
Tamoxifen Ameliorates Cholestatic Liver Fibrosis in Mice: Upregulation of TGFβ and IL6 Is a Potential Protective Mechanism

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. DDC-Food Supplementation
2.3. Fibrosis Induction and Experimental Design
2.4. Determination of Hydroxyproline Content in Liver Tissue
2.5. Histology—Sirius Red Staining
2.6. Immunohistochemistry
2.7. Determination of Serum Activity of Aminotransferases
2.8. Quantitative PCR Gene Expression Analysis
2.9. Statistical Analysis
3. Results
3.1. DDC-Supplementation Model—Dose Adjustment
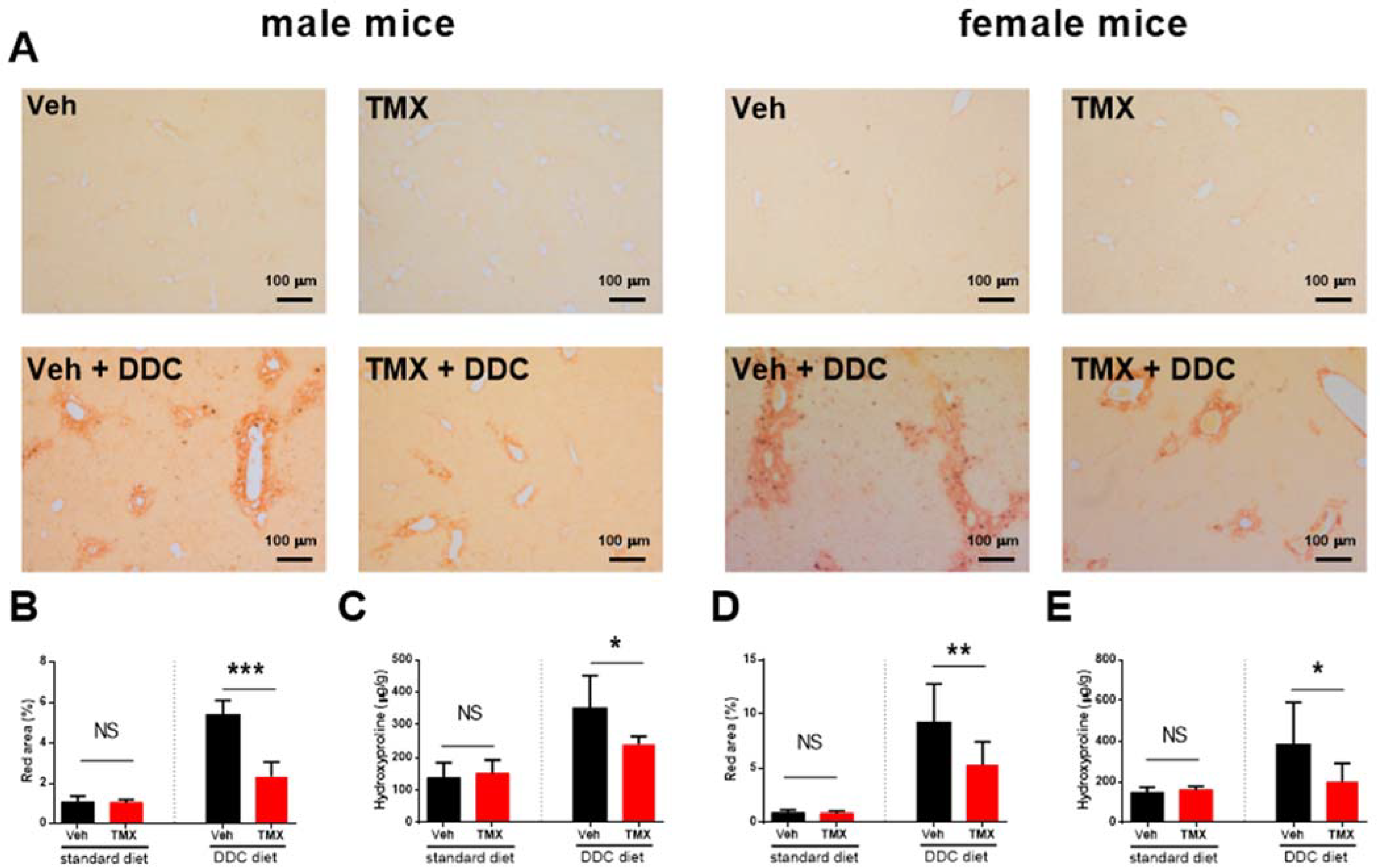
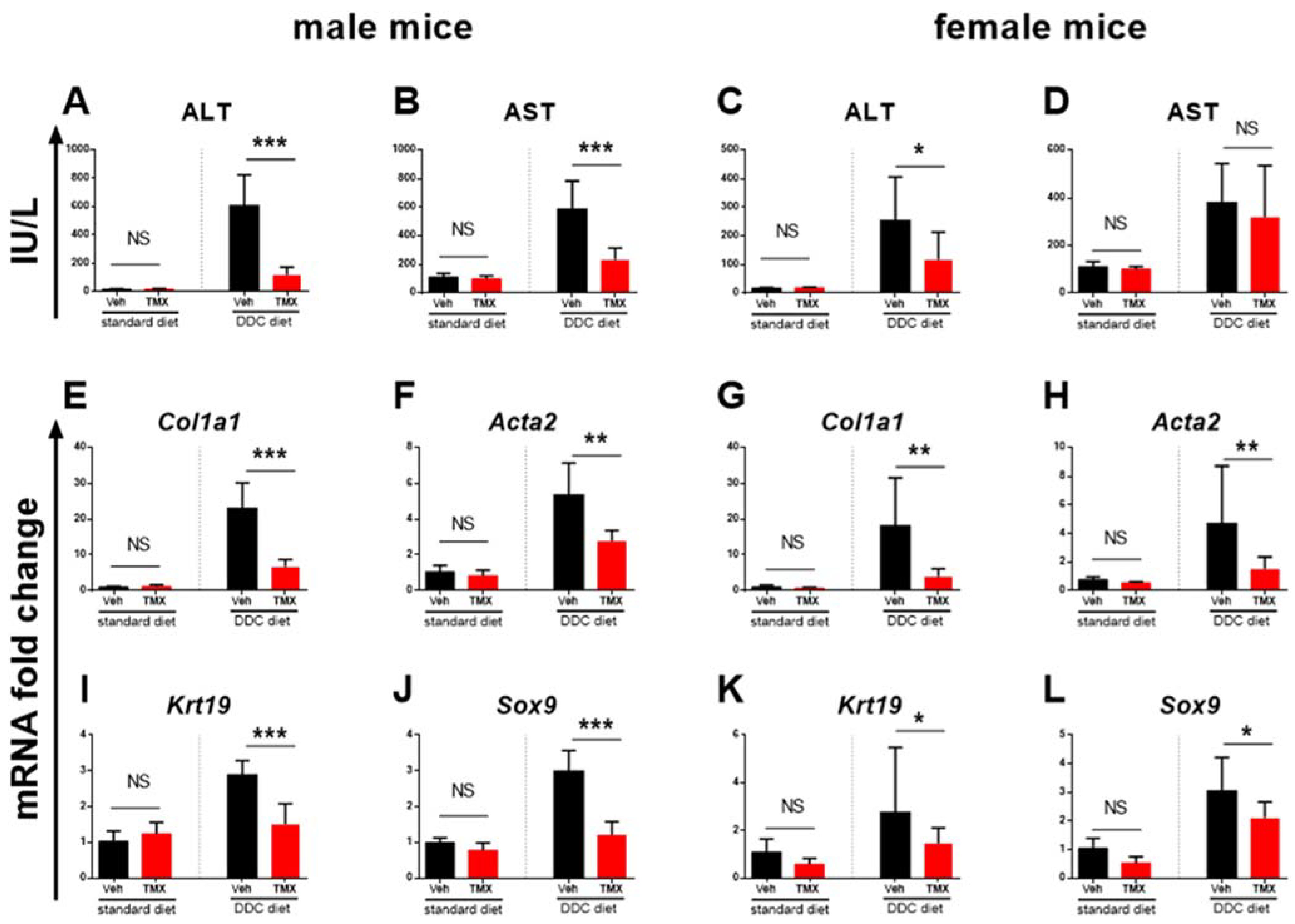
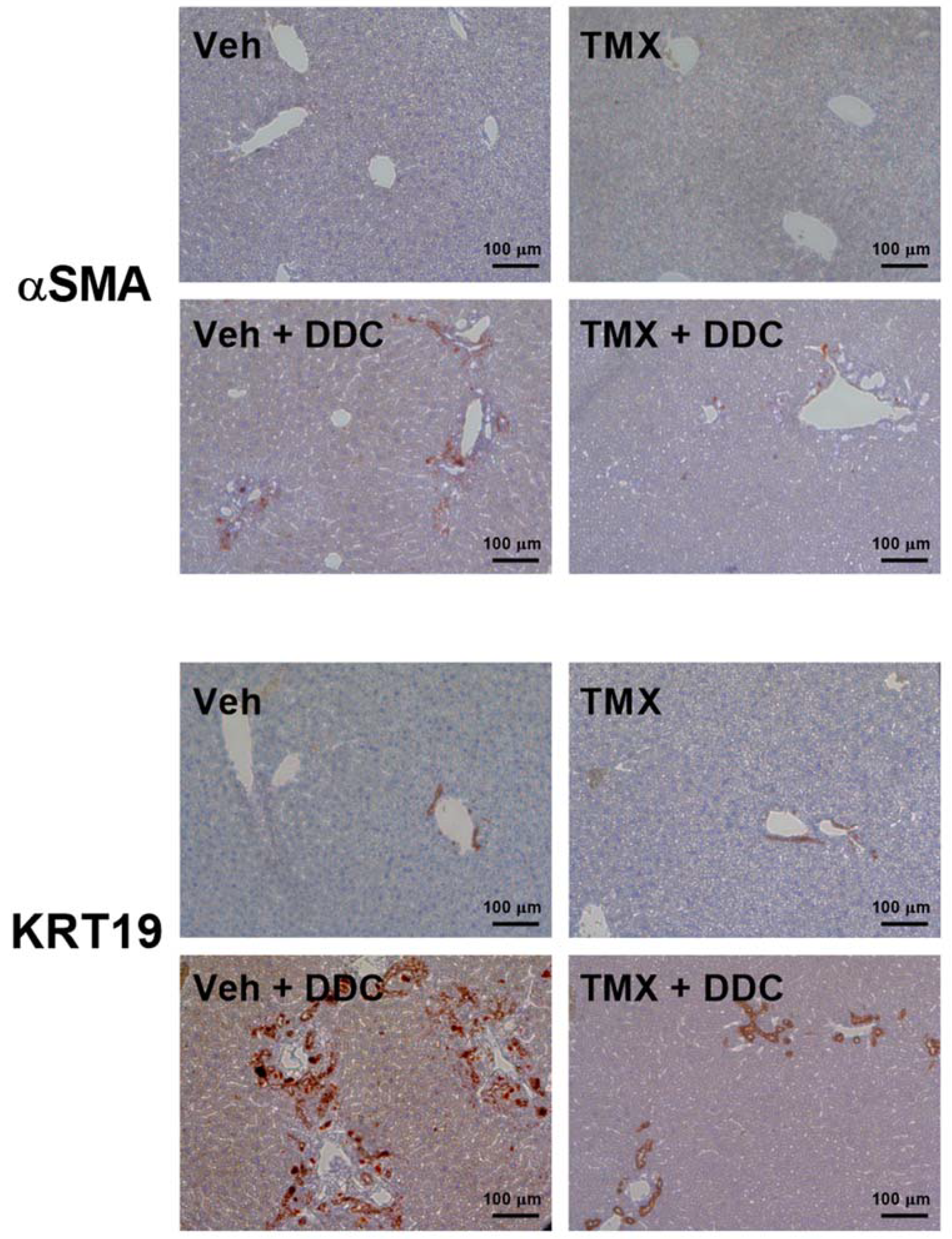
3.2. Tamoxifen Treatment Ameliorates Liver Fibrosis Development
3.3. Tamoxifen Increases Expression of Il6 and Tgfβ in the Livers of Standard Diet-Fed Mice
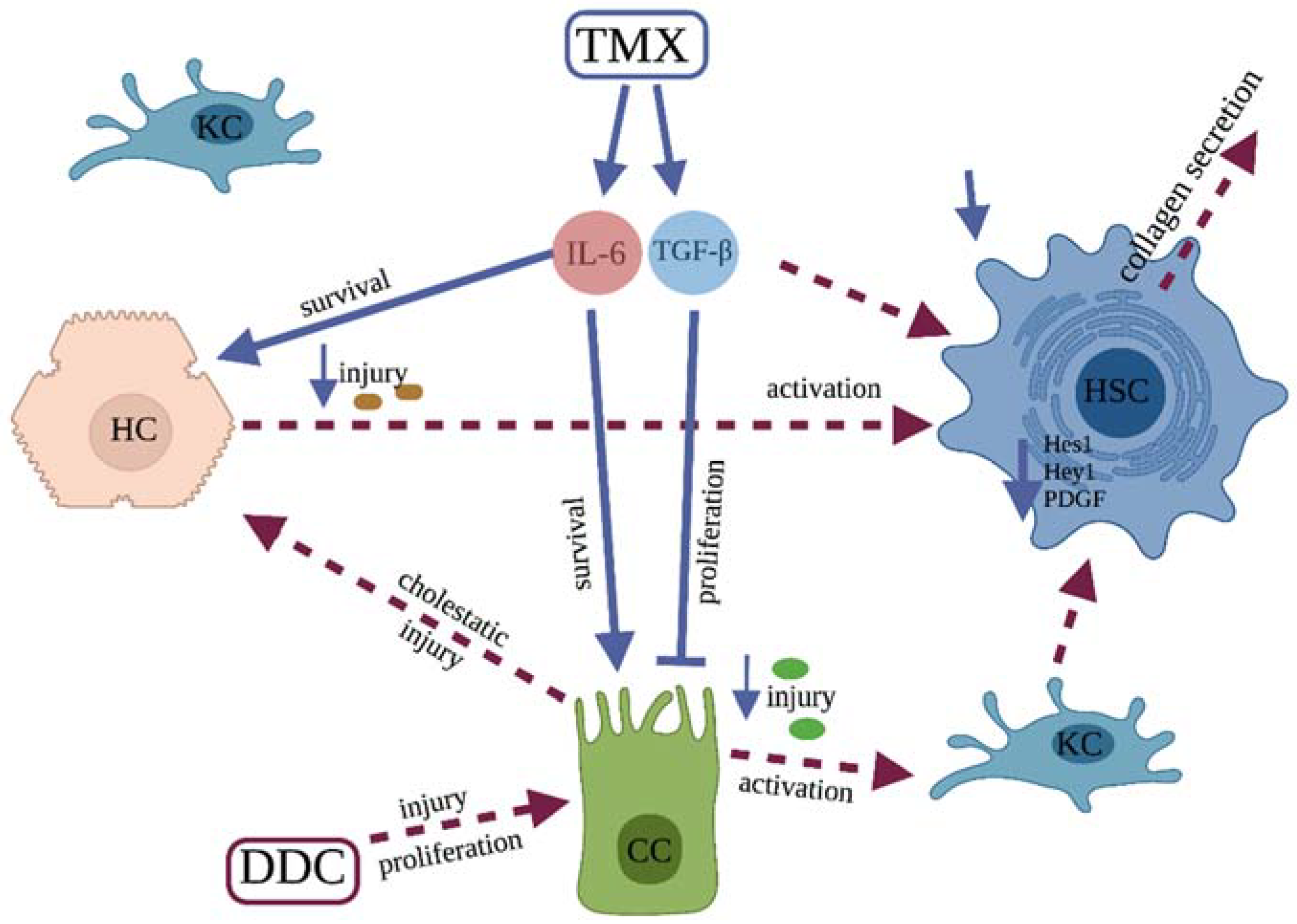
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gossard, A.A.; Talwalkar, J.A. Cholestatic liver disease. Med. Clin. N. Am. 2014, 98, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Aydın, M.M.; Akçalı, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M.; Luong, T.V. Pathogenesis of biliary fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Jang, H.J.; Leem, J.; Kim, G.M. Protective Effects of Bee Venom-Derived Phospholipase A2 against Cholestatic Liver Disease in Mice. Biomedicines 2021, 9, 992. [Google Scholar] [CrossRef]
- Fickert, P.; Stöger, U.; Fuchsbichler, A.; Moustafa, T.; Marschall, H.U.; Weiglein, A.H.; Tsybrovskyy, O.; Jaeschke, H.; Zatloukal, K.; Denk, H.; et al. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am. J. Pathol. 2007, 171, 525–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meerman, L.; Koopen, N.R.; Bloks, V.; Van Goor, H.; Havinga, R.; Wolthers, B.G.; Kramer, W.; Stengelin, S.; Müller, M.; Kuipers, F.; et al. Biliary fibrosis associated with altered bile composition in a mouse model of erythropoietic protoporphyria. Gastroenterology 1999, 117, 696–705. [Google Scholar] [CrossRef]
- Ahmad, I. Tamoxifen a pioneering drug: An update on the therapeutic potential of tamoxifen derivatives. Eur. J. Med. Chem. 2018, 143, 515–531. [Google Scholar] [CrossRef]
- Cortes, E.; Lachowski, D.; Rice, A.; Thorpe, S.D.; Robinson, B.; Yeldag, G.; Lee, D.A.; Ghemtio, L.; Rombouts, K.; Del Río Hernández, A.E. Tamoxifen mechanically deactivates hepatic stellate cells via the G protein-coupled estrogen receptor. Oncogene 2019, 38, 2910–2922. [Google Scholar] [CrossRef] [Green Version]
- Kulcsár, A.; Kulcsár-Gergely, J. Effects of tamoxifen and levonorgestrel treatment on carbon tetrachloride induced alterations in rats. Arzneimittelforschung 1991, 41, 1298–1301. [Google Scholar]
- Xu, J.W.; Gong, J.; Chang, X.M.; Luo, J.Y.; Dong, L.; Hao, Z.M.; Jia, A.; Xu, G.P. Estrogen reduces CCL4-induced liver fibrosis in rats. World J. Gastroenterol. 2002, 8, 883–887. [Google Scholar] [CrossRef]
- Gong, X.; Cao, P.; Liu, L.; Lin, Y.; Yang, Q.; Zhou, L.; Wu, T.; Luo, M. Tamoxifen Prevents D-galactosamine/Lipopolysaccharide-Induced Murine Acute Hepatic Failure through Inhibition of Oxidative Stress and Mmd-2 Upregulation. Immunol. Investig. 2018, 47, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Toyoda, Y.; Tsuneyama, K.; Fukami, T.; Nakajima, M.; Yokoi, T. Hepatoprotective effect of tamoxifen on steatosis and non-alcoholic steatohepatitis in mouse models. J. Toxicol. Sci. 2012, 37, 931–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, S.; Valtcheva, N.; Feil, R. Inducible Cre mice. Methods Mol. Biol. 2009, 530, 343–363. [Google Scholar] [CrossRef] [PubMed]
- Troeger, J.S.; Mederacke, I.; Gwak, G.Y.; Dapito, D.H.; Mu, X.; Hsu, C.C.; Pradere, J.P.; Friedman, R.A.; Schwabe, R.F. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012, 143, 1073–1083.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koli, K.M.; Ramsey, T.T.; Ko, Y.; Dugger, T.C.; Brattain, M.G.; Arteaga, C.L. Blockade of transforming growth factor-beta signaling does not abrogate antiestrogen-induced growth inhibition of human breast carcinoma cells. J. Biol. Chem. 1997, 272, 8296–8302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainger, D.J.; Witchell, C.M.; Metcalfe, J.C. Tamoxifen elevates transforming growth factor-beta and suppresses diet-induced formation of lipid lesions in mouse aorta. Nat. Med. 1995, 1, 1067–1073. [Google Scholar] [CrossRef]
- Qian, J.; Jiao, Y.; Wang, G.; Liu, H.; Cao, X.; Yang, H. Mechanism of TGF-β1 inhibiting Kupffer cell immune responses in cholestatic cirrhosis. Exp. Ther. Med. 2020, 20, 1541–1549. [Google Scholar] [CrossRef]
- Mu, X.; Pradere, J.P.; Affò, S.; Dapito, D.H.; Friedman, R.; Lefkovitch, J.H.; Schwabe, R.F. Epithelial Transforming Growth Factor-β Signaling Does Not Contribute to Liver Fibrosis but Protects Mice From Cholangiocarcinoma. Gastroenterology 2016, 150, 720–733. [Google Scholar] [CrossRef]
- Blum, K.M.; Roby, L.C.; Zbinden, J.C.; Chang, Y.C.; Mirhaidari, G.J.M.; Reinhardt, J.W.; Yi, T.; Barker, J.C.; Breuer, C.K. Sex and Tamoxifen confound murine experimental studies in cardiovascular tissue engineering. Sci. Rep. 2021, 11, 8037. [Google Scholar] [CrossRef]
- Liu, J.; Kasai, S.; Tatara, Y.; Yamazaki, H.; Mimura, J.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; Sato, T.; Ozaki, T.; et al. Inducible Systemic Gcn1 Deletion in Mice Leads to Transient Body Weight Loss upon Tamoxifen Treatment Associated with Decrease of Fat and Liver Glycogen Storage. Int. J. Mol. Sci. 2022, 23, 3201. [Google Scholar] [CrossRef]
- Park, Y.J.; An, H.T.; Park, J.S.; Park, O.; Duh, A.J.; Kim, K.; Chung, K.H.; Lee, K.C.; Oh, Y.; Lee, S. Tyrosine kinase inhibitor neratinib attenuates liver fibrosis by targeting activated hepatic stellate cells. Sci. Rep. 2020, 10, 14756. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Zatloukal, K.; Stumptner, C.; Fuchsbichler, A.; Fickert, P.; Lackner, C.; Trauner, M.; Denk, H. The keratin cytoskeleton in liver diseases. J. Pathol. 2004, 204, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Markotic, A.; Flegar, D.; Grcevic, D.; Sucur, A.; Lalic, H.; Turcic, P.; Kovacic, N.; Lukac, N.; Pravdic, D.; Vukojevic, K.; et al. LPS-induced inflammation desensitizes hepatocytes to Fas-induced apoptosis through Stat3 activation-The effect can be reversed by ruxolitinib. J. Cell. Mol. Med. 2020, 24, 2981–2992. [Google Scholar] [CrossRef]
- Bentzen, S.M.; Skoczylas, J.Z.; Overgaard, M.; Overgaard, J. Radiotherapy-related lung fibrosis enhanced by tamoxifen. J. Natl. Cancer Inst. 1996, 88, 918–922. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Xiang, D.M.; Sun, W.; Ning, B.F.; Zhou, T.F.; Li, X.F.; Zhong, W.; Cheng, Z.; Xia, M.Y.; Wang, X.; Deng, X.; et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut 2018, 67, 1704–1715. [Google Scholar] [CrossRef]
- Mair, M.; Zollner, G.; Schneller, D.; Musteanu, M.; Fickert, P.; Gumhold, J.; Schuster, C.; Fuchsbichler, A.; Bilban, M.; Tauber, S.; et al. Signal transducer and activator of transcription 3 protects from liver injury and fibrosis in a mouse model of sclerosing cholangitis. Gastroenterology 2010, 138, 2499–2508. [Google Scholar] [CrossRef]
- Plum, W.; Tschaharganeh, D.F.; Kroy, D.C.; Corsten, E.; Erschfeld, S.; Dierssen, U.; Wasmuth, H.; Trautwein, C.; Streetz, K.L. Lack of glycoprotein 130/signal transducer and activator of transcription 3-mediated signaling in hepatocytes enhances chronic liver injury and fibrosis progression in a model of sclerosing cholangitis. Am. J. Pathol. 2010, 176, 2236–2246. [Google Scholar] [CrossRef] [Green Version]
- Borkham-Kamphorst, E.; Weiskirchen, R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics. Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.M.; Jafar-Nejad, H. The Roles of Notch Signaling in Liver Development and Disease. Biomolecules 2019, 9, 608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmail, M.M.; Saeed, N.M.; Michel, H.E.; El-Naga, R.N. The ameliorative effect of niclosamide on bile duct ligation induced liver fibrosis via suppression of NOTCH and Wnt pathways. Toxicol. Lett. 2021, 347, 23–35. [Google Scholar] [CrossRef]
- Takahashi, K.; Sato, Y.; Yamamura, M.; Nakada, S.; Tamano, Y.; Sasaki, M.; Harada, K. Notch-Hes1 signaling activation in Caroli disease and polycystic liver disease. Pathol. Int. 2021, 71, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Kohut, T.J.; Gilbert, M.A.; Loomes, K.M. Alagille Syndrome: A Focused Review on Clinical Features, Genetics, and Treatment. Semin. Liver Dis. 2021, 41, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Bainrauch, A.; Šisl, D.; Markotić, A.; Ostojić, A.; Gašparov, S.; Bralić Lang, V.; Kovačić, N.; Grčević, D.; Mrzljak, A.; Kelava, T. NOTCH3 rs1043996 Polymorphism Is Associated with the Occurrence of Alcoholic Liver Cirrhosis Independently of PNPLA3 and TM6SF2 Polymorphisms. J. Clin. Med. 2021, 10, 4621. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Okajima, T.; Takeuchi, H. Significant Roles of Notch O-Glycosylation in Cancer. Molecules 2022, 27, 1783. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, S.; Qi, D.; Zheng, S.; Guo, J.; Zhang, S.; Weng, Z. Inhibition of Notch signaling by a γ-secretase inhibitor attenuates hepatic fibrosis in rats. PLoS ONE 2012, 7, e46512. [Google Scholar] [CrossRef]
- Ceasrine, A.M.; Ruiz-Otero, N.; Lin, E.E.; Lumelsky, D.N.; Boehm, E.D.; Kuruvilla, R. Tamoxifen Improves Glucose Tolerance in a Delivery-, Sex-, and Strain-Dependent Manner in Mice. Endocrinology 2019, 160, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Falke, L.L.; Broekhuizen, R.; Huitema, A.; Maarseveen, E.; Nguyen, T.Q.; Goldschmeding, R. Tamoxifen for induction of Cre-recombination may confound fibrosis studies in female mice. J. Cell Commun. Signal. 2017, 11, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jiang, Z.; Ruan, B.; Duan, J.; Song, P.; Liu, J.; Han, H.; Wang, L. Disruption of myofibroblastic Notch signaling attenuates liver fibrosis by modulating fibrosis progression and regression. Int. J. Biol. Sci. 2021, 17, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.L.; Ruan, B.; Yan, X.C.; Liang, L.; Song, P.; Yang, Z.Y.; Liu, Y.; Dou, K.F.; Han, H.; Wang, L. Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 2018, 68, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersell, K.; Choudhury, S.; Mollova, M.; Polizzotti, B.D.; Ganapathy, B.; Walsh, S.; Wadugu, B.; Arab, S.; Kühn, B. Moderate and high amounts of tamoxifen in αMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis. Models Mech. 2013, 6, 1459–1469. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šisl, D.; Flegar, D.; Filipović, M.; Turčić, P.; Planinić, P.; Šućur, A.; Kovačić, N.; Grčević, D.; Kelava, T. Tamoxifen Ameliorates Cholestatic Liver Fibrosis in Mice: Upregulation of TGFβ and IL6 Is a Potential Protective Mechanism. Biomedicines 2022, 10, 1209. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051209
Šisl D, Flegar D, Filipović M, Turčić P, Planinić P, Šućur A, Kovačić N, Grčević D, Kelava T. Tamoxifen Ameliorates Cholestatic Liver Fibrosis in Mice: Upregulation of TGFβ and IL6 Is a Potential Protective Mechanism. Biomedicines. 2022; 10(5):1209. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051209
Chicago/Turabian StyleŠisl, Dino, Darja Flegar, Maša Filipović, Petra Turčić, Pavao Planinić, Alan Šućur, Nataša Kovačić, Danka Grčević, and Tomislav Kelava. 2022. "Tamoxifen Ameliorates Cholestatic Liver Fibrosis in Mice: Upregulation of TGFβ and IL6 Is a Potential Protective Mechanism" Biomedicines 10, no. 5: 1209. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10051209