
Oxidative Stress and Deregulated DNA Damage Response Network in Lung Cancer Patients

 , , , ,
, , , ,
Abstract
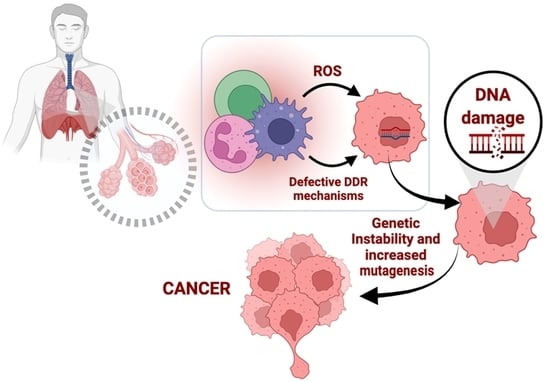
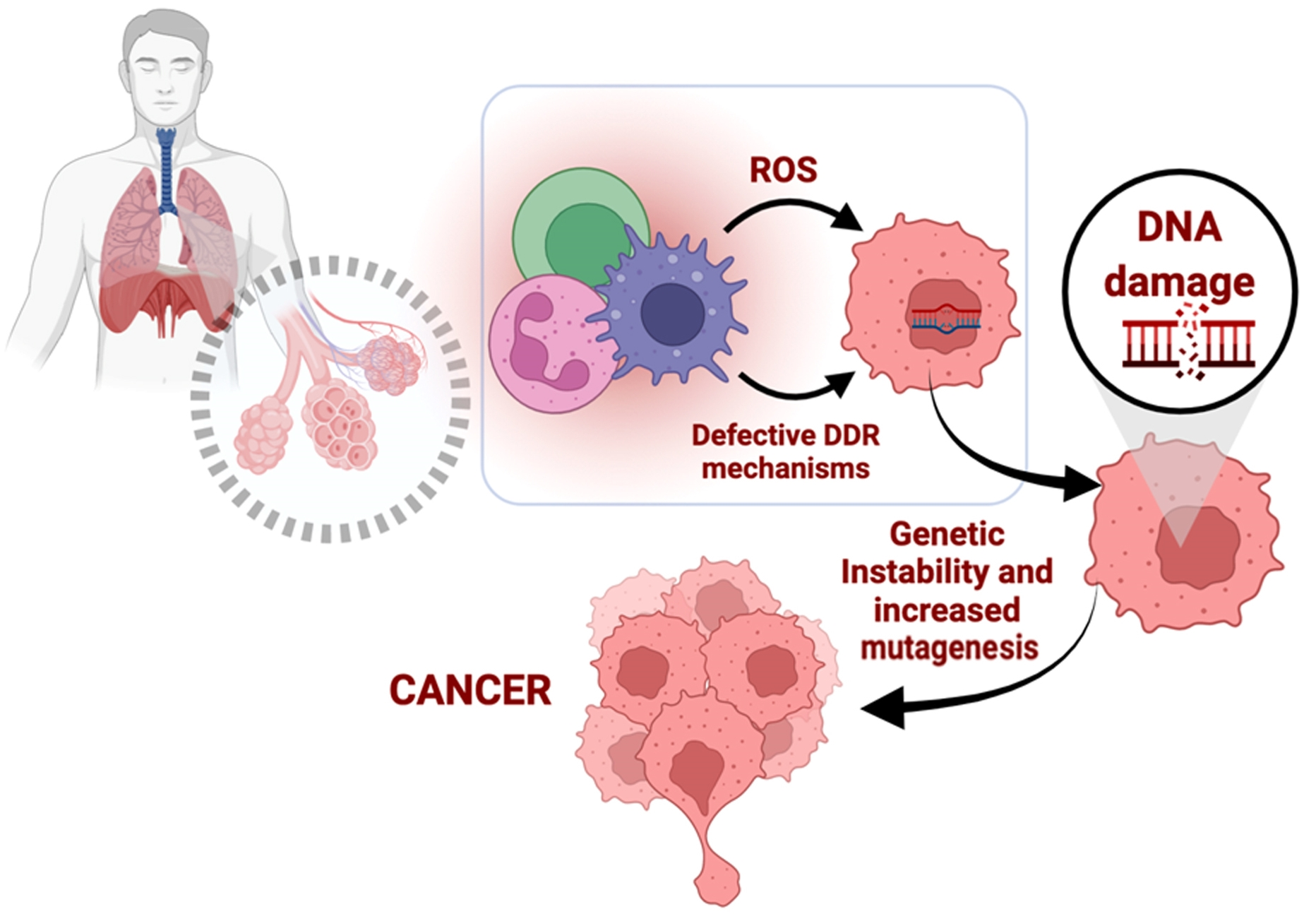
:
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Single-Cell Gel Electrophoresis (Comet Assay)
2.3. Nucleotide Excision Repair Measurement
2.4. Immunofluorescence Detection of γH2AX Foci
2.5. Assessment of Oxidative Stress and Abasic Sites
2.6. Apoptosis
2.7. Expression of DDR-Related Genes
2.8. Systems Biology Statistical Analysis
2.9. Statistical Analysis
3. Results
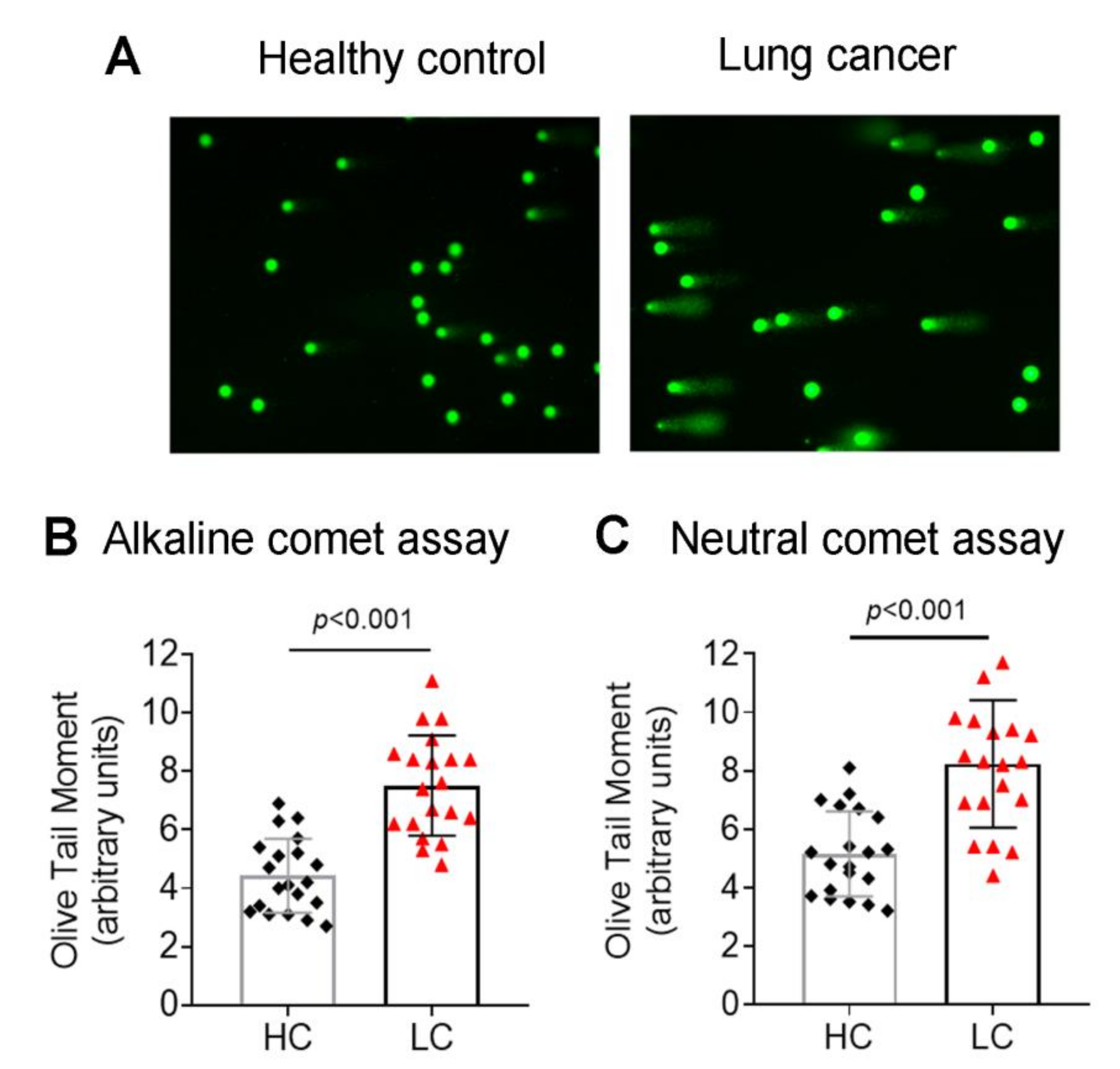
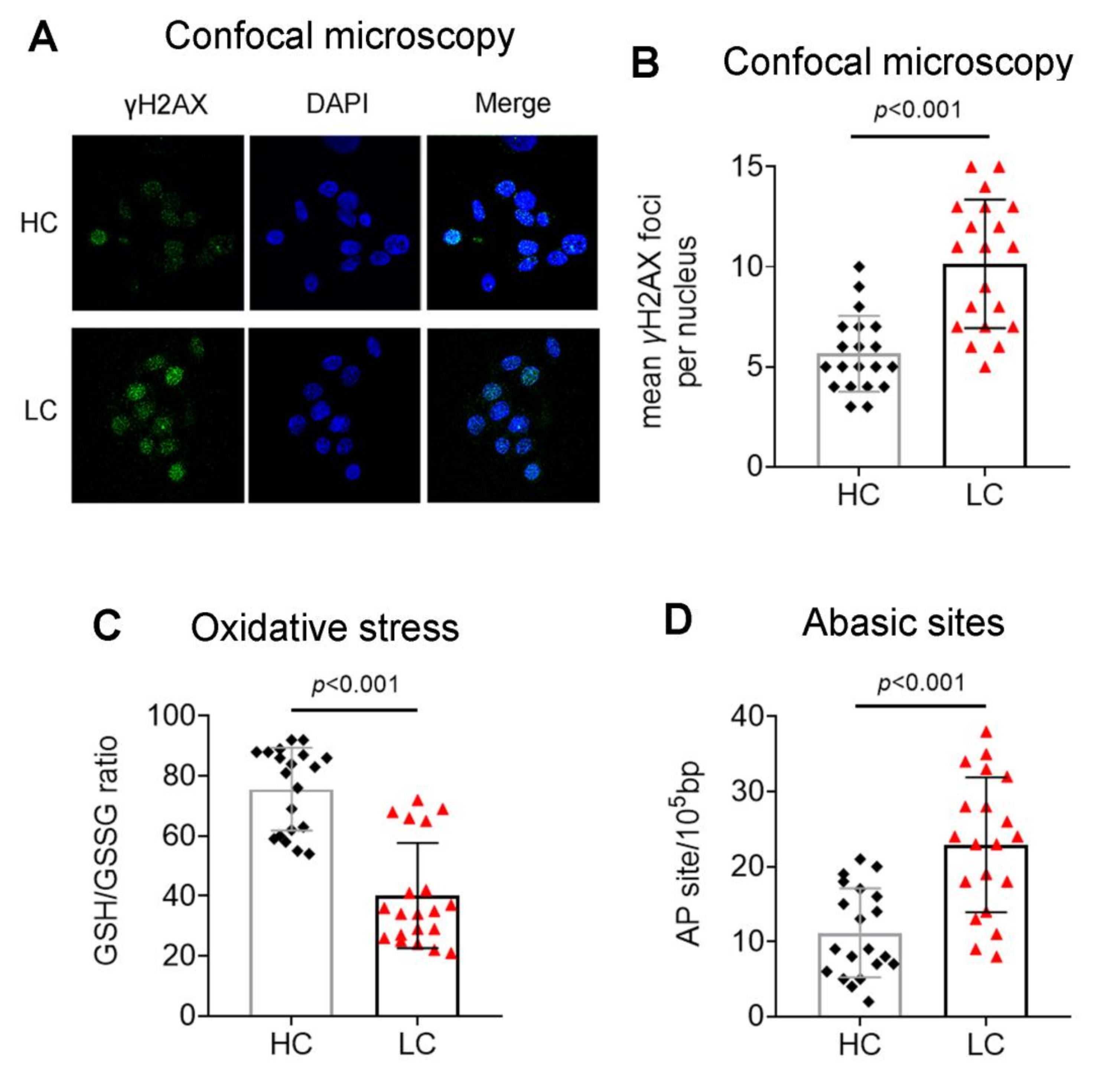
3.1. Accumulation of Endogenous DNA Damage in Lung Cancer Patients
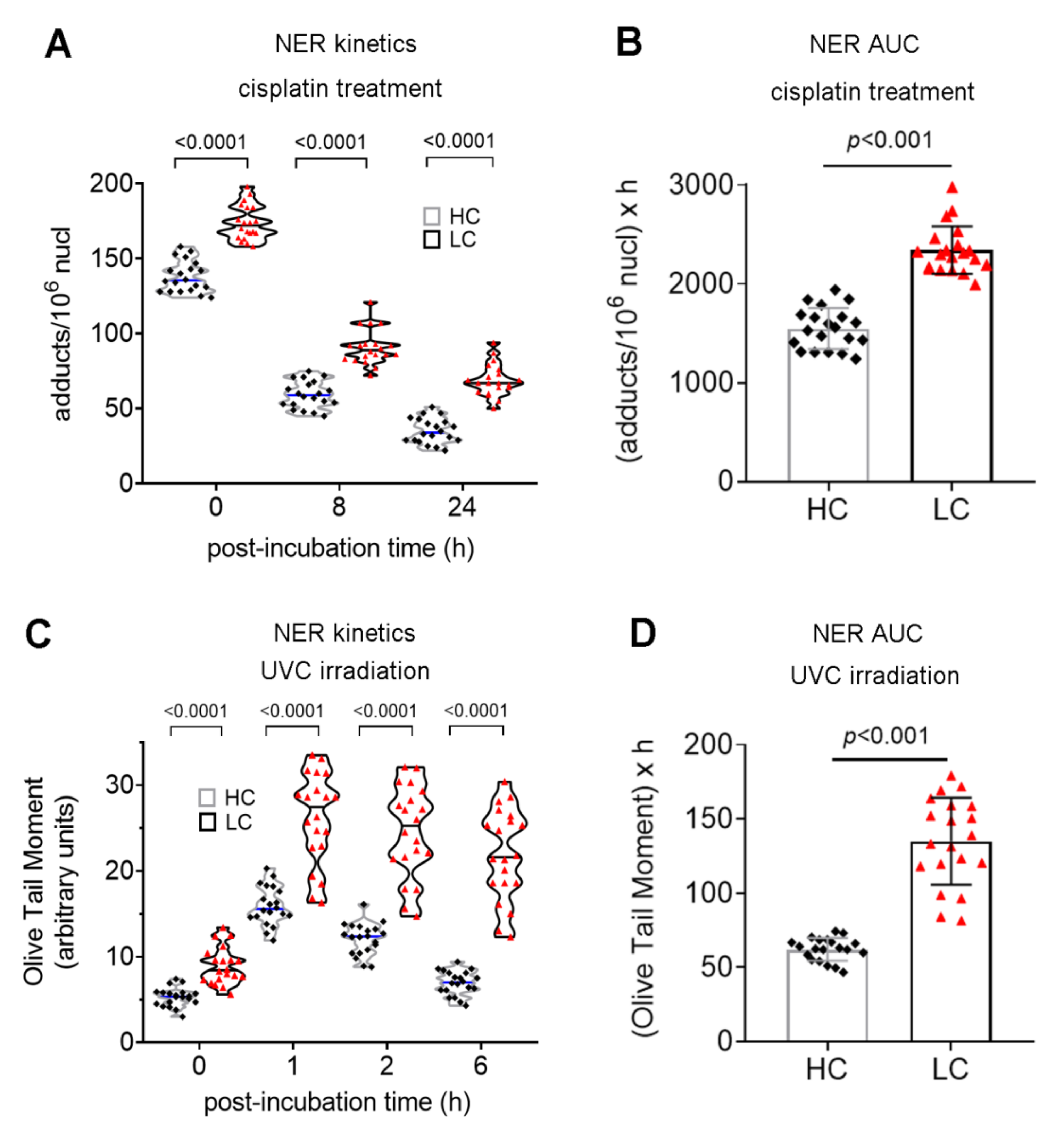
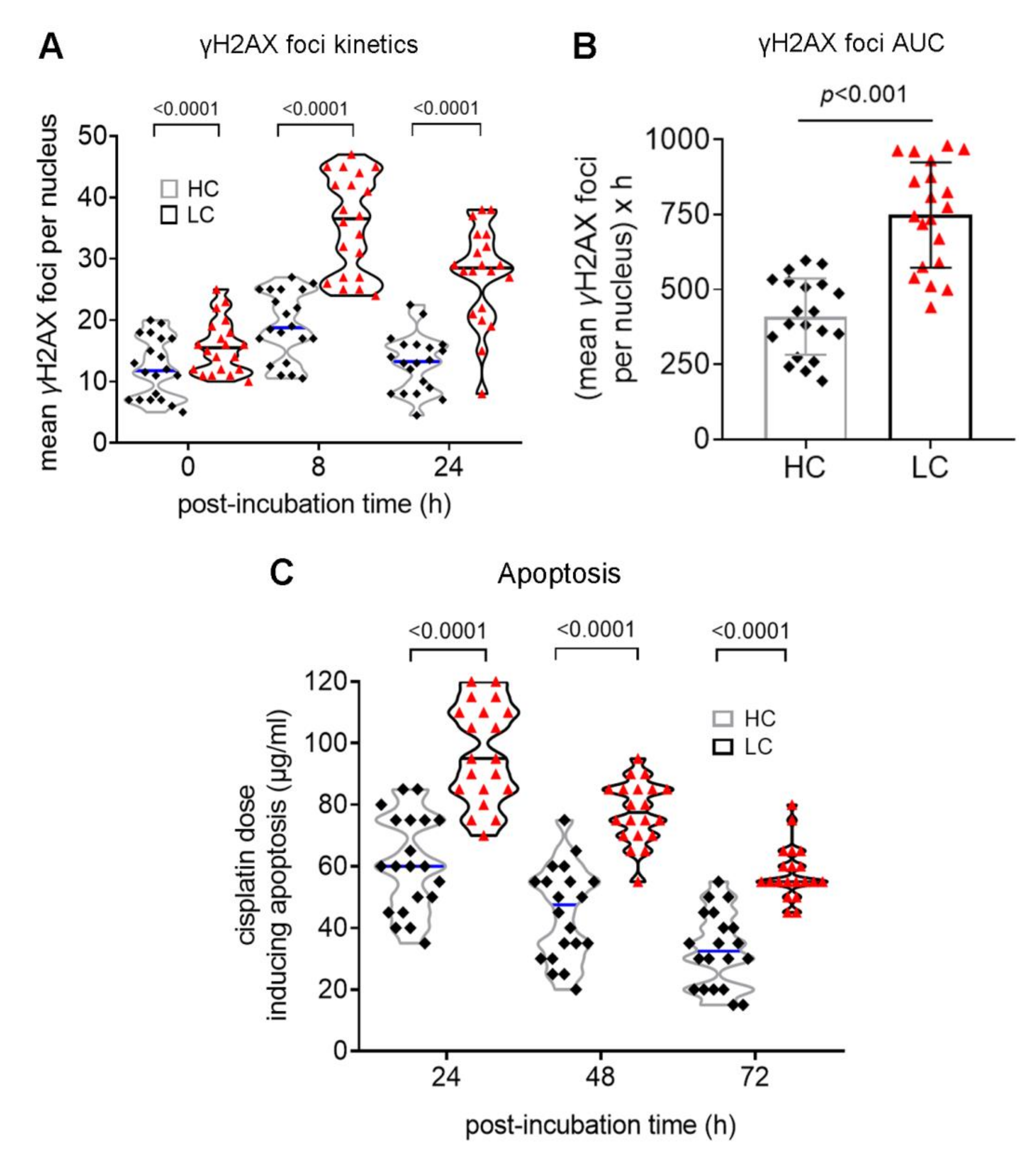
3.2. Defective DDR Parameters in Cancer Patients
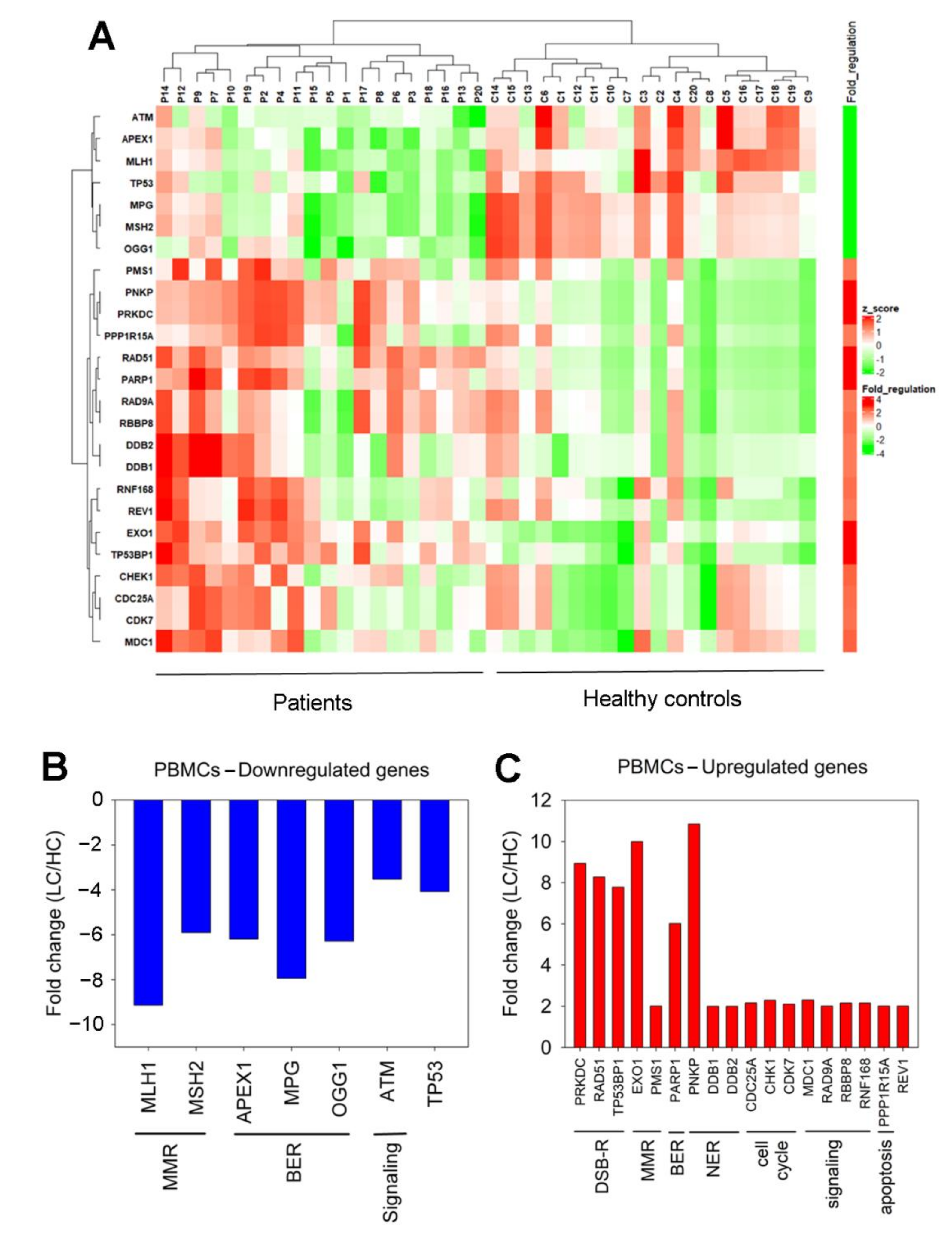
3.3. Expression of DDR-Associated Genes in Cancer Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global Epidemiology of Lung Cancer. Ann. Glob. Health 2019, 85, 8. [Google Scholar] [CrossRef] [Green Version]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubaux, R.; Becker-Santos, D.D.; Enfield, K.S.S.; Lam, S.; Lam, W.L.; Martinez, V.D. Arsenic, asbestos and radon: Emerging players in lung tumorigenesis. Environ. Health 2012, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Souliotis, V.L.; Vlachogiannis, N.I.; Pappa, M.; Argyriou, A.; Ntouros, P.A.; Sfikakis, P.P. DNA Damage Response and Oxidative Stress in Systemic Autoimmunity. Int. J. Mol. Sci. 2019, 21, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.S.; Kim, S.R.; Lee, Y.C. Impact of oxidative stress on lung diseases. Respirology 2009, 14, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Oxidatively induced DNA damage: Mechanisms, repair and disease. Cancer Lett. 2012, 327, 26–47. [Google Scholar] [CrossRef]
- Zhao, H.; Shen, J.; Deininger, P.; Hunt, J.D. Abasic sites and survival in resected patients with non-small cell lung cancer. Cancer Lett. 2007, 246, 47–53. [Google Scholar] [CrossRef]
- Hsiehchen, D.; Hsieh, A.; Samstein, R.M.; Lu, T.; Beg, M.S.; Gerber, D.E.; Wang, T.; Morris, L.G.T.; Zhu, H. DNA Repair Gene Mutations as Predictors of Immune Checkpoint Inhibitor Response beyond Tumor Mutation Burden. Cell Rep. Med. 2020, 1, 100034. [Google Scholar] [CrossRef]
- Rossi, A.; Di Maio, M. Platinum-based chemotherapy in advanced non-small-cell lung cancer: Optimal number of treatment cycles. Expert Rev. Anticancer Ther. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, H.M.; Yan, W.J.; Qin, Y. Identification of the Signature Genes and Network of Reactive Oxygen Species Related Genes and DNA Repair Genes in Lung Adenocarcinoma. Front. Med. 2022, 9, 833829. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Gong, T.; Sun, B.; Zhang, Z.; Zhong, D.; Wang, C. Identification of a DNA damage repair gene-related signature for lung squamous cell carcinoma prognosis. Thorac. Cancer 2022, 13, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Souliotis, V.L.; Vlachogiannis, N.I.; Pappa, M.; Argyriou, A.; Sfikakis, P.P. DNA damage accumulation, defective chromatin organization and deficient DNA repair capacity in patients with rheumatoid arthritis. Clin. Immunol. 2019, 203, 28–36. [Google Scholar] [CrossRef]
- Vlachogiannis, N.I.; Pappa, M.; Ntouros, P.A.; Nezos, A.; Mavragani, C.P.; Souliotis, V.L.; Sfikakis, P.P. Association Between DNA Damage Response, Fibrosis and Type I Interferon Signature in Systemic Sclerosis. Front. Immunol. 2020, 11, 582401. [Google Scholar] [CrossRef] [PubMed]
- Stellas, D.; Souliotis, V.L.; Bekyrou, M.; Smirlis, D.; Kirsch-Volders, M.; Degrassi, F.; Cundari, E.; Kyrtopoulos, S.A. Benzo[a]pyrene-induced cell cycle arrest in HepG2 cells is associated with delayed induction of mitotic instability. Mutat. Res. 2014, 769, 59–68. [Google Scholar] [CrossRef]
- Cheng, S.-B.; Liu, H.-T.; Chen, S.-Y.; Lin, P.-T.; Lai, C.-Y.; Huang, Y.-C. Changes of Oxidative Stress, Glutathione, and Its Dependent Antioxidant Enzyme Activities in Patients with Hepatocellular Carcinoma before and after Tumor Resection. PLoS ONE 2017, 12, e0170016. [Google Scholar] [CrossRef]
- Peddireddy, V.; Prasad, B.S.; Gundimeda, S.D.; Penagaluru, P.R.; Mundluru, H.P. Assessment of 8-oxo-7, 8-dihydro-2′-deoxyguanosine and malondialdehyde levels as oxidative stress markers and antioxidant status in non-small cell lung cancer. Biomarkers 2012, 17, 261–268. [Google Scholar] [CrossRef]
- Korkmaz, G.G.; Inal, B.B.; Ortakoylu, G.M.; Irmak, H.; Kara, A.A.; Gelisgen, R.; Ogurlu, O.; Uzun, H. Changes in oxidative stress parameters and antioxidant status in lung cancer: Western blot analysis of nitrotyrosine and protein carbonyls content. Clin. Lab. 2014, 60, 599–607. [Google Scholar] [CrossRef]
- Gào, X.; Brenner, H.; Holleczek, B.; Cuk, K.; Zhang, Y.; Anusruti, A.; Xuan, Y.; Xu, Y.; Schöttker, B. Urinary 8-isoprostane levels and occurrence of lung, colorectal, prostate, breast and overall cancer: Results from a large, population-based cohort study with 14 years of follow-up. Free Radic. Biol. Med. 2018, 123, 20–26. [Google Scholar] [CrossRef]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic inflammation: Key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019, 10, 365–381. [Google Scholar] [CrossRef] [Green Version]
- Doz, E.; Noulin, N.; Boichot, E.; Guénon, I.; Fick, L.; Le Bert, M.; Lagente, V.; Ryffel, B.; Schnyder, B.; Quesniaux, V.F.J.; et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J. Immunol. 2008, 180, 1169–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz-Elizur, T.; Leitner-Dagan, Y.; Meyer, K.B.; Markus, B.; Giorgi, F.M.; O’Reilly, M.; Kim, H.; Evgy, Y.; Fluss, R.; Freedman, L.S.; et al. DNA Repair Biomarker for Lung Cancer Risk and its Correlation With Airway Cells Gene Expression. JNCI Cancer Spectr. 2019, 4, pkz067. [Google Scholar] [CrossRef] [PubMed]
- Hocsak, E.; Szabo, V.; Kalman, N.; Antus, C.; Cseh, A.; Sumegi, K.; Eros, K.; Hegedus, Z.; Gallyas, F., Jr.; Sumegi, B.; et al. PARP inhibition protects mitochondria and reduces ROS production via PARP-1-ATF4-MKP-1-MAPK retrograde pathway. Free Radic. Biol. Med. 2017, 108, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, C.R.; Zhou, H.; Justice, M.J.; Fisher, A.J.; Saliba, J.; Lamb, I.; Wicker, J.; Schweitzer, K.S.; Petrache, I. Xeroderma Pigmentosum Group C Deficiency Alters Cigarette Smoke DNA Damage Cell Fate and Accelerates Emphysema Development. Am. J. Respir. Cell Mol. Biol. 2018, 58, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holcomb, N.; Goswami, M.; Han, S.G.; Clark, S.; Orren, D.K.; Gairola, C.G.; Mellon, I. Exposure of Human Lung Cells to Tobacco Smoke Condensate Inhibits the Nucleotide Excision Repair Pathway. PLoS ONE 2016, 11, e0158858. [Google Scholar] [CrossRef] [PubMed]
- Wittschieben, B.Ø.; Iwai, S.; Wood, R.D. DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. J. Biol. Chem. 2005, 280, 39982–39989. [Google Scholar] [CrossRef] [Green Version]
- Mo, X. Low Expression of 12 DNA repair genes was associated with better disease-free survival in non-small cell lung cancer patients having adjuvant chemotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 237. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Qiao, G.B.; Wu, Y.L.; Yang, X.N.; Zhong, W.Z.; Xie, D.; Guan, X.Y.; Fischer, D.; Kolberg, H.-C.; Kruger, S.; Stuerzbecher, H.-W. High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br. J. Cancer 2005, 93, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medová, M.; Medo, M.; Hovhannisyan, L.; Muñoz-Maldonado, C.; Aebersold, D.M.; Zimmer, Y. DNA-PK in human malignant disorders: Mechanisms and implications for pharmacological interventions. Pharmacol. Ther. 2020, 215, 107617. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, P.; Lv, X.; Zhang, L.; Zhang, J. The role of the ataxia telangiectasia mutated gene in lung cancer: Recent advances in research. Ther. Adv. Respir. Dis. 2017, 11, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Tong, X.; Ma, Y.; Liu, S.; Yang, L.; Yang, X.; Yang, X.; Bai, M.; Fan, H. Association between ATM gene polymorphisms, lung cancer susceptibility and radiation-induced pneumonitis: A meta-analysis. BMC Pulm. Med. 2017, 17, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Lin, H.; He, P.; He, L.; Chen, J.; Lin, L.; Chen, Y. A TP53-associated gene signature for prediction of prognosis and therapeutic responses in lung squamous cell carcinoma. Oncoimmunology 2020, 9, 1731943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, C.; Davis, C.W.; Mick, R.; Thompson, J.C.; Ahmed, S.; Jeffries, S.; Bagley, S.; Gabriel, P.; Evans, T.L.; Bauml, J.M.; et al. Influence of TP53 Mutation on Survival in Patients With Advanced EGFR-Mutant Non-Small-Cell Lung Cancer. JCO Precis. Oncol. 2018, 2018, PO.18.00107. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, G.; Li, J.; Huang, Y.Y.; Li, Y.; Lin, J.; Chen, L.Z.; Lu, J.P.; Wang, U.Q.; Wang, C.X.; et al. Association of Tumor Protein p53 and Ataxia-Telangiectasia Mutated Comutation With Response to Immune Checkpoint Inhibitors and Mortality in Patients with Non–Small Cell Lung Cancer. JAMA Netw. Open 2019, 2, e1911895. [Google Scholar] [CrossRef]
- Hsu, H.S.; Wen, C.K.; Tang, Y.A.; Lin, R.K.; Li, W.Y.; Hsu, W.H.; Wang, Y.C. Promoter hypermethylation is the predominant mechanism in hMLH1 and hMSM2 deregulation and is a poor prognostic factor in nonsmoking lung cancer. Clin. Cancer Res. 2005, 11, 5410–5416. [Google Scholar] [CrossRef] [Green Version]
- Kanellis, G.; Chatzistamou, I.; Koutselini, H.; Politi, E.; Gouliamos, A.; Vlahos, L.; Koutselinis, A. Expression of DNA mismatch repair gene MSH2 in cytological material from lung cancer patients. Diagn. Cytopathol. 2006, 34, 463–466. [Google Scholar] [CrossRef]
- Keijzers, G.; Bakula, D.; Petr, M.A.; Madsen, N.G.K.; Teklu, A.; Mkrtchyan, G.; Osborne, B.; Scheibye-Knudsen, M. Human Exonuclease 1 (EXO1) Regulatory Functions in DNA Replication with Putative Roles in Cancer. Int. J. Mol. Sci. 2018, 20, 74. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Tang, S.; Liu, J.; Wu, Q.; Wan, L.; Xu, Q. Genetic risk of lung cancer associated with a single nucleotide polymorphism from EXO1: A meta analysis. Int. J. Clin. Exp. Med. 2015, 8, 11132–11138. [Google Scholar] [PubMed]
- Dong, J.; Hu, Z.; Shu, Y.; Pan, S.; Chen, W.; Wang, Y.; Hu, L.; Jiang, K.; Dai, J.; Ma, H.; et al. Potentially functional polymorphisms in DNA repair genes and nonsmall-cell lung cancer survival: A pathway-based analysis. Mol. Carcinog. 2012, 51, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Younis, R.H.; Cao, W.; Lin, R.; Xia, R.; Liu, Z.; Edelman, M.J.; Mei, Y.; Mao, L.; Ren, H. CDC25AQ110del: A Novel Cell Division Cycle 25A Isoform Aberrantly Expressed in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e46464. [Google Scholar] [CrossRef] [PubMed]
- Grabauskiene, S.; Bergeron, E.J.; Chen, G.; Chang, A.C.; Lin, J.; Thomas, D.G.; Giordano, T.J.; Beer, D.G.; Morgan, M.A.; Reddy, R.M. CHK1 levels correlate with sensitization to pemetrexed by CHK1 inhibitors in non-small cell lung cancer cells. Lung Cancer 2013, 82, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Broustas, C.G.; Hopkins, K.M.; Panigrahi, S.K.; Wang, L.; Virk, R.K.; Lieberman, H.B. RAD9A promotes metastatic phenotypes through transcriptional regulation of anterior gradient 2 (AGR2). Carcinogenesis 2019, 40, 164–172. [Google Scholar] [CrossRef]
- Ahmed, K.M.; Tsai, C.Y.; Lee, W.H. Derepression of HMGA2 via removal of ZBRK1/BRCA1/CtIP complex enhances mammary tumorigenesis. J. Biol. Chem. 2010, 285, 4464–4471. [Google Scholar] [CrossRef] [Green Version]
- Eshan, P.; Alexandru, A.; Arishya, S. RNF168 is a Potential Therapeutic Target in Non-Small Cell Lung Cancer. Cancer Ther. Oncol. Int. J. 2021, 18, 555999. [Google Scholar] [CrossRef]
- Liu, L.; Ito, S.; Nishio, N.; Sun, Y.; Chen, N.; Tanaka, Y.; Isobe, K.I. GADD34 Facilitates Cell Death Resulting from Proteasome Inhibition. Anticancer Res. 2015, 35, 5317–5324. [Google Scholar]
- Sasatani, M.; Xi, Y.; Kajimura, J.; Kawamura, T.; Piao, J.; Masuda, Y.; Honda, H.; Kubo, K.; Mikamoto, T.; Watanabe, H.; et al. Overexpression of Rev1 promotes the development of carcinogen-induced intestinal adenomas via accumulation of point mutation and suppression of apoptosis proportionally to the Rev1 expression level. Carcinogenesis 2017, 38, 570–578. [Google Scholar] [CrossRef]
- Sharma, A.; Boise, L.H.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | |||
|---|---|---|---|
| Characteristic | N | Years | % of Total |
| Sex | |||
| Male | 17 | - | 85 |
| Female | 3 | - | 15 |
| Age | |||
| Median | - | 71 | - |
| Range | - | 57–84 | - |
| Histology | |||
| Squamous | 4 | - | 20 |
| Nonsquamous | 10 | - | 50 |
| Small cell Lung Cancer | 5 | - | 25 |
| EGFR mutation status | |||
| Positive | 1 | - | 5 |
| Negative | 9 | - | 45 |
| EML4-ALK rearrrangement status | |||
| Positive | 0 | - | 0 |
| Negative | 10 | - | 50 |
| BRAF mutation status | |||
| Positive | 1 | - | 5 |
| Negative | 9 | - | 45 |
| History of tobacco use | |||
| Never | 3 | - | 15 |
| Current | 3 | - | 15 |
| Previous | 14 | - | 70 |
| PD-L1 | |||
| <1% | 4 | - | 20 |
| 1–50% | 8 | - | 40 |
| >50% | 2 | - | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefanou, D.T.; Kouvela, M.; Stellas, D.; Voutetakis, K.; Papadodima, O.; Syrigos, K.; Souliotis, V.L. Oxidative Stress and Deregulated DNA Damage Response Network in Lung Cancer Patients. Biomedicines 2022, 10, 1248. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061248
Stefanou DT, Kouvela M, Stellas D, Voutetakis K, Papadodima O, Syrigos K, Souliotis VL. Oxidative Stress and Deregulated DNA Damage Response Network in Lung Cancer Patients. Biomedicines. 2022; 10(6):1248. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061248
Chicago/Turabian StyleStefanou, Dimitra T., Marousa Kouvela, Dimitris Stellas, Konstantinos Voutetakis, Olga Papadodima, Konstantinos Syrigos, and Vassilis L. Souliotis. 2022. "Oxidative Stress and Deregulated DNA Damage Response Network in Lung Cancer Patients" Biomedicines 10, no. 6: 1248. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061248