
Temporal Characterization of Behavioral and Hippocampal Dysfunction in the YAC128 Mouse Model of Huntington’s Disease

 , ,
, ,  ,
,  ,
,
Abstract
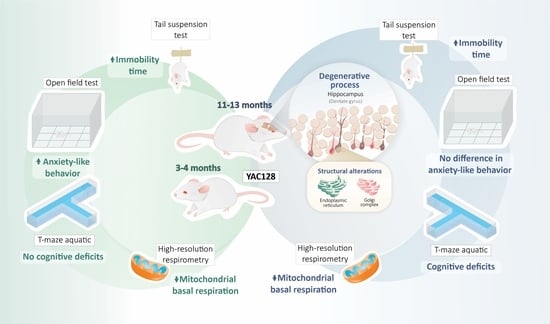
:
1. Introduction
2. Materials and Methods
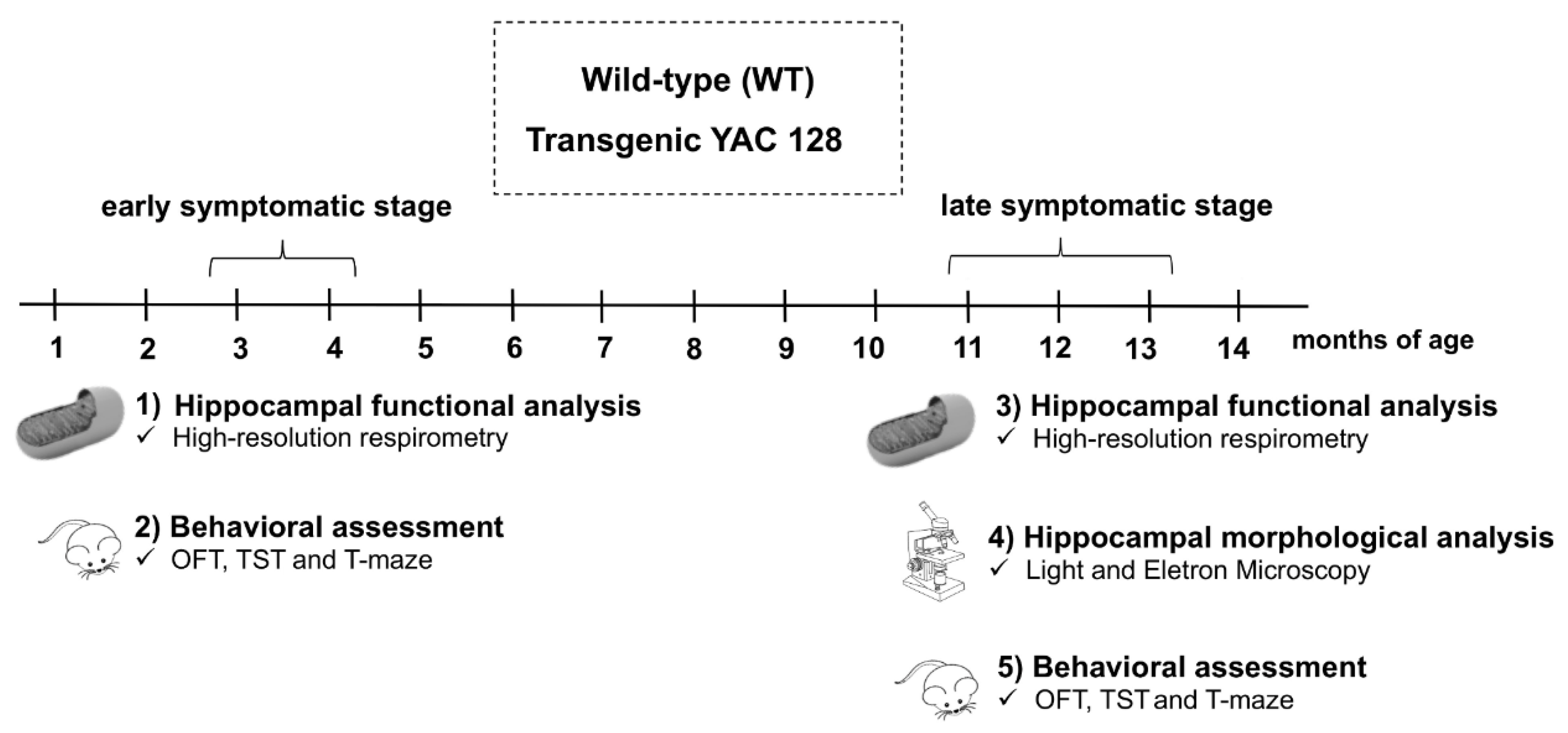
2.1. Animals
2.2. Experimental Protocol
2.3. Behavioral Analyses
- (a)
- Open-Field Test (OFT).
- (b)
- Tail Suspension Test (TST).
- (c)
- Normal Phase Swimming T-maze Test.
- (d)
- Reversal Phase Swimming T-maze Test.
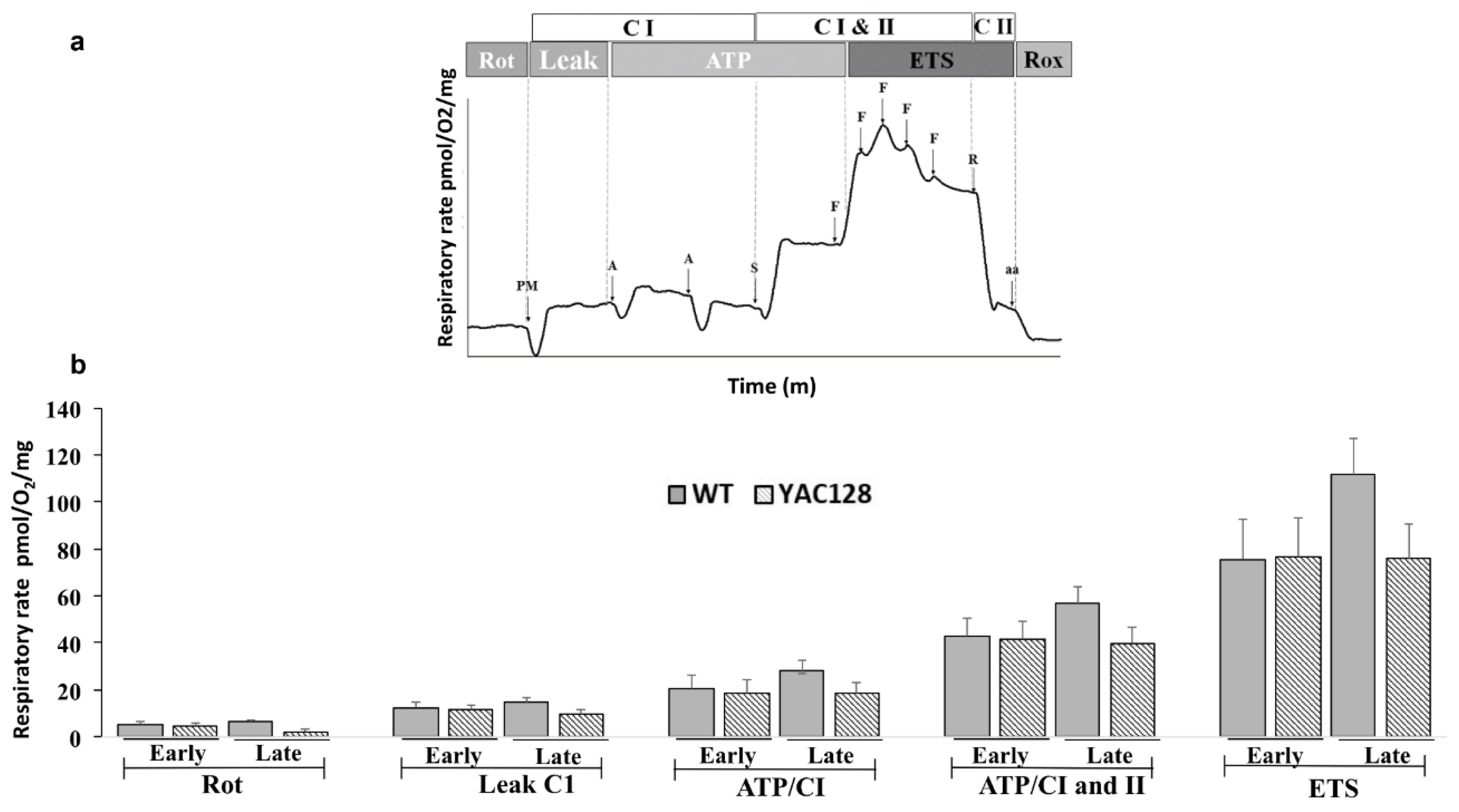
2.4. High-Resolution Respirometry (Oroboros Oxygraph-O2K)
2.5. Morphological Analysis (Light and Electron Microscopy)
2.6. Image Capture and Analysis
2.7. Statistical Analysis
3. Results
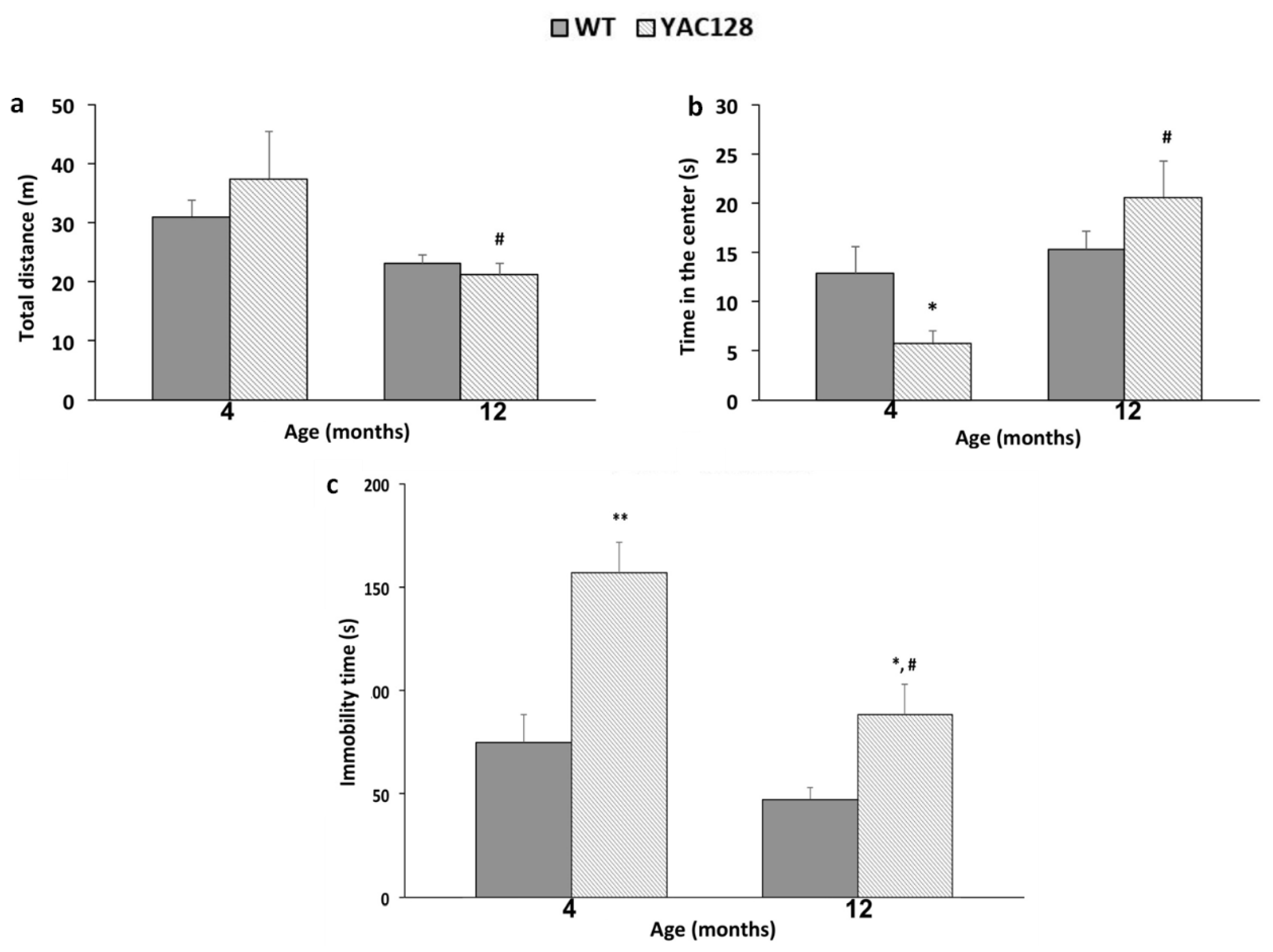
3.1. Effect of Genotype and Stage of Disease Progression on Locomotor Activity and on the Occurrence of Depressive-Like Behaviors
3.2. Effect of Genotype and Stage of Disease Progression on Procedural and Spatial Learning
3.3. Effect of Genotype and Stage of Disease Progression on the Evaluation of Mitochondrial Oxygen Consumption in the Hippocampus
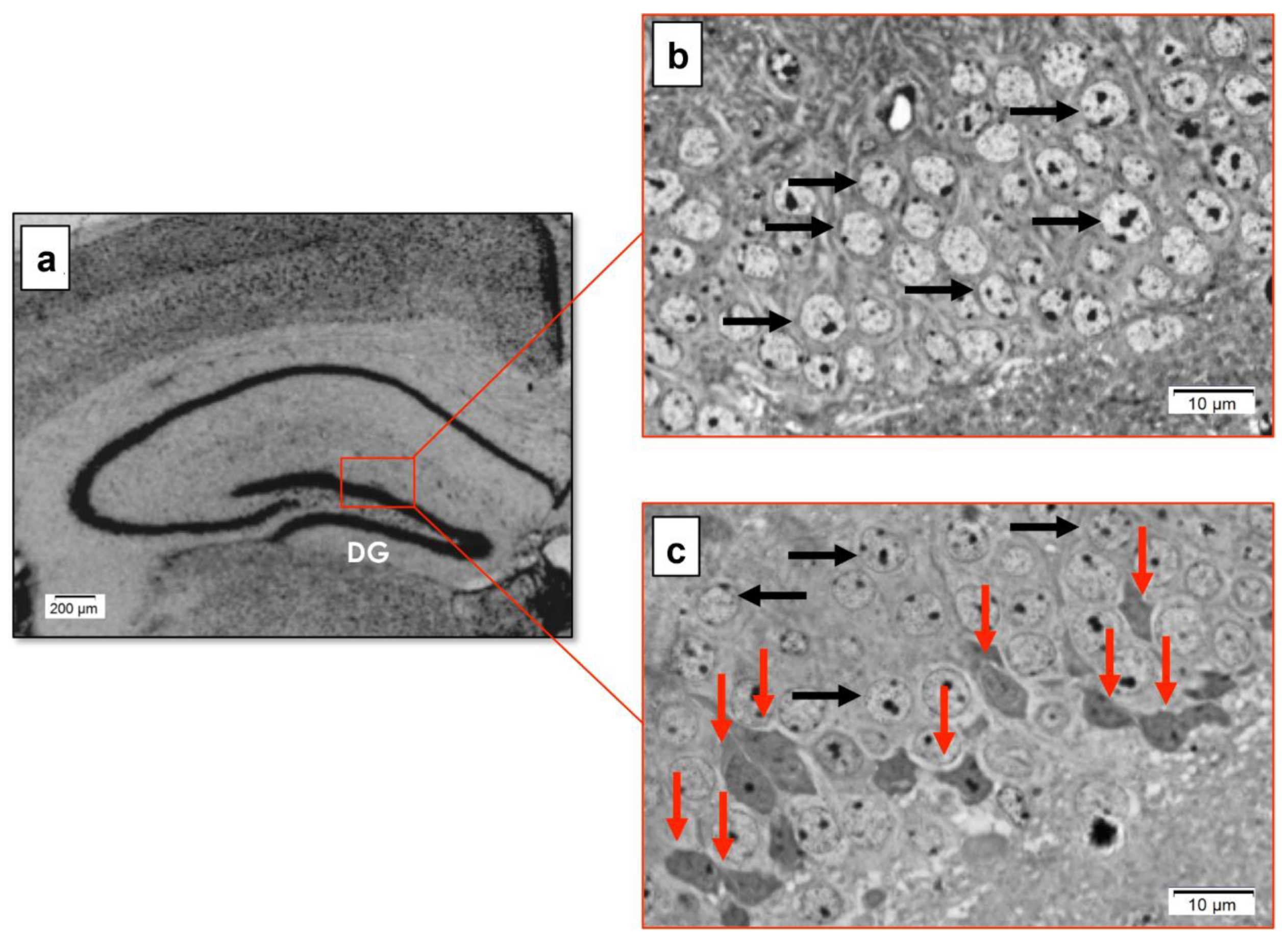
3.4. Effect of Genotype on the Morphology of the Hippocampal DG in Late Symptomatic YAC128 Mice
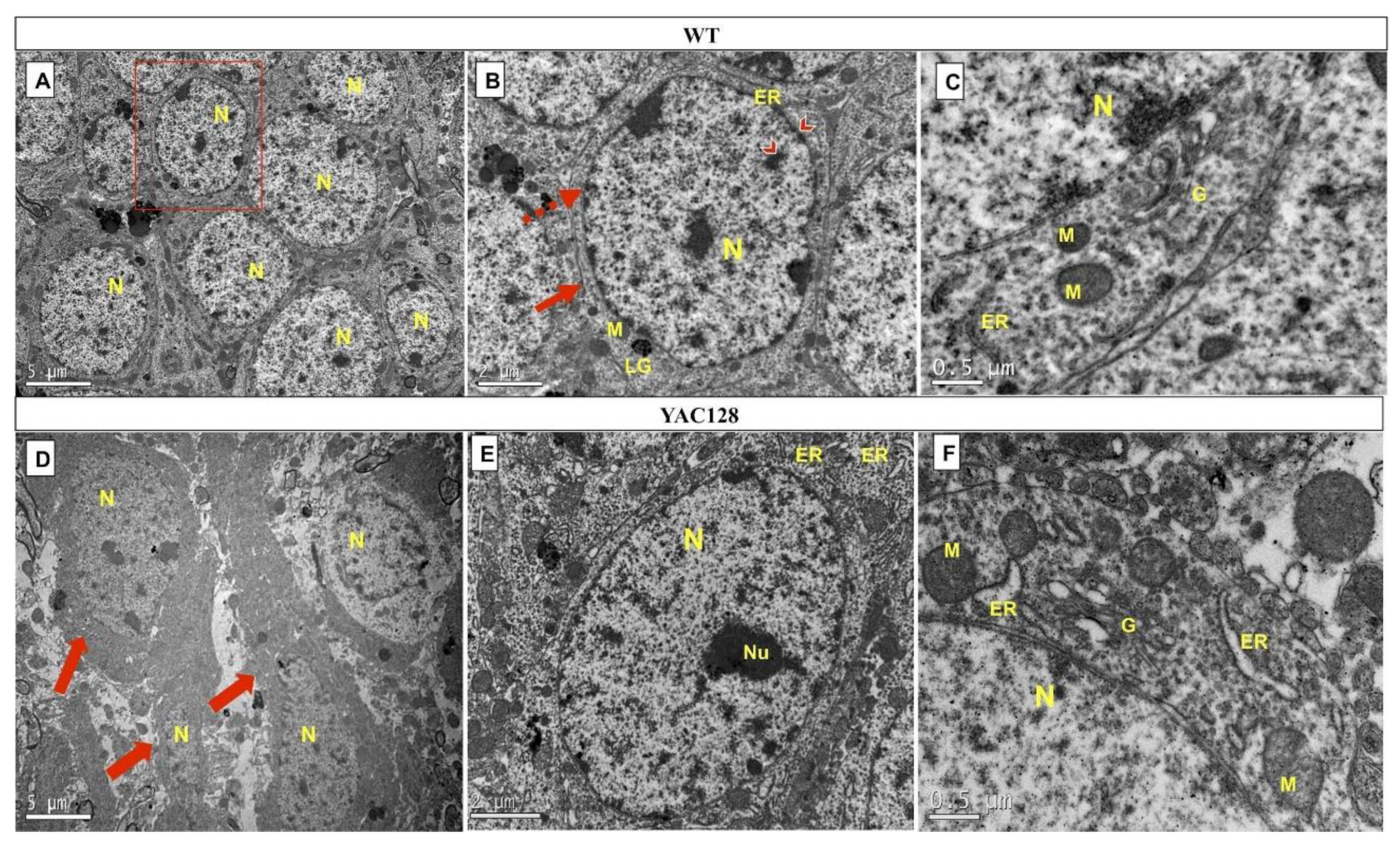
3.5. Effect of Genotype on the Ultrastructure of the Hippocampal DG in Late Symptomatic YAC128 Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Legend to the Graphical Abstract
References
- Roos, R.A.C. Huntington’s Disease: A Clinical Review. Orphanet J. Rare Dis. 2010, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.W.; Carmichael, J.; Swartz, J.; Wytternbach, A.; Rankin, J.; Rubinsztein, D.C. The Molecular Biology of Huntington’s Disease. Psychol. Med. 2001, 31, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spargo, E.; Everall, I.P.; Lantos, P.L. Neuronal Loss in the Hippocampus in Huntington’s Disease: A Comparison with HIV Infection. J. Neurol. Neurosurg. Psychiatry 1993, 56, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Vonsattel, J.P.G.; DiFiglia, M. Huntington Disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef] [Green Version]
- Rosas, H.D.; Koroshetz, W.J.; Chen, Y.I.; Skeuse, C.; Vangel, M.; Cudkowicz, M.E.; Caplan, K.; Marek, K.; Seidman, L.J.; Makris, N.; et al. Evidence for More Widespread Cerebral Pathology in Early HD: An MRI-Based Morphometric Analysis. Neurology 2003, 60, 1615–1620. [Google Scholar] [CrossRef]
- Ille, R.; Schäfer, A.; Scharmüller, W.; Enzinger, C.; Schöggl, H.; Kapfhammer, H.P.; Schienle, A. Emotion Recognition and Experience in Huntington Disease: A Voxel-Based Morphometry Study. J. Psychiatry Neurosci. 2011, 36, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Coppen, E.M.; Jacobs, M.; van den Berg-Huysmans, A.A.; van der Grond, J.; Roos, R.A.C. Grey Matter Volume Loss Is Associated with Specific Clinical Motor Signs in Huntington’s Disease. Parkinsonism Relat. Disord. 2018, 46, 56–61. [Google Scholar] [CrossRef]
- Cheng, Y.; Peng, Q.; Hou, Z.; Aggarwal, M.; Zhang, J.; Mori, S.; Ross, C.A.; Duan, W. Structural MRI Detects Progressive Regional Brain Atrophy and Neuroprotective Effects in N171-82Q Huntington’s Disease Mouse Model. Neuroimage 2011, 56, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Peng, Q.; Liu, X.; Jin, J.; Hou, Z.; Zhang, J.; Mori, S.; Ross, C.A.; Ye, K.; Duan, W. Small-Molecule TrkB Receptor Agonists Improve Motor Function and Extend Survival in a Mouse Model of Huntington’s Disease. Hum. Mol. Genet. 2013, 22, 2462. [Google Scholar] [CrossRef] [Green Version]
- Steventon, J.J.; Trueman, R.C.; Ma, D.; Yhnell, E.; Bayram-Weston, Z.; Modat, M.; Cardoso, J.; Ourselin, S.; Lythgoe, M.; Stewart, A.; et al. Longitudinal In Vivo MRI in a Huntington’s Disease Mouse Model: Global Atrophy in the Absence of White Matter Microstructural Damage. Sci. Rep. 2016, 6, 32423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattray, I.; Smith, E.; Gale, R.; Matsumoto, K.; Bates, G.P.; Modo, M. Correlations of Behavioral Deficits with Brain Pathology Assessed through Longitudinal MRI and Histopathology in the R6/2 Mouse Model of HD. PLoS ONE 2013, 8, e60012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelhano, M.; Ribeiro, M.; Petrella, L.I.; Sereno, V.; Gonc, I.; Hayden, M.R.; Rego, A.C.; Castelo-branco, M.; Castelhano, J.M.; Ribeiro, M.; et al. A Whole Brain Longitudinal Study in the YAC128 Mouse Model of Huntington’s Disease Shows Distinct Trajectories of Neurochemical, Structural Connectivity and Volumetric Changes. Hum. Mol. Genet. 2018, 27, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Iannicola, C.; Moreno, S.; Oliverio, S.; Nardacci, R.; Ciofi-Luzzatto, A.; Piacentini, M. Early Alterations in Gene Expression and Cell Morphology in a Mouse Model of Huntington’s Disease. J. Neurochem. 2000, 75, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Paldino, E.; Giampà, C.; Montagna, E.; Angeloni, C.; Fusco, F.R. Modulation of Phospho-CREB by Systemically Administered Recombinant BDNF in the Hippocampus of the R6/2 Mouse Model of Huntington’s Disease. Neurosci. J. 2019, 2019, 8363274. [Google Scholar] [CrossRef] [Green Version]
- Simpson, J.M.; Gil-Mohapel, J.; Pouladi, M.A.; Ghilan, M.; Xie, Y.; Hayden, M.R.; Christie, B.R. Altered Adult Hippocampal Neurogenesis in the YAC128 Transgenic Mouse Model of Huntington Disease. Neurobiol. Dis. 2011, 41, 249–260. [Google Scholar] [CrossRef]
- Ismailoglu, I.; Chen, Q.; Popowski, M.; Yang, L.; Gross, S.S.; Brivanlou, A.H. Huntingtin protein is essential for mitochondrial metabolism, bioenergetics and structure in murine embryonic stem cells. Dev. Biol. 2014, 391, 230–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Mao, P.; Manczak, M. Mitochondrial Structural and Functional Dynamics in Huntington’s Disease. Brain Res. Rev. 2009, 61, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Björkqvist, M.; Fex, M.; Renström, E.; Wierup, N.; Petersén, A.; Gil, J.; Bacos, K.; Popovic, N.; Li, J.Y.; Sundler, F.; et al. The R6/2 transgenic mouse model of Huntington’s disease develops diabetes due to deficient beta-cell mass and exocytosis. Hum. Mol. Genet. 2005, 14, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Mielcarek, M. Huntington’s disease is a multi-system disorder. Rare Dis. 2015, 3, e1058464. [Google Scholar] [CrossRef]
- Zielonka, D.; Piotrowska, I.; Marcinkowski, J.T.; Mielcarek, M. Skeletal muscle pathology in Huntington’s disease. Front. Physiol. 2014, 5, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomczyk, M.; Glaser, T.; Ulrich, H.; Slominska, E.M.; Smolenski, R.T. Huntingtin protein maintains balanced energetics in mouse cardiomyocytes. Nucleosides Nucleotides Nucleic Acids 2022, 41, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Dyer, R.B.; Badger, J.D.; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant Huntingtin Impairs Axonal Trafficking in Mammalian Neurons In Vivo and In Vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, V.; Giacomello, M.; Hudec, R.; Lopreiato, R.; Ermak, G.; Lim, D.; Malorni, W.; Davies, K.J.A.; Carafoli, E.; Scorrano, L. Mitochondrial Fission and Cristae Disruption Increase the Response of Cell Models of Huntington’s Disease to Apoptotic Stimuli. EMBO Mol. Med. 2010, 2, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant Huntingtin Binds the Mitochondrial Fission GTPase Dynamin-Related Protein-1 and Increases Its Enzymatic Activity. Nat. Med. 2011, 17, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant Huntingtin’s Interaction with Mitochondrial Protein Drp1 Impairs Mitochondrial Biogenesis and Causes Defective Axonal Transport and Synaptic Degeneration in Huntington’s Disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef]
- Seong, I.S.; Ivanova, E.; Lee, J.M.; Choo, Y.S.; Fossale, E.; Anderson, M.A.; Gusella, J.F.; Laramie, J.M.; Myers, R.H.; Lesort, M.; et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005, 14, 2871–2880. [Google Scholar] [CrossRef]
- Brennan, W.A.; Bird, E.D.; Aprille, J.R. Regional Mitochondrial Respiratory Activity in Huntington’s Disease Brain. J. Neurochem. 1985, 44, 1948–1950. [Google Scholar] [CrossRef]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H.V. Mitochondrial Defect in Huntington’s Disease Caudate Nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; Macgarvey, U.; Baik, M.J.; Berger, S.C.; Muquit, M.M.K.; Bird, E.D.; Beal, M.F. Oxidative Damage and Metabolic Dysfunction in Huntington’s Disease: Selective Vulnerability of the Basal Ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Guidetti, P.; Charles, V.; Chen, E.-Y.; Reddy, P.H.; Kordower, J.H.; Whetsell, W.O.; Schwarcz, R.; Tagle, D.A. Early Degenerative Changes in Transgenic Mice Expressing Mutant Huntingtin Involve Dendritic Abnormalities but No Impairment of Mitochondrial Energy Production. Exp. Neurol. 2001, 169, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of Mitochondrial Protein Import by Mutant Huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.; Pellman, J.J.; Brustovetsky, T.; Harris, R.A.; Brustovetsky, N. Oxidative Metabolism in YAC128 Mouse Model of Huntington’s Disease. Hum. Mol. Genet. 2015, 24, 4862–4878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. Mutant Huntingtin Fails to Directly Impair Brain Mitochondria. J. Neurochem. 2019, 151, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Da Fonsêca, V.S.; da Silva Colla, A.R.; de Paula Nascimento-Castro, C.; Plácido, E.; Rosa, J.M.; Farina, M.; Gil-Mohapel, J.; Rodrigues, A.L.S.; Brocardo, P.S. Brain-Derived Neurotrophic Factor Prevents Depressive-Like Behaviors in Early-Symptomatic YAC128 Huntington’s Disease Mice. Mol. Neurobiol. 2018, 55, 7201–7215. [Google Scholar] [CrossRef] [PubMed]
- Slow, E.J.; van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.Z.; et al. Selective Striatal Neuronal Loss in a YAC128 Mouse Model of Huntington Disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef]
- Pouladi, M.A.; Graham, R.K.; Karasinska, J.M.; Xie, Y.; Santos, R.D.; Petersn, Â.; Hayden, M.R.; Petersen, A.; Hayden, M.R. Prevention of Depressive Behaviour in the YAC128 Mouse Model of Huntington Disease by Mutation at Residue 586 of Huntingtin. Brain 2008, 132, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.N.; Cummins, R.A. The Open-Field Test: A Critical Review. Psychol. Bull. 1976, 83, 482–504. [Google Scholar] [CrossRef]
- Steru, L.; Chermat, R.; Thierry, B.; Simon, P. The Tail Suspension Test: A New Method for Screening Antidepressants in Mice. Psychopharmacology 1985, 85, 367–370. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Pearson, J.; Slow, E.J.; Hossain, S.M.; Leavitt, B.R.; Hayden, M.R. Cognitive Dysfunction Precedes Neuropathology and Motor Abnormalities in the YAC128 Mouse Model of Huntington’s Disease. J. Neurosci. 2005, 25, 4169–4180. [Google Scholar] [CrossRef] [Green Version]
- Taketo, M.; Schroeder, A.C.; Mobraaten, L.E.; Gunning, K.B.; Hanten, G.; Fox, R.R.; Roderick, T.H.; Stewart, C.L.; Lilly, F.; Hansen, C.T. FVB/N: An Inbred Mouse Strain Preferable for Transgenic Analyses. Proc. Natl. Acad. Sci. USA 1991, 88, 2065–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta, J.J.; Llamosas, M.M.; Cernuda-Cernuda, R.; García-Fernández, J.M. Spatio-Temporal Analysis of Light-Induced Fos Expression in the Retina of Rd Mutant Mice. Brain Res. 1999, 834, 122–127. [Google Scholar] [CrossRef]
- Burtscher, J.; Zangrandi, L.; Schwarzer, C.; Gnaiger, E. Differences in Mitochondrial Function in Homogenated Samples from Healthy and Epileptic Specific Brain Tissues Revealed by High-Resolution Respirometry. Mitochondrion 2015, 25, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Murphy, Z.; Slow, E.J.; Leavitt, B.R.; Hayden, M.R. Selective Degeneration and Nuclear Localization of Mutant Huntingtin in the YAC128 Mouse Model of Huntington Disease. Hum. Mol. Genet. 2005, 14, 3823–3835. [Google Scholar] [CrossRef] [PubMed]
- Bayram-Weston, Z.; Jones, L.; Dunnett, S.B.; Brooks, S.P. Light and Electron Microscopic Characterization of the Evolution of Cellular Pathology in YAC128 Huntington’s Disease Transgenic Mice. Brain Res. Bull. 2012, 88, 137–147. [Google Scholar] [CrossRef]
- Franklin, K.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates, Compact, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Van Raamsdonk, J.M.; Warby, S.C.; Hayden, M.R. Selective Degeneration in YAC Mouse Models of Huntington Disease. Brain Res. Bull. 2007, 72, 124–131. [Google Scholar] [CrossRef]
- Bayram-Weston, Z.; Torres, E.M.; Jones, L.; Dunnett, S.B.; Brooks, S.P. Light and Electron Microscopic Characterization of the Evolution of Cellular Pathology in the Hdh (CAG)150 Huntington’s Disease Knock-in Mouse. Brain Res. Bull. 2012, 88, 189–198. [Google Scholar] [CrossRef]
- Duff, K.; Paulsen, J.S.; Beglinger, L.J.; Langbehn, D.R.; Stout, J.C. Psychiatric Symptoms in Huntington’s Disease before Diagnosis: The Predict-HD Study. Biol. Psychiatry 2007, 62, 1341–1346. [Google Scholar] [CrossRef]
- Julien, C.L.; Thompson, J.C.; Wild, S.; Yardumian, P.; Snowden, J.S.; Turner, G.; Craufurd, D. Psychiatric Disorders in Preclinical Huntington’s Disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 939–943. [Google Scholar] [CrossRef] [Green Version]
- Renoir, T.; Pang, T.Y.C.; Mo, C.; Chan, G.; Chevarin, C.; Lanfumey, L.; Hannan, A.J. Differential Effects of Early Environmental Enrichment on Emotionality Related Behaviours in Huntington’s Disease Transgenic Mice. J. Physiol. 2013, 591, 41–55. [Google Scholar] [CrossRef]
- De Paula Nascimento-Castro, C.; Wink, A.C.; Da Fônseca, V.S.; Bianco, C.D.; Winkelmann-Duarte, E.C.; Farina, M.; Rodrigues, A.L.S.; Gil-Mohapel, J.; De Bem, A.F.; Brocardo, P.S. Antidepressant Effects of Probucol on Early-Symptomatic YAC128 Transgenic Mice for Huntington’s Disease. Neural Plast. 2018, 2018, 4056383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvoen, S.; Pla, P.; Gardier, A.M.; Saudou, F.; David, D.J. Huntington’s Disease Knock-in Male Mice Show Specific Anxiety-like Behaviour and Altered Neuronal Maturation. Neurosci. Lett. 2012, 507, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Lundh, S.H.; Nilsson, N.; Soylu, R.; Kirik, D.; Petersén, Å. Hypothalamic Expression of Mutant Huntingtin Contributes to the Development of Depressive-like Behavior in the BAC Transgenic Mouse Model of Huntington’s Disease. Hum. Mol. Genet. 2013, 22, 3485–3497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldo, B.; Cheong, R.Y.; Petersén, Å. Effects of Deletion of Mutant Huntingtin in Steroidogenic Factor 1 Neurons on the Psychiatric and Metabolic Phenotype in the BACHD Mouse Model of Huntington Disease. PLoS ONE 2014, 9, e107691. [Google Scholar] [CrossRef]
- Aharony, I.; Ehrnhoefer, D.E.; Shruster, A.; Qiu, X.; Franciosi, S.; Hayden, M.R.; Offen, D. A Huntingtin-Based Peptide Inhibitor of Caspase-6 Provides Protection from Mutant Huntingtin-Induced Motor and Behavioral Deficits. Hum. Mol. Genet. 2015, 24, 2604–2614. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Pang, T.Y.; Mo, C.; Renoir, T.; Wright, D.J.; Hannan, A.J. The Influence of the HPG Axis on Stress Response and Depressive-like Behaviour in a Transgenic Mouse Model of Huntington’s Disease. Exp. Neurol. 2015, 263, 63–71. [Google Scholar] [CrossRef]
- Wright, D.J.; Gray, L.J.; Finkelstein, D.I.; Crouch, P.J.; Pow, D.; Pang, T.Y.; Li, S.; Smith, Z.M.; Francis, P.S.; Renoir, T.; et al. N-Acetylcysteine Modulates Glutamatergic Dysfunction and Depressive Behavior in Huntington’s Disease. Hum. Mol. Genet. 2016, 25, 2923–2933. [Google Scholar] [CrossRef] [Green Version]
- Van Duijn, E.; Kingma, E.M.; Van Der Mast, R.C. Psychopathology in Verified Huntington’s Disease Gene Carriers. J. Neuropsychiatry Clin. Neurosci. 2007, 19, 441–448. [Google Scholar] [CrossRef]
- Martinez-Horta, S.; Perez-Perez, J.; van Duijn, E.; Fernandez-Bobadilla, R.; Carceller, M.; Pagonabarraga, J.; Pascual-Sedano, B.; Campolongo, A.; Ruiz-Idiago, J.; Sampedro, F.; et al. Neuropsychiatric Symptoms Are Very Common in Premanifest and Early Stage Huntington’s Disease. Parkinsonism Relat. Disord. 2016, 25, 58–64. [Google Scholar] [CrossRef]
- Galts, C.P.C.; Bettio, L.E.B.; Jewett, D.C.; Yang, C.C.; Brocardo, P.S.; Rodrigues, A.L.S.; Thacker, J.S.; Gil-Mohapel, J. Depression in Neurodegenerative Diseases: Common Mechanisms and Current Treatment Options. Neurosci. Biobehav. Rev. 2019, 102, 56–84. [Google Scholar] [CrossRef]
- Dale, M.; van Duijn, E. Anxiety in Huntington’s Disease. J. Neuropsychiatry Clin. Neurosci. 2015, 27, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Crawley, J.N. Exploratory Behavior Models of Anxiety in Mice. Neurosci. Biobehav. Rev. 1985, 9, 37–44. [Google Scholar] [CrossRef]
- Chiu, C.-T.; Liu, G.; Leeds, P.; Chuang, D.-M. Combined Treatment with the Mood Stabilizers Lithium and Valproate Produces Multiple Beneficial Effects in Transgenic Mouse Models of Huntington’s Disease. Neuropsychopharmacology 2011, 36, 2406–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menalled, L.; El-Khodor, B.F.; Patry, M.; Suárez-Fariñas, M.; Orenstein, S.J.; Zahasky, B.; Leahy, C.; Wheeler, V.; Yang, X.W.; MacDonald, M.; et al. Systematic Behavioral Evaluation of Huntington’s Disease Transgenic and Knock-in Mouse Models. Neurobiol. Dis. 2009, 35, 319–336. [Google Scholar] [CrossRef] [Green Version]
- Southwell, A.L.; Ko, J.; Patterson, P.H. Intrabody Gene Therapy Ameliorates Motor, Cognitive, and Neuropathological Symptoms in Multiple Mouse Models of Huntington’s Disease. J. Neurosci. 2009, 29, 13589–13602. [Google Scholar] [CrossRef] [Green Version]
- Hahn-Barma, V.; Deweer, B.; Durr, A.; Dode, C.; Feingold, J.; Pillon, B.; Agid, Y.; Brice, A.; Dubois, B. Are Cognitive Changes the First Symptoms of Huntington’s Disease? A Study of Gene Carriers. J. Neurol. Neurosurg. Psychiatry 1998, 64, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, A.D.; Hodges, J.R.; Rosser, A.E.; Kershaw, A.; Ffrench-Constant, C.; Rubinsztein, D.C.; Robbins, T.W.; Sahakian, B.J. Evidence for Specific Cognitive Deficits in Preclinical Huntington’s Disease. Brain 1998, 121, 1329–1341. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, J.S.; Zhao, H.; Stout, J.C.; Brinkman, R.R.; Guttman, M.; Ross, C.A.; Como, P.; Manning, C.; Hayden, M.R.; Shoulson, I. Clinical Markers of Early Disease in Persons near Onset of Huntington’s Disease. Neurology 2001, 57, 658–662. [Google Scholar] [CrossRef]
- Berrios, G.E.; Wagle, A.C.; Marková, I.S.; Wagle, S.A.; Rosser, A.; Hodges, J.R. Psychiatric Symptoms in Neurologically Asymptomatic Huntington’s Disease Gene Carriers: A Comparison with Gene Negative at Risk Subjects. Acta Psychiatr. Scand. 2002, 105, 224–230. [Google Scholar] [CrossRef]
- Brooks, S.P.; Janghra, N.; Higgs, G.V.; Bayram-Weston, Z.; Heuer, A.; Jones, L.; Dunnett, S.B. Selective Cognitive Impairment in the YAC128 Huntington’s Disease Mouse. Brain Res. Bull. 2012, 88, 121–129. [Google Scholar] [CrossRef]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine Improves Mitochondrial Function and Ameliorates Behavioral Deficits in the R6/1 Mouse Model of Huntington’s Disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oláh, J.; Klivényi, P.; Gardián, G.; Vécsei, L.; Orosz, F.; Kovacs, G.G.; Westerhoff, H.V.; Ovádi, J. Increased Glucose Metabolism and ATP Level in Brain Tissue of Huntington’s Disease Transgenic Mice. FEBS J. 2008, 275, 4740–4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milakovic, T.; Johnson, G.V.W. Mitochondrial Respiration and ATP Production Are Significantly Impaired in Striatal Cells Expressing Mutant Huntingtin. J. Biol. Chem. 2005, 280, 30773–30782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastroberardino, P.G.; Iannicola, C.; Nardacci, R.; Bernassola, F.; De Laurenzi, V.; Melino, G.; Moreno, S.; Pavone, F.; Oliverio, S.; Fesus, L.; et al. “Tissue” Transglutaminase Ablation Reduces Neuronal Death and Prolongs Survival in a Mouse Model of Huntington’s Disease. Cell Death Differ. 2002, 9, 873–880. [Google Scholar] [CrossRef] [Green Version]
- Stack, E.C.; Kubilus, J.K.; Smith, K.; Cormier, K.; Del Signore, S.J.; Guelin, E.; Ryu, H.; Hersch, S.M.; Ferrante, R.J. Chronology of Behavioral Symptoms and Neuropathological Sequela in R6/2 Huntington’s Disease Transgenic Mice. J. Comp. Neurol. 2005, 490, 354–370. [Google Scholar] [CrossRef]
- Turmaine, M.; Raza, A.; Mahal, A.; Mangiarini, L.; Bates, G.P.; Davies, S.W. Nonapoptotic Neurodegeneration in a Transgenic Mouse Model of Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 8093–8097. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.; Shirasaki, D.I.; Cepeda, C.; André, V.M.; Wilburn, B.; Lu, X.H.; Tao, J.; Yamazaki, I.; Li, S.H.; Sun, Y.E.; et al. Full-Length Human Mutant Huntingtin with a Stable Polyglutamine Repeat Can Elicit Progressive and Selective Neuropathogenesis in BACHD Mice. J. Neurosci. 2008, 28, 6182–6195. [Google Scholar] [CrossRef] [Green Version]
- Roos, R.A.C.; Bots, G.T.A.M. Nuclear Membrane Indentations in Huntington’s Chorea. J. Neurol. Sci. 1983, 61, 37–47. [Google Scholar] [CrossRef]
- Tellez-Nagel, I.; Johnson, A.B.; Terry, R.D. Studies on Brain Biopsies of Patients with Huntington’s Chorea. J. Neuropathol. Exp. Neurol. 1974, 33, 308–332. [Google Scholar] [CrossRef]
- Yamanishi, E.; Hasegawa, K.; Fujita, K.; Ichinose, S.; Yagishita, S.; Murata, M.; Tagawa, K.; Akashi, T.; Eishi, Y.; Okazawa, H. A Novel Form of Necrosis, TRIAD, Occurs in Human Huntington’s Disease. Acta Neuropathol. Commun. 2017, 5, 19. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Structures | WT | YAC128 |
|---|---|---|
| Nucleus | - | + |
| Nuclear membrane | - | + |
| Plasma membrane | - | + |
| Cytoplasmic organelles: | ||
| Mitochondria | - | + |
| Golgi apparatus | + | ++ |
| Rough endoplasmic reticulum | - | +++ |
| Lipofuscin granules | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Paula Nascimento-Castro, C.; Winkelmann-Duarte, E.C.; Mancini, G.; Welter, P.G.; Plácido, E.; Farina, M.; Gil-Mohapel, J.; Rodrigues, A.L.S.; de Bem, A.F.; Brocardo, P.S. Temporal Characterization of Behavioral and Hippocampal Dysfunction in the YAC128 Mouse Model of Huntington’s Disease. Biomedicines 2022, 10, 1433. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061433
de Paula Nascimento-Castro C, Winkelmann-Duarte EC, Mancini G, Welter PG, Plácido E, Farina M, Gil-Mohapel J, Rodrigues ALS, de Bem AF, Brocardo PS. Temporal Characterization of Behavioral and Hippocampal Dysfunction in the YAC128 Mouse Model of Huntington’s Disease. Biomedicines. 2022; 10(6):1433. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061433
Chicago/Turabian Stylede Paula Nascimento-Castro, Cristine, Elisa C. Winkelmann-Duarte, Gianni Mancini, Priscilla Gomes Welter, Evelini Plácido, Marcelo Farina, Joana Gil-Mohapel, Ana Lúcia S. Rodrigues, Andreza Fabro de Bem, and Patricia S. Brocardo. 2022. "Temporal Characterization of Behavioral and Hippocampal Dysfunction in the YAC128 Mouse Model of Huntington’s Disease" Biomedicines 10, no. 6: 1433. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061433