
Generation of hiPSC-Derived Skeletal Muscle Cells: Exploiting the Potential of Skeletal Muscle-Derived hiPSCs

 ,
,  , ,
, ,
Abstract
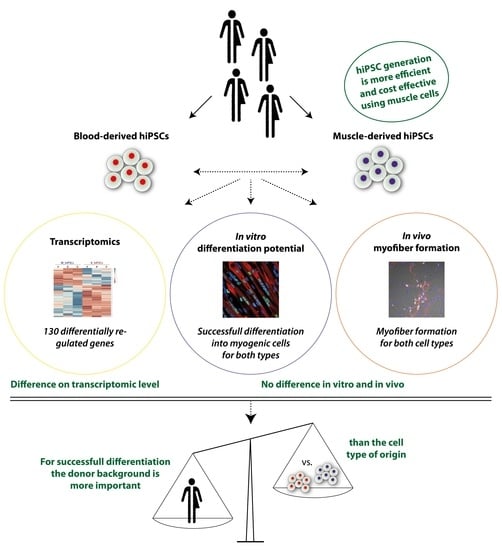
:
1. Introduction
2. Materials and Methods
2.1. Donors
2.2. Primary MuSC Isolation and Culture
2.3. PBMC Isolation and Culture
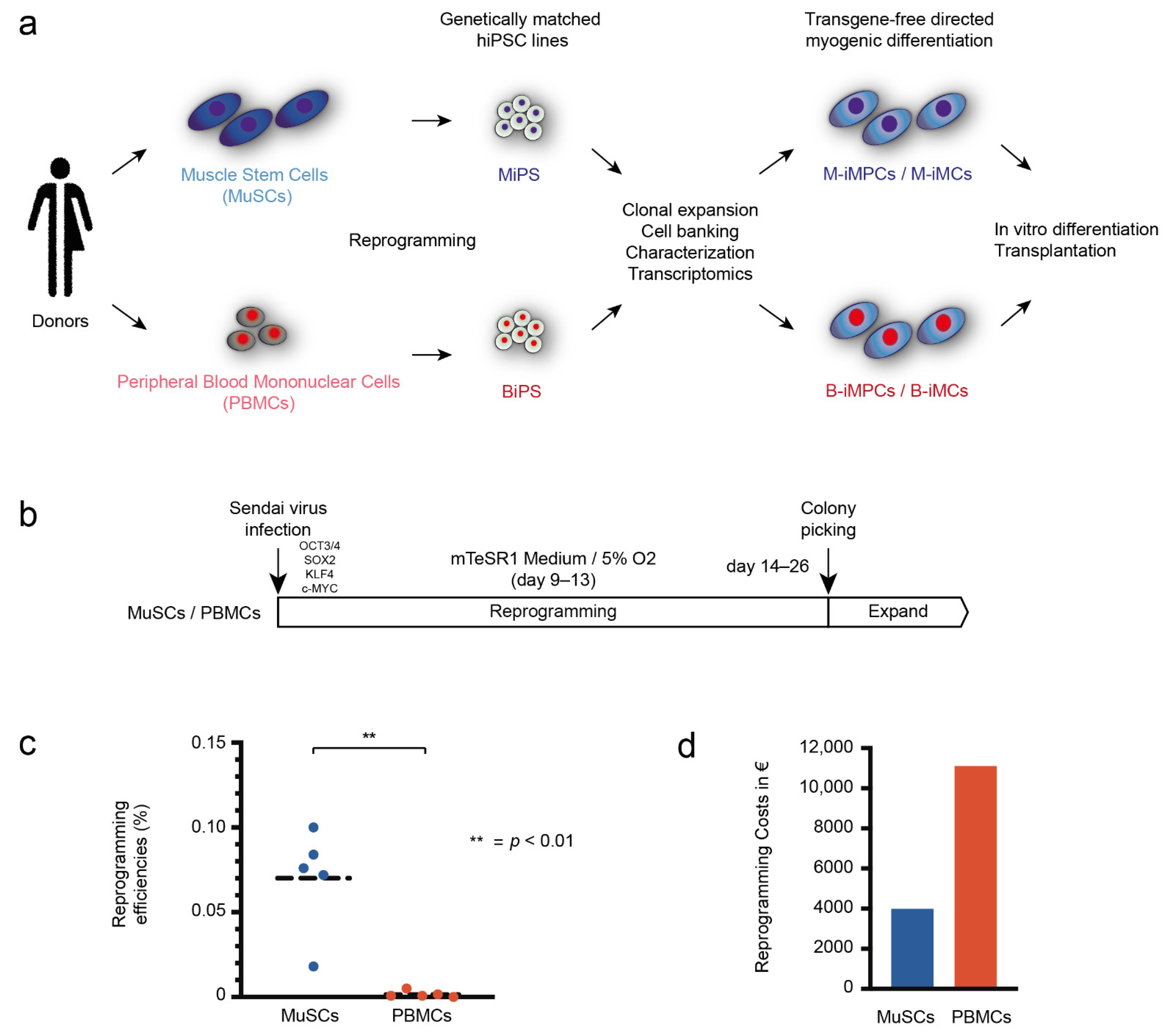
2.4. Reprogramming
2.5. hiPSC Culture
2.6. RNA Sequencing and Data Analysis
2.7. Myogenic Differentiation
2.8. Immunofluorescence Staining in Cultured Cells
2.9. RT-qPCR
2.10. Mouse Strain and Irradiation of Hind Limbs
2.11. Transplantation of iMPCs
2.12. Histological Sections
2.13. Immunofluorescence Staining in Histological Tissue Sections
2.14. Statistics
3. Results
3.1. MuSCs Reprogram more Efficiently into hiPSCs Than PBMCs
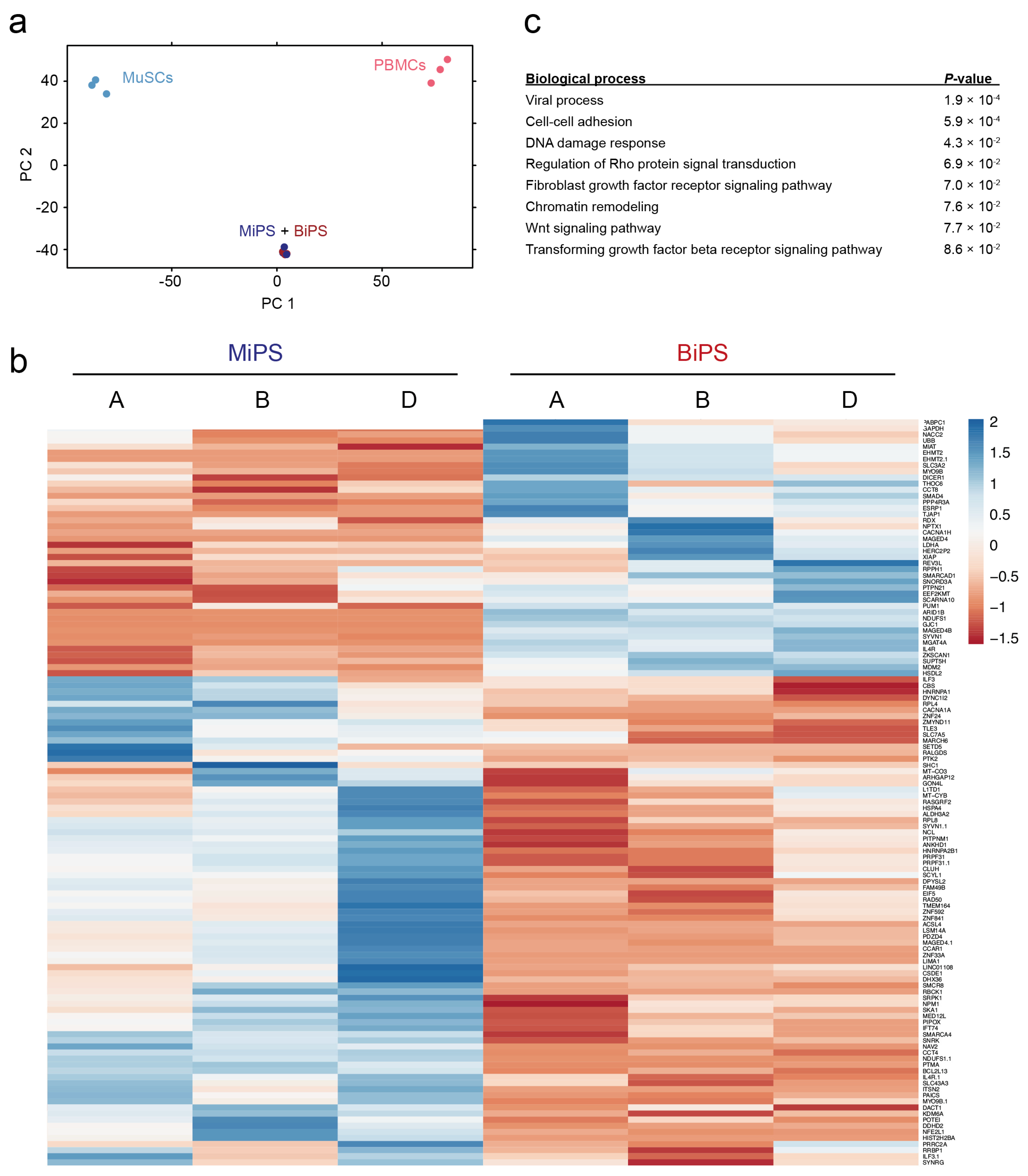
3.2. Global Transcriptomic Analysis of the Generated MiPS and BiPS
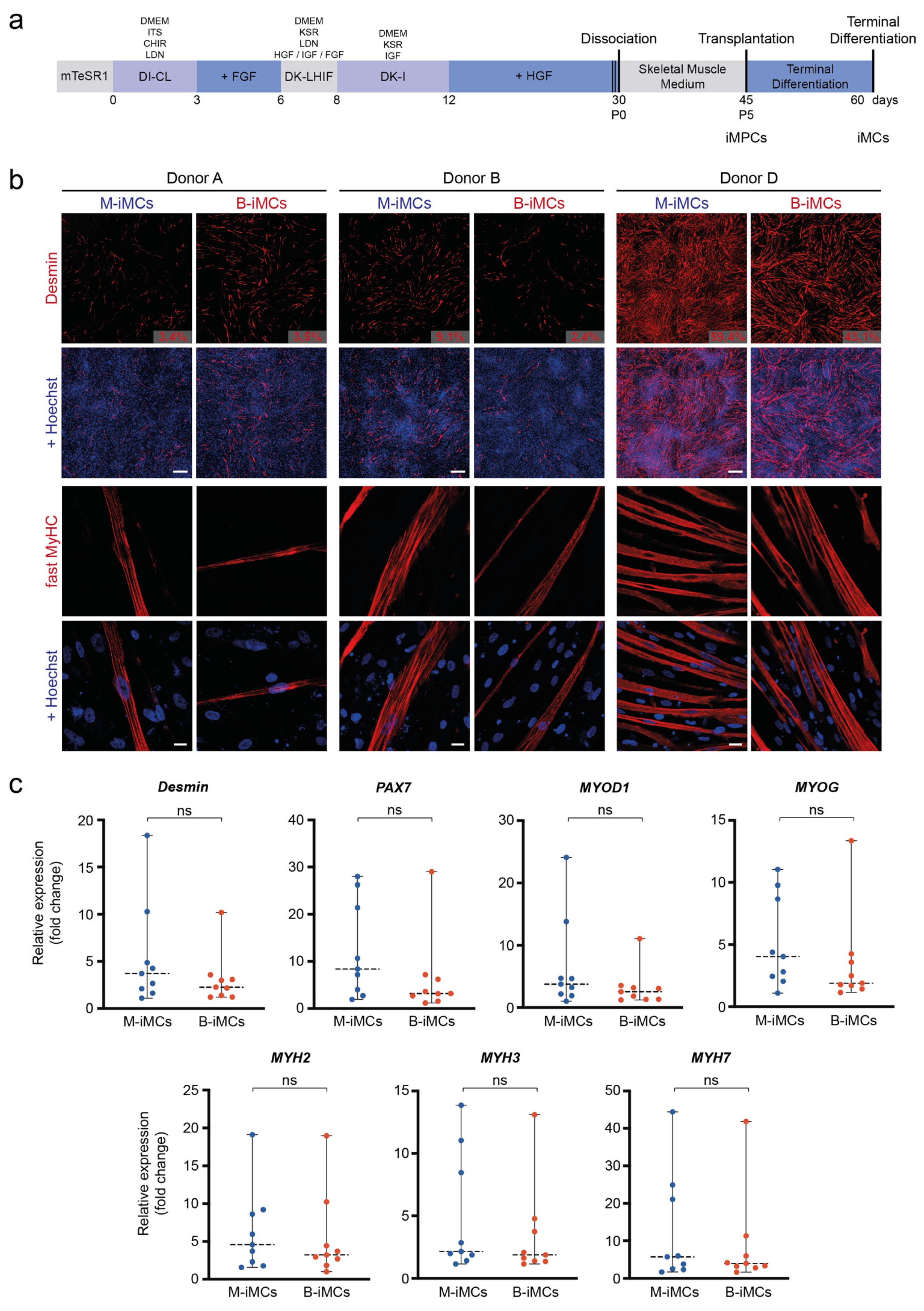
3.3. In Vitro Myogenic Differentiation Capacity of MiPS and BiPS
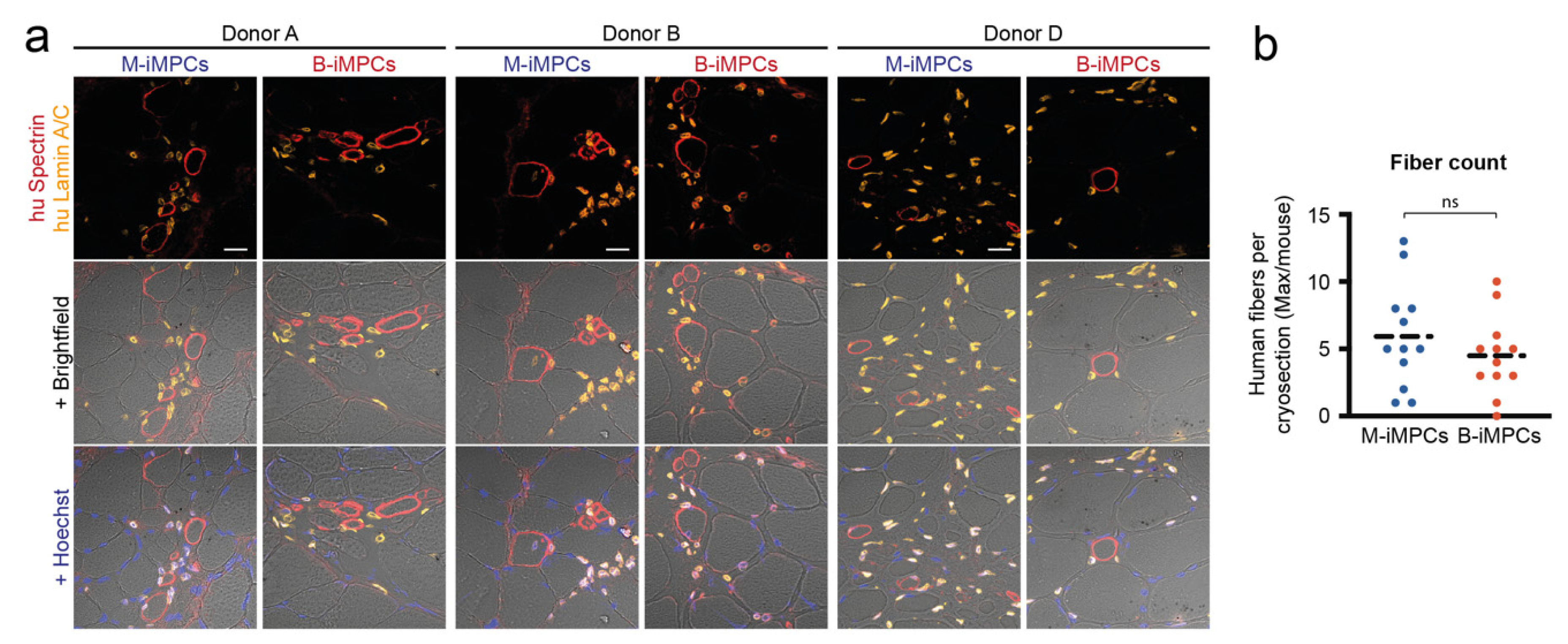
3.4. In Vivo Myofiber Formation Potential of Induced Myogenic Progenitor Cells (iMPCs) Generated from MiPS and BiPS
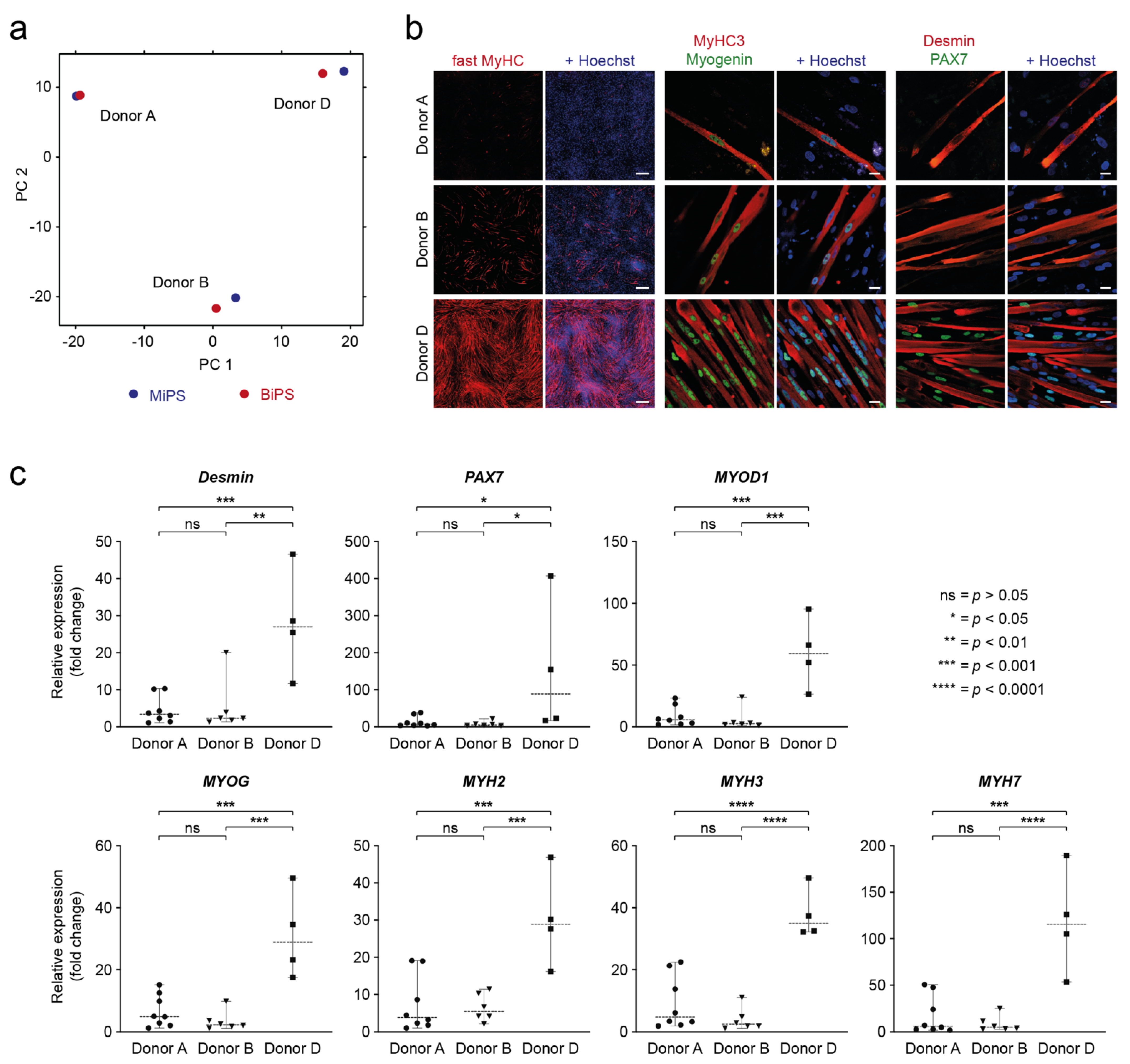
3.5. Analysis of Donor Background versus Cell Type of Origin
4. Discussion
5. Conclusions
- hiPSCs can be generated highly efficiently from muscle stem cells
- Muscle-derived hiPSCs save costs and time as compared to PBMC-derived hiPSCs
- Muscle-derived hiPSCs are yet to be improved as compared to primary human muscle stem cells
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biressi, S.; Filareto, A.; Rando, T.A. Stem Cell Therapy for Muscular Dystrophies. J. Clin. Investig. 2020, 130, 5652–5664. [Google Scholar] [CrossRef] [PubMed]
- Boyer, O.; Butler-Brown, G.; Chinoy, H.; Cossu, G.; Galli, F.; Lilleker, J.B.; Magli, A.; Mouly, V.; Perlingeiro, R.C.R.; Previtali, S.C.; et al. Myogenic Cell Transplantation in Genetic and Acquired Diseases of Skeletal Muscle. Front. Genet. 2021, 12, 1226. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite Cell of Skeletal Muscle Fibers. J. Cell Biol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Schüler, S.C.; Hüttner, S.S.; von Eyss, B.; Maltzahn, J. Von Adult Stem Cells at Work: Regenerating Skeletal Muscle. Cell. Mol. Life Sci. 2019, 76, 2559–2570. [Google Scholar] [CrossRef] [Green Version]
- Lepper, C.; Partridge, T.A.; Fan, C.-M. An Absolute Requirement for Pax7-Positive Satellite Cells in Acute Injury-Induced Skeletal Muscle Regeneration. Development 2011, 138, 3639–3646. [Google Scholar] [CrossRef] [Green Version]
- Relaix, F.; Zammit, P.S. Satellite Cells Are Essential for Skeletal Muscle Regeneration: The Cell on the Edge Returns Centre Stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef] [Green Version]
- Marg, A.; Escobar, H.; Gloy, S.; Kufeld, M.; Zacher, J.; Spuler, A.; Birchmeier, C.; Izsvák, Z.; Spuler, S.; Izsvak, Z.; et al. Human Satellite Cells Have Regenerative Capacity and Are Genetically Manipulatable. J. Clin. Investig. 2014, 124, 4257–4265. [Google Scholar] [CrossRef] [Green Version]
- Marg, A.; Escobar, H.; Karaiskos, N.; Grunwald, S.A.; Metzler, E.; Kieshauer, J.; Sauer, S.; Pasemann, D.; Malfatti, E.; Mompoint, D.; et al. Human Muscle-Derived CLEC14A-Positive Cells Regenerate Muscle Independent of PAX7. Nat. Commun. 2019, 10, 5776. [Google Scholar] [CrossRef] [Green Version]
- Périé, S.; Trollet, C.; Mouly, V.; Vanneaux, V.; Mamchaoui, K.; Bouazza, B.; Marolleau, J.P.; Laforêt, P.; Chapon, F.; Eymard, B.; et al. Autologous Myoblast Transplantation for Oculopharyngeal Muscular Dystrophy: A Phase I/IIa Clinical Study. Mol. Ther. 2014, 22, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Kajbafzadeh, A.M.; Elmi, A.; Payabvash, S.; Salmasi, A.H.; Saeedi, P.; Mohamadkhani, A.; Sadeghi, Z.; Nikfarjam, L. Transurethral Autologous Myoblast Injection for Treatment of Urinary Incontinence in Children with Classic Bladder Exstrophy. J. Urol. 2008, 180, 1098–1105. [Google Scholar] [CrossRef]
- Elmi, A.; Kajbafzadeh, A.M.; Tourchi, A.; Talab, S.S.; Esfahani, S.A. Safety, Efficacy and Health Related Quality of Life of Autologous Myoblast Transplantation for Treatment of Urinary Incontinence in Children with Bladder Exstrophy-Epispadias Complex. J. Urol. 2011, 186, 2021–2026. [Google Scholar] [CrossRef] [PubMed]
- Ortuño-Costela, M.D.C.; Cerrada, V.; García-López, M.; Gallardo, M.E. The Challenge of Bringing IPSCs to the Patient. Int. J. Mol. Sci. 2019, 20, 6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Rodríguez-delaRosa, A.; Pourquié, O. Human Muscle Production in Vitro from Pluripotent Stem Cells: Basic and Clinical Applications. Semin. Cell Dev. Biol. 2021, 119, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Gerli, M.F.M.; Perani, L.; Benedetti, S.; Ungaro, F.; Cassano, M.; Antonini, S.; Tagliafico, E.; Artusi, V.; Longa, E.; et al. Transplantation of Genetically Corrected Human IPSC-Derived Progenitors in Mice with Limb-Girdle Muscular Dystrophy. Sci. Transl. Med. 2012, 4, 140ra89. [Google Scholar] [CrossRef]
- Darabi, R.; Arpke, R.R.W.; Irion, S.; Dimos, J.T.J.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C.R. Human ES- and IPS-Derived Myogenic Progenitors Restore DYSTROPHIN and Improve Contractility upon Transplantation in Dystrophic Mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Chal, J.; Oginuma, M.; Al Tanoury, Z.; Gobert, B.; Sumara, O.; Hick, A.; Bousson, F.; Zidouni, Y.; Mursch, C.; Moncuquet, P.; et al. Differentiation of Pluripotent Stem Cells to Muscle Fiber to Model Duchenne Muscular Dystrophy. Nat. Biotechnol. 2015, 33, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Chal, J.; Tanoury, Z.A.; Hestin, M.; Gobert, B.; Aivio, S.; Hick, A.; Cherrier, T.; Nesmith, A.P.; Parker, K.K.; Pourquié, O. Generation of Human Muscle Fibers and Satellite-like Cells from Human Pluripotent Stem Cells in Vitro. Nat. Protoc. 2016, 11, 1833–1850. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.Y.; Lim, H.; Estrellas, K.; Mula, J.; Cohen, T.V.; Zhang, Y.; Donnelly, C.J.; Richard, J.-P.; Kim, Y.J.; Kim, H.; et al. Concordant but Varied Phenotypes among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived Using a Human IPSC-Based Model. Cell Rep. 2016, 15, 2301–2312. [Google Scholar] [CrossRef] [Green Version]
- Shelton, M.; Metz, J.; Liu, J.; Carpenedo, R.L.; Demers, S.P.; Stanford, W.L.; Skerjanc, I.S. Derivation and Expansion of PAX7-Positive Muscle Progenitors from Human and Mouse Embryonic Stem Cells. Stem Cell Rep. 2014, 3, 516–529. [Google Scholar] [CrossRef] [Green Version]
- Caron, L.; Kher, D.; Lee, K.L.; McKernan, R.; Dumevska, B.; Hidalgo, A.; Li, J.; Yang, H.; Main, H.; Ferri, G.; et al. A Human Pluripotent Stem Cell Model of Facioscapulohumeral Muscular Dystrophy-Affected Skeletal Muscles. Stem Cells Transl. Med. 2016, 5, 1145–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Daman, K.; Chen, J.J.; Shi, M.J.; Yan, J.; Matijasevic, Z.; Rickard, A.M.; Bennett, M.H.; Kiselyov, A.; Zhou, H.; et al. IMyoblasts for Ex Vivo and in Vivo Investigations of Human Myogenesis and Disease Modeling. eLife 2022, 11, e70341. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Fujiwara, W.; Gonzalez, K.; Jan, M.; Liebscher, S.; Van Handel, B.; Schenke-Layland, K.; Pyle, A.D. In Vivo Human Somitogenesis Guides Somite Development from HPSCs. Cell Rep. 2017, 18, 1573–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanoury, Z.A.; Rao, J.; Tassy, O.; Gobert, B.; Gapon, S.; Garnier, J.M.; Wagner, E.; Hick, A.; Hall, A.; Gussoni, E.; et al. Differentiation of the Human PAX7-Positive Myogenic Precursors/Satellite Cell Lineage In Vitro. Development 2020, 147, dev187344. [Google Scholar] [CrossRef] [PubMed]
- Hicks, M.R.; Hiserodt, J.; Paras, K.; Fujiwara, W.; Eskin, A.; Jan, M.; Xi, H.; Young, C.S.; Evseenko, D.; Nelson, S.F.; et al. ERBB3 and NGFR Mark a Distinct Skeletal Muscle Progenitor Cell in Human Development and HPSCs. Nat. Cell Biol. 2018, 20, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Nalbandian, M.; Zhao, M.; Sasaki-Honda, M.; Jonouchi, T.; Lucena-Cacace, A.; Mizusawa, T.; Yasuda, M.; Yoshida, Y.; Hotta, A.; Sakurai, H. Characterization of HiPSC-Derived Muscle Progenitors Reveals Distinctive Markers for Myogenic Cell Purification toward Cell Therapy. Stem Cell Rep. 2021, 16, 883–898. [Google Scholar] [CrossRef]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced Pluripotent Stem Cells: Applications in Regenerative Medicine, Disease Modeling and Drug Discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Kajiwara, M.; Aoi, T.; Okita, K.; Takahashi, R.; Inoue, H.; Takayama, N.; Endo, H.; Eto, K.; Toguchida, J.; Uemoto, S.; et al. Donor-Dependent Variations in Hepatic Differentiation from Human-Induced Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12538–12543. [Google Scholar] [CrossRef] [Green Version]
- Kyttälä, A.; Moraghebi, R.; Valensisi, C.; Kettunen, J.; Andrus, C.; Pasumarthy, K.K.; Nakanishi, M.; Nishimura, K.; Ohtaka, M.; Weltner, J.; et al. Genetic Variability Overrides the Impact of Parental Cell Type and Determines IPSC Differentiation Potential. Stem Cell Rep. 2016, 6, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Kilpinen, H.; Goncalves, A.; Leha, A.; Afzal, V.; Alasoo, K.; Ashford, S.; Bala, S.; Bensaddek, D.; Casale, F.P.; Culley, O.J.; et al. Common Genetic Variation Drives Molecular Heterogeneity in Human IPSCs. Nature 2017, 546, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Rouhani, F.; Kumasaka, N.; de Brito, M.C.; Bradley, A.; Vallier, L.; Gaffney, D. Genetic Background Drives Transcriptional Variation in Human Induced Pluripotent Stem Cells. PLoS Genet. 2014, 10, e1004432. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell Type of Origin Influences the Molecular and Functional Properties of Mouse Induced Pluripotent Stem Cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Hongguang, H.; Loh, Y.; Aryee, M.J.; Lensch, M.W.; Li, H.; et al. Donor Cell Type Can Influence the Epigenome and Differentiation Potential of Human Induced Pluripotent Stem Cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; Ogmalley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of Aberrant Epigenomic Reprogramming in Human Induced Pluripotent Stem Cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic Memory in Induced Pluripotent Stem Cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Ohi, Y.; Qin, H.; Hong, C.; Blouin, L.; Polo, J.M.; Guo, T.; Qi, Z.; Downey, S.L.; Manos, P.D.; Rossi, D.J.; et al. Incomplete DNA Methylation Underlies a Transcriptional Memory of Somatic Cells in Human IPS Cells. Nat. Cell Biol. 2011, 13, 541–549. [Google Scholar] [CrossRef]
- Hu, Q.; Friedrich, A.M.; Johnson, L.V.; Clegg, D.O. Memory in Induced Pluripotent Stem Cells: Reprogrammed Human Retinal-Pigmented Epithelial Cells Show Tendency for Spontaneous Redifferentiation. Stem Cells 2010, 28, 1981–1991. [Google Scholar] [CrossRef]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic Memory and Preferential Lineage-Specific Differentiation in Induced Pluripotent Stem Cells Derived from Human Pancreatic Islet Beta Cells. Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Freire, V.; Lee, A.S.; Hu, S.; Abilez, O.J.; Liang, P.; Lan, F.; Huber, B.C.; Ong, S.G.; Hong, W.X.; Huang, M.; et al. Effect of Human Donor Cell Source on Differentiation and Function of Cardiac Induced Pluripotent Stem Cells. J. Am. Coll. Cardiol. 2014, 64, 436–448. [Google Scholar] [CrossRef] [Green Version]
- Chlebanowska, P.; Sułkowski, M.; Skrzypek, K.; Tejchman, A.; Muszyńska, A.; Noroozi, R.; Majka, M. Origin of the Induced Pluripotent Stem Cells Affects Their Differentiation into Dopaminergic Neurons. Int. J. Mol. Sci. 2020, 21, 5705. [Google Scholar] [CrossRef]
- Sareen, D.; Mehrnoosh, S.; Ornelas, L.; Winkler, M.A.; Narwani, K.; Sahabian, A.; Funari, V.A.; Tang, J.; Spurka, L.; Punj, V.; et al. Differentiation of Human Limbal-Derived Induced Pluripotent Stem Cells Into Limbal-Like Epithelium. Stem Cells Transl. Med. 2014, 3, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Zhao, M.; Jahanbani, F.; Shao, N.; Lee, W.H.; Chen, H.; Snyder, M.P.; Wu, J.C. Effects of Cellular Origin on Differentiation of Human Induced Pluripotent Stem Cell–Derived Endothelial Cells. JCI Insight 2016, 1, e85558. [Google Scholar] [CrossRef] [PubMed]
- Pianezzi, E.; Altomare, C.; Bolis, S.; Balbi, C.; Torre, T.; Rinaldi, A.; Camici, G.G.; Barile, L.; Vassalli, G. Role of Somatic Cell Sources in the Maturation Degree of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118538. [Google Scholar] [CrossRef] [PubMed]
- Dorn, I.; Klich, K.; Arauzo-Bravo, M.J.; Radstaak, M.; Santourlidis, S.; Ghanjati, F.; Radke, T.F.; Psathaki, O.E.; Hargus, G.; Kramer, J.; et al. Erythroid Differentiation of Human Induced Pluripotent Stem Cells Is Independent of Donor Cell Type of Origin. Haematologica 2014, 100, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Escobar, H.; Krause, A.; Keiper, S.; Kieshauer, J.; Müthel, S.; de Paredes, M.G.; Metzler, E.; Kühn, R.; Heyd, F.; Spuler, S. Base Editing Repairs an SGCA Mutation in Human Primary Muscle Stem Cells. JCI Insight 2021, 6, e145994. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Di Francescantonio, S.; Marg, A.; Müthel, S.; Spuler, S.; Escobar, H. MRNA-Mediated Delivery of Gene Editing Tools to Human Primary Muscle Stem Cells. Mol. Ther.-Nucleic Acids 2022, 28, 47–57. [Google Scholar] [CrossRef]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia Enhances the Generation of Induced Pluripotent Stem Cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Metzler, E.; Telugu, N.; Diecke, S.; Spuler, S.; Escobar, H. Generation of Three Age and Gender Matched Pairs of Human Induced Pluripotent Stem Cells Derived from Myoblasts (MDCi011-A, MDCi012-A, MDCi013-A) and from Peripheral Blood Mononuclear Cells (MDCi011-B, MDCi012-B, MDCi013-B) from the Same Donor. Stem Cell Res. 2020, 48, 101987. [Google Scholar] [CrossRef]
- Metzler, E.; Telugu, N.; Diecke, S.; Spuler, S.; Escobar, H. Generation of Two Human Induced Pluripotent Stem Cell Lines Derived from Myoblasts (MDCi014-A) and from Peripheral Blood Mononuclear Cells (MDCi014-B) from the Same Donor. Stem Cell Res. 2020, 48, 101998. [Google Scholar] [CrossRef]
- Kuijk, E.; Jager, M.; van der Roest, B.; Locati, M.D.; Van Hoeck, A.; Korzelius, J.; Janssen, R.; Besselink, N.; Boymans, S.; van Boxtel, R.; et al. The Mutational Impact of Culturing Human Pluripotent and Adult Stem Cells. Nat. Commun. 2020, 11, 2493. [Google Scholar] [CrossRef]
- Thompson, O.; von Meyenn, F.; Hewitt, Z.; Alexander, J.; Wood, A.; Weightman, R.; Gregory, S.; Krueger, F.; Andrews, S.; Barbaric, I.; et al. Low Rates of Mutation in Clinical Grade Human Pluripotent Stem Cells under Different Culture Conditions. Nat. Commun. 2020, 11, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential Expression Analysis for Sequence Count Data. Genome Biol. 2010, 98, 14973–14978. [Google Scholar] [CrossRef] [Green Version]
- Kufeld, M.; Escobar, H.; Marg, A.; Pasemann, D.; Budach, V.; Spuler, S. Localized Irradiation of Mouse Legs Using an Image-Guided Robotic Linear Accelerator. Ann. Transl. Med. 2017, 5, 156. [Google Scholar] [CrossRef] [Green Version]
- Trokovic, R.; Weltner, J.; Manninen, T.; Mikkola, M.; Lundin, K.; Hämäläinen, R.; Suomalainen, A.; Otonkoski, T. Small Molecule Inhibitors Promote Efficient Generation of Induced Pluripotent Stem Cells from Human Skeletal Myoblasts. Stem Cells Dev. 2013, 22, 120730075754007. [Google Scholar] [CrossRef]
- Trokovic, R.; Weltner, J.; Nishimura, K.; Ohtaka, M.; Nakanishi, M.; Salomaa, V.; Jalanko, A.; Otonkoski, T.; Kyttälä, A. Advanced Feeder-Free Generation of Induced Pluripotent Stem Cells Directly From Blood Cells. Stem Cells Transl. Med. 2014, 3, 1402–1409. [Google Scholar] [CrossRef]
- Byers, C.; Spruce, C.; Fortin, H.J.; Hartig, E.I.; Czechanski, A.; Munger, S.C.; Reinholdt, L.G.; Skelly, D.A.; Baker, C.L. Genetic Control of the Pluripotency Epigenome Determines Differentiation Bias in Mouse Embryonic Stem Cells. EMBO J. 2022, 41, e109445. [Google Scholar] [CrossRef]
- Xi, H.; Young, C.S.; Pyle, A.D. Generation of PAX7 Reporter Cells to Investigate Skeletal Myogenesis from Human Pluripotent Stem Cells. STAR Protoc. 2020, 1, 100158. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Sex/Age | Cell Type | Cell Line (hPSCreg) | Cell Type of Origin | Passage | Successful Reprogramming | RNA-Seq | In Vitro Myogenic Differentiation | Transplantation |
|---|---|---|---|---|---|---|---|---|---|
| A | female, 47 | MuSCs | yes | yes | |||||
| PBMCs | yes | yes | |||||||
| hiPSCs | MDCi011-A | MuSCs | 13 | yes | yes | yes | |||
| hiPSCs | MDCi011-B | PBMCs | 15 | yes | yes | yes | |||
| B | female, 50 | MuSCs | yes | yes | |||||
| PBMCs | yes | yes | |||||||
| hiPSCs | MDCi012-A | MuSCs | 15 | yes | yes | yes | |||
| hiPSCs | MDCi012-B | PBMCs | 15 | yes | yes | yes | |||
| C | male, 18 | MuSCs | yes | ||||||
| PBMCs | yes | ||||||||
| D | female, 47 | MuSCs | yes | yes | |||||
| PBMCs | yes | yes | |||||||
| hiPSCs | MDCi013-A | MuSCs | 13 | yes | yes | yes | |||
| hiPSCs | MDCi013-B | PBMCs | 15 | yes | yes | yes | |||
| E | male, 58 | MuSCs | yes | ||||||
| PBMCs | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metzler, E.; Escobar, H.; Sunaga-Franze, D.Y.; Sauer, S.; Diecke, S.; Spuler, S. Generation of hiPSC-Derived Skeletal Muscle Cells: Exploiting the Potential of Skeletal Muscle-Derived hiPSCs. Biomedicines 2022, 10, 1204. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051204
Metzler E, Escobar H, Sunaga-Franze DY, Sauer S, Diecke S, Spuler S. Generation of hiPSC-Derived Skeletal Muscle Cells: Exploiting the Potential of Skeletal Muscle-Derived hiPSCs. Biomedicines. 2022; 10(5):1204. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051204
Chicago/Turabian StyleMetzler, Eric, Helena Escobar, Daniele Yumi Sunaga-Franze, Sascha Sauer, Sebastian Diecke, and Simone Spuler. 2022. "Generation of hiPSC-Derived Skeletal Muscle Cells: Exploiting the Potential of Skeletal Muscle-Derived hiPSCs" Biomedicines 10, no. 5: 1204. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051204