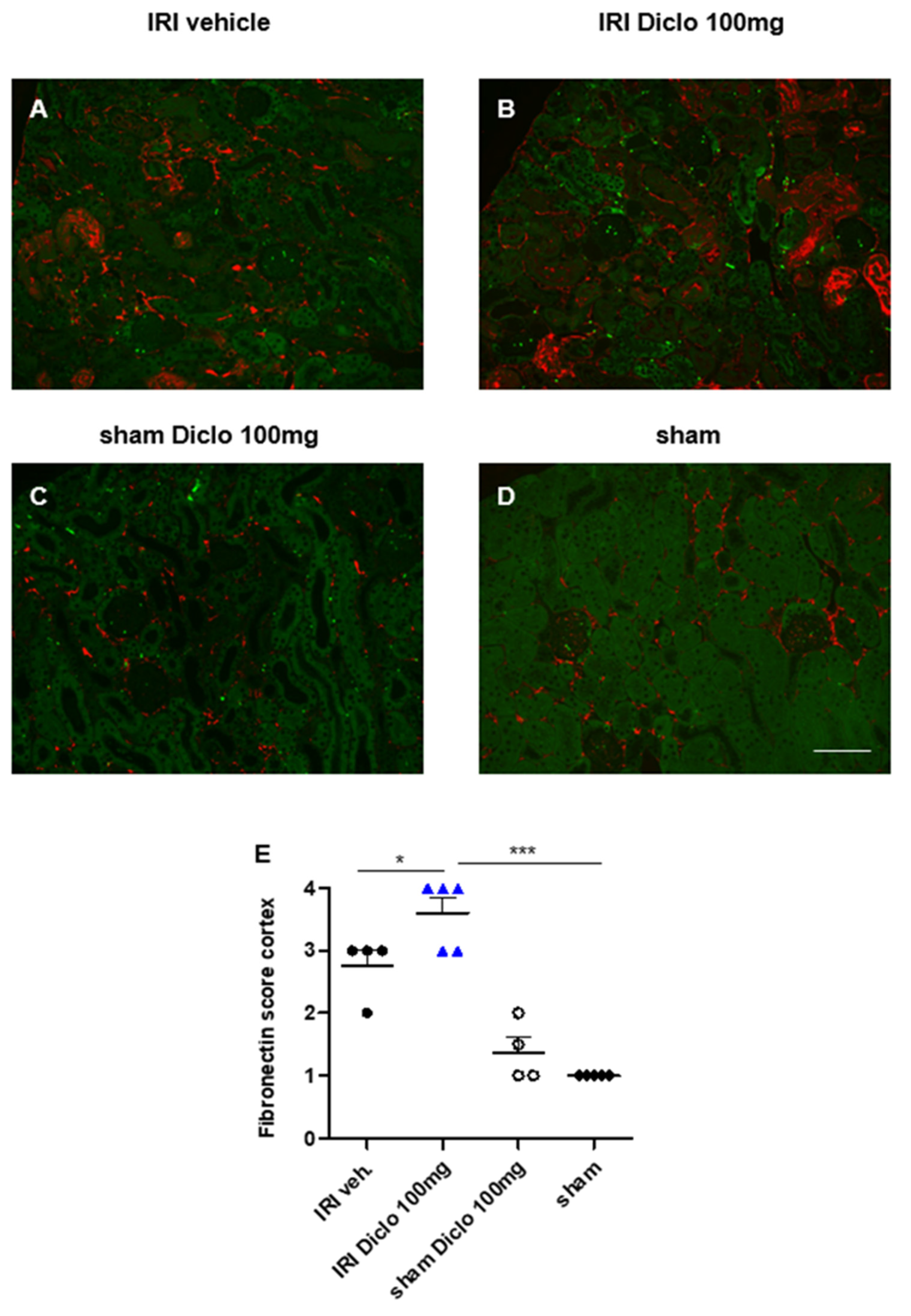
3.3. Acute and Chronic Renal Effects of Repetitive Diclofenac Treatment in Subclinical AKI
3.3.1. Renal Morphology and Tubular Injury Three Days after Renal IRI and Repetitive Diclofenac Treatment
3.3.2. Neutrophil Infiltration in Diclofenac Treatment after Renal IRI
3.3.3. Early Upregulation of Fibronectin in Repetitive Diclofenac Treatment after Renal IRI
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chawla, L.S.; Bellomo, R.; Bihorac, A.; Goldstein, S.L.; Siew, E.D.; Bagshaw, S.M.; Bittleman, D.; Cruz, D.; Endre, Z.; Fitzgerald, R.L.; et al. Acute kidney disease and renal recovery: Consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat. Rev. Nephrol. 2017, 13, 241–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Succar, L.; Pianta, T.J.; Davidson, T.; Pickering, J.W.; Endre, Z.H. Subclinical chronic kidney disease modifies the diagnosis of experimental acute kidney injury. Kidney Int. 2017, 92, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Manrique-Caballero, C.L.; del Rio-Pertuz, G.; Gomez, H. Sepsis-associated acute kidney injury. Crit. Care Clin. 2021, 37, 279–301. [Google Scholar] [CrossRef]
- Fähling, M.; Seeliger, E.; Patzak, A.; Persson, P. Understanding and preventing contrast-induced acute kidney injury. Nat. Rev. Nephrol. 2017, 13, 169–180. [Google Scholar] [CrossRef]
- Messerer, D.A.C.; Halbgebauer, R.; Nilsson, B.; Pavenstädt, H.; Radermacher, P.; Huber-Lang, M. Immunopathophysiology of trauma-related acute kidney injury. Nat. Rev. Nephrol. 2020, 17, 91–111. [Google Scholar] [CrossRef]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Nat. Rev. Dis. Primers 2021, 7, 52. [Google Scholar]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Han, Y.; Balkrishnan, R.; Hirth, R.A.; Hutton, D.W.; He, K.; Steffick, D.E.; Saran, R. Assessment of prescription analgesic use in older adults with and without chronic kidney disease and outcomes. JAMA Netw. Open 2020, 3, e2016839. [Google Scholar] [CrossRef]
- Barbieri, M.A.; Rottura, M.; Cicala, G.; Mandraffino, R.; Marino, S.; Irrera, N.; Mannucci, C.; Santoro, D.; Squadrito, F.; Arcoraci, V. Chronic kidney disease management in general practice: A focus on inappropriate drugs prescriptions. J. Clin. Med. 2020, 9, 1346. [Google Scholar] [CrossRef]
- Drożdżal, S.; Lechowicz, K.; Szostak, B.; Rosik, J.; Kotfis, K.; Machoy-Mokrzyńska, A.; Białecka, M.; Ciechanowski, K.; Gawrońska-Szklarz, B. Kidney damage from nonsteroidal anti-inflammatory drugs—Myth or truth? Review of selected literature. Pharmacol. Res. Perspect. 2021, 9, e00817. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Bhala, N.; Emberson, J.; Merhi, A.; Abramson, S.; Arber, N.; Baron, J.A.; Bombardier, C.; Cannon, C.; Yau, F.; et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: Meta-analyses of individual participant data from randomised trials. Lancet 2013, 382, 769–779. [Google Scholar]
- Ng, L.E.; Halliwell, B.; Wong, K.P. Nephrotoxic cell death by diclofenac and meloxicam. Biochem. Biophys. Res. Commun. 2008, 369, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Meng, Q.; Wang, C.; Wu, J.; Wang, C.; Zhu, Y.; Ma, X.; Sun, H.; Liu, K. Protective effect of cilastatin against diclofenac-induced nephrotoxicity through interaction with diclofenac acyl glucuronide via organic anion transporters. Br. J. Pharmacol. Metab. 2020, 177, 1933–1948. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Ge, Y.; Zhuang, S.; Dworkin, L.D.; Liu, Z.; Gong, R. Inhibition of glycogen synthase kinase-3beta prevents NSAID-induced acute kidney injury. Kidney Int. 2012, 81, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Hickey, E.J.; Raje, R.R.; Reid, V.E.; Gross, S.M.; Ray, S.D. Diclofenac induced in vivo nephrotoxicity may involve oxidative stress-mediated massive genomic DNA fragmentation and apoptotic cell death. Free Radic. Biol. Med. 2001, 31, 139–152. [Google Scholar] [CrossRef]
- Ungprasert, P.; Cheungpasitporn, W.; Crowson, C.S.; Matteson, E.L. Individual non-steroidal anti-inflammatory drugs and risk of acute kidney injury: A systematic review and meta-analysis of observational studies. Eur. J. Intern. Med. 2015, 26, 285–291. [Google Scholar] [CrossRef]
- Zhang, X.; Donnan, P.T.; Bell, S.; Guthrie, B. Non-steroidal anti-inflammatory drug induced acute kidney injury in the community dwelling general population and people with chronic kidney disease: Systematic review and meta-analysis. BMC Nephrol. 2017, 18, 256. [Google Scholar] [CrossRef] [Green Version]
- Nderitu, P.; Doos, L.; Jones, P.W.; Davies, S.J.; Kadam, U.T. Non-steroidal anti-inflammatory drugs and chronic kidney disease progression: A systematic review. Fam. Pract. 2013, 30, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Davidson, I.; Parker, J.; Beliles, R. Biological basis for extrapolation across mammalian species. Regul. Toxicol. Pharmacol. 1986, 6, 211–237. [Google Scholar] [CrossRef]
- Boizet-Bonhoure, B.; Déjardin, S.; Rossitto, M.; Poulat, F.; Philibert, P. Using experimental models to decipher the effects of acetaminophen and NSAIDs on reproductive development and health. Front. Toxicol. 2022, 4, 835360. [Google Scholar] [CrossRef] [PubMed]
- Shakibaie, M.; Forootanfar, H.; Ghaseminejad, A.; Salimi, A.; Ameri, A.; Doostmohammadi, M.; Jafari, E.; Rahimi, H. Ondansetron enhanced diclofenac-induced nephrotoxicity in mice. J. Biochem. Mol. Toxicol. 2019, 33, e22378. [Google Scholar] [CrossRef] [PubMed]
- Greite, R.; Thorenz, A.; Chen, R.; Jang, M.-S.; Rong, S.; Brownstein, M.J.; Tewes, S.; Wang, L.; Baniassad, B.; Kirsch, T.; et al. Renal ischemia-reperfusion injury causes hypertension and renal perfusion impairment in the CD1 mice which promotes progressive renal fibrosis. Am. J. Physiol. Physiol. 2018, 314, F881–F892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerink, L.J.S.; de Kok, M.J.; Mulvey, J.F.; Le Dévédec, S.E.; Markovski, A.A.; Wüst, R.C.I.; Alwayn, I.P.J.; Ploeg, R.J.; Schaapherder, A.F.M.; Bakker, J.A.; et al. Preclinical models versus clinical renal ischemia reperfusion injury: A systematic review based on metabolic signatures. Am. J. Transplant. 2021, 22, 344–370. [Google Scholar] [CrossRef]
- Navar, L.G.; Inscho, E.W.; Majid, S.A.; Imig, J.; Harrison-Bernard, L.M.; Mitchell, K.D. Paracrine regulation of the renal microcirculation. Physiol. Rev. 1996, 76, 425–536. [Google Scholar] [CrossRef]
- Villa, E.; Garcia-Robles, R.; Haas, J.; Romero, J.C. Comparative effect of PGE2 and PGI2 on renal function. Hypertension 1997, 30 Pt 2, 664–666. [Google Scholar] [CrossRef]
- Hellms, S.; Gueler, F.; Gutberlet, M.; Schebb, N.H.; Rund, K.; Kielstein, J.T.; VoChieu, V.; Rauhut, S.; Greite, R.; Martirosian, P.; et al. Single-dose diclofenac in healthy volunteers can cause decrease in renal perfusion measured by functional magnetic resonance imaging†. J. Pharm. Pharmacol. 2019, 71, 1262–1270. [Google Scholar] [CrossRef]
- Imamura, H.; Hata, J.; Iida, A.; Manabe, N.; Haruma, K. Evaluating the effects of diclofenac sodium and etodolac on renal hemodynamics with contrast-enhanced ultrasonography: A pilot study. Eur. J. Clin. Pharmacol. 2012, 69, 161–165. [Google Scholar] [CrossRef]
- Burne, M.J.; Haq, M.; Matsuse, H.; Mohapatra, S.; Rabb, H. Genetic susceptibility to renal ischemia reperfusion injury revealed in a murine model. Transplantation 2000, 69, 1023–1025. [Google Scholar] [CrossRef]
- Tewes, S.; Gueler, F.; Chen, R.; Gutberlet, M.; Jang, M.-S.; Meier, M.; Mengel, M.; Hartung, D.; Wacker, F.; Rong, S.; et al. Functional MRI for characterization of renal perfusion impairment and edema formation due to acute kidney injury in different mouse strains. PLoS ONE 2017, 12, e0173248. [Google Scholar] [CrossRef]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- James, M.T.; Grams, M.E.; Woodward, M.; Elley, C.R.; Green, J.A.; Wheeler, D.C.; de Jong, P.; Gansevoort, R.T.; Levey, A.S.; Warnock, D.G.; et al. A Meta-analysis of the association of estimated GFR, albuminuria, diabetes mellitus, and hypertension with acute kidney injury. Am. J. Kidney Dis. 2015, 66, 602–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, V.-C.; Huang, T.-M.; Lai, C.-F.; Shiao, C.-C.; Lin, Y.-F.; Chu, T.-S.; Wu, P.-C.; Chao, C.-T.; Wang, J.-Y.; Kao, T.-W.; et al. Acute-on-chronic kidney injury at hospital discharge is associated with long-term dialysis and mortality. Kidney Int. 2011, 80, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polichnowski, A.J.; Lan, R.; Geng, H.; Griffin, K.A.; Venkatachalam, M.A.; Bidani, A.K. Severe renal mass reduction impairs recovery and promotes fibrosis after AKI. J. Am. Soc. Nephrol. 2014, 25, 1496–1507. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.M.-W.; Bonventre, J.V. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 310–318. [Google Scholar] [CrossRef]
- Yin, Q.; Liu, H. Connective tissue growth factor and renal fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 365–380. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of renal fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Lan, R.; Geng, H.; Polichnowski, A.J.; Singha, P.K.; Saikumar, P.; McEwen, D.G.; Griffin, K.A.; Koesters, R.; Weinberg, J.M.; Bidani, A.K.; et al. PTEN loss defines a TGF-beta-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am. J. Physiol. Renal. Physiol. 2012, 302, F1210–F1223. [Google Scholar] [CrossRef]
- Yokoi, H.; Sugawara, A.; Mukoyama, M.; Mori, K.; Makino, H.; Suganami, T.; Nagae, T.; Yahata, K.; Fujinaga, Y.; Tanaka, I.; et al. Role of connective tissue growth factor in profibrotic action of transforming growth factor-beta: A potential target for preventing renal fibrosis. Am. J. Kidney Dis. 2001, 38 (Suppl. 1), S134–S138. [Google Scholar] [CrossRef]
- Gewin, L.; Zent, R. How does TGF-beta mediate tubulointerstitial fibrosis? Semin. Nephrol. 2012, 32, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Bowers, S.L.K.; Davis-Rodriguez, S.; Thomas, Z.; Rudomanova, V.; Bacon, W.C.; Beiersdorfer, A.; Ma, Q.; Devarajan, P.; Blaxall, B.C.; Rudomanova, V. Inhibition of fibronectin polymerization alleviates kidney injury due to ischemia-reperfusion. Am. J. Physiol. Physiol. 2019, 316, F1293–F1298. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Shirai, Y.; Akai, S.; Nakajima, A.; Tsuneyama, K.; Yokoi, T. Toxicological role of an acyl glucuronide metabolite in diclofenac-induced acute liver injury in mice. J. Appl. Toxicol. 2016, 37, 545–553. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Störmer, J.; Gwinner, W.; Derlin, K.; Immenschuh, S.; Rong, S.; Jang, M.-S.; Shushakova, N.; Haller, H.; Gueler, F.; Greite, R. A Single Oral Dose of Diclofenac Causes Transition of Experimental Subclinical Acute Kidney Injury to Chronic Kidney Disease. Biomedicines 2022, 10, 1198. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051198
Störmer J, Gwinner W, Derlin K, Immenschuh S, Rong S, Jang M-S, Shushakova N, Haller H, Gueler F, Greite R. A Single Oral Dose of Diclofenac Causes Transition of Experimental Subclinical Acute Kidney Injury to Chronic Kidney Disease. Biomedicines. 2022; 10(5):1198. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051198
Chicago/Turabian StyleStörmer, Johanna, Wilfried Gwinner, Katja Derlin, Stephan Immenschuh, Song Rong, Mi-Sun Jang, Nelli Shushakova, Hermann Haller, Faikah Gueler, and Robert Greite. 2022. "A Single Oral Dose of Diclofenac Causes Transition of Experimental Subclinical Acute Kidney Injury to Chronic Kidney Disease" Biomedicines 10, no. 5: 1198. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051198