
Mitochondrial Phenotype as a Driver of the Racial Dichotomy in Obesity and Insulin Resistance

 , ,
, , 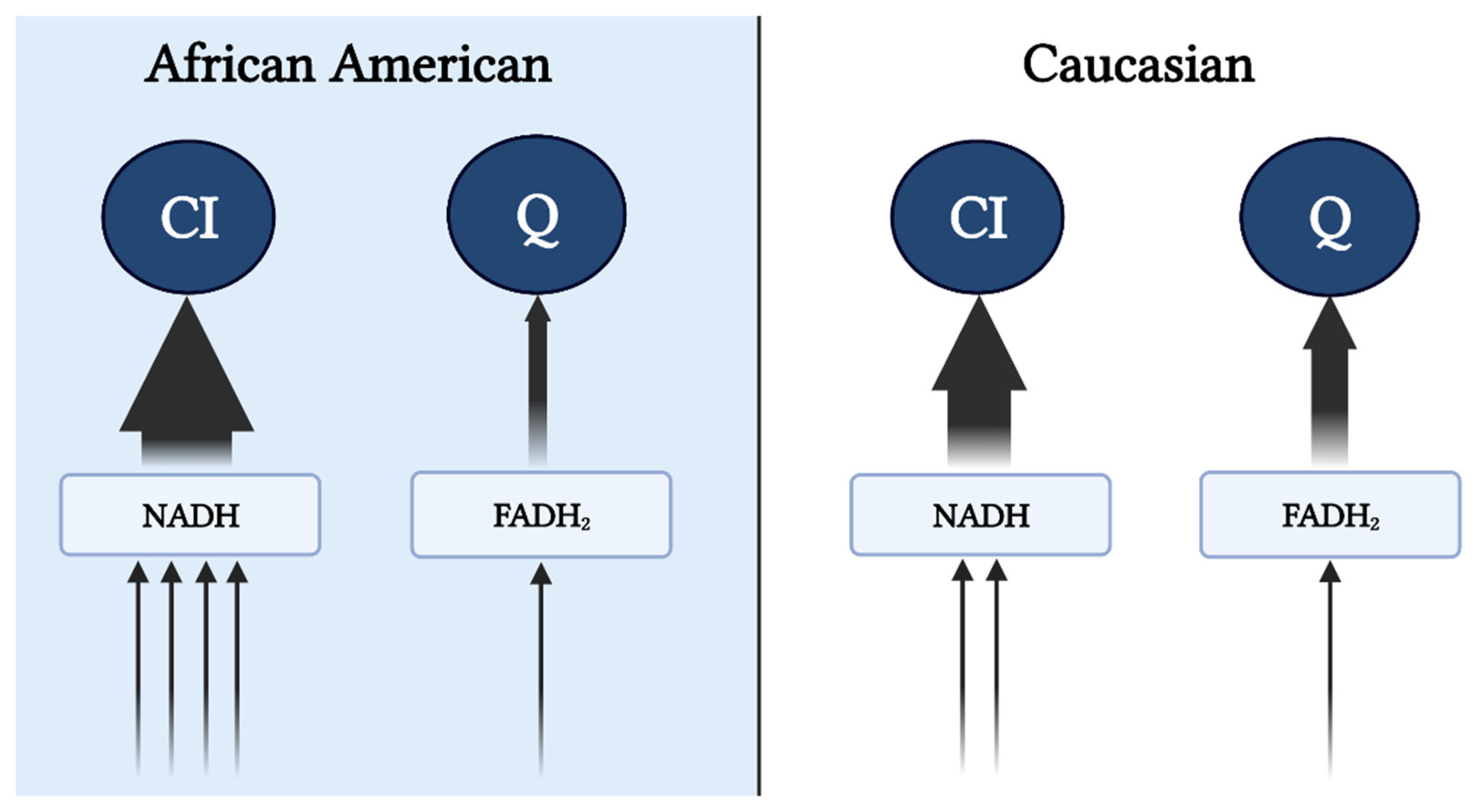{kind=link}
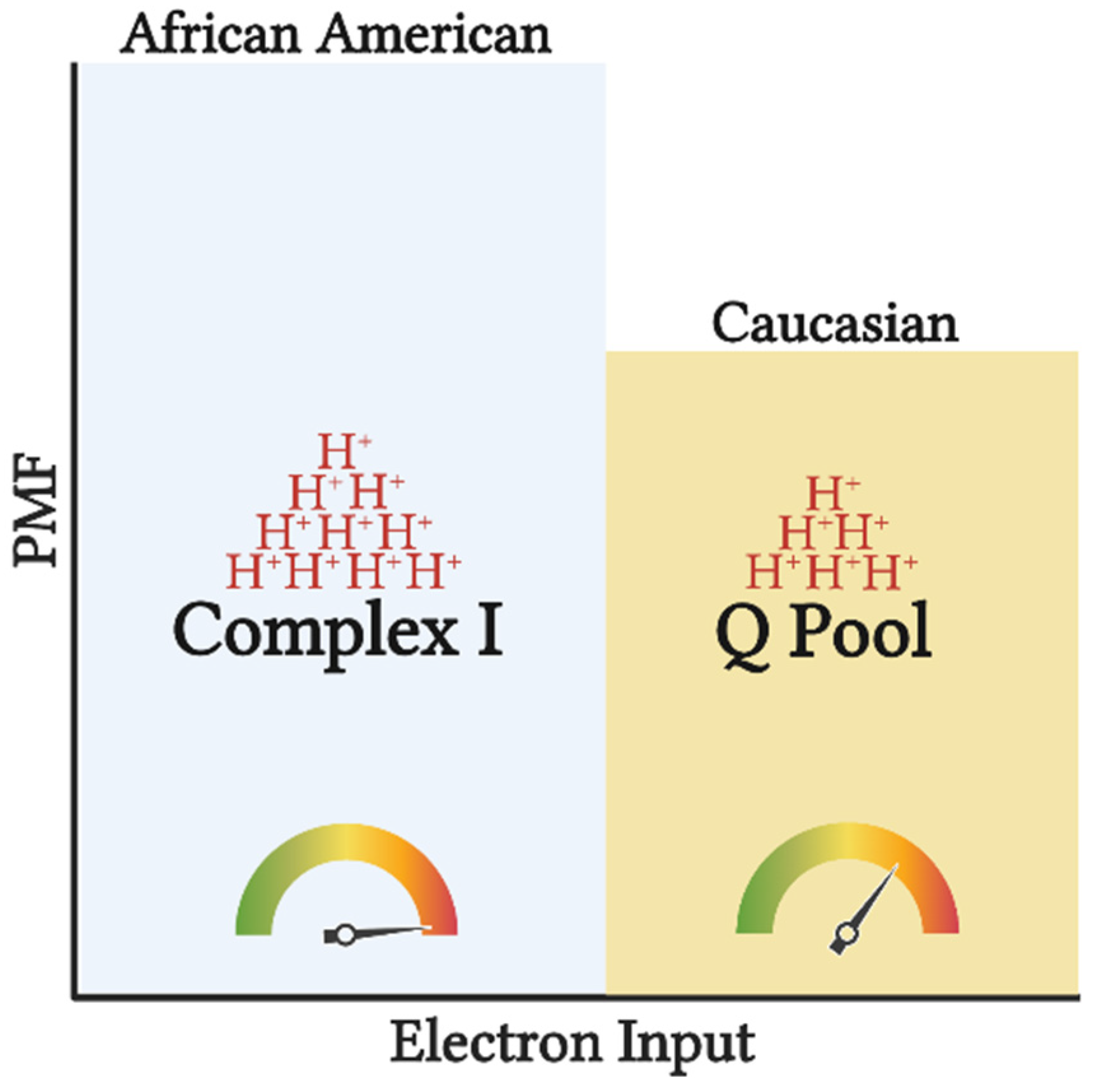{kind=link}
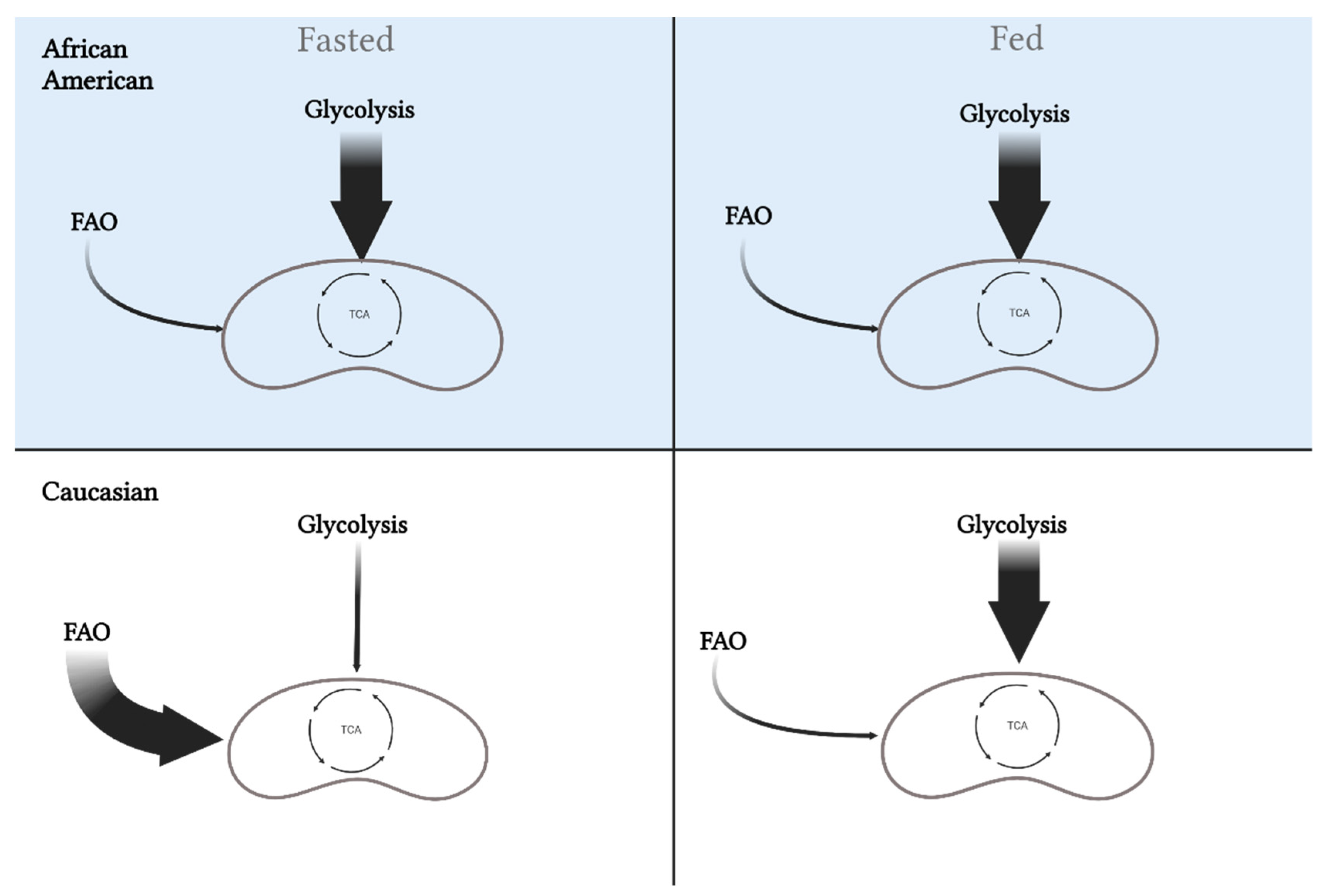{kind=link}
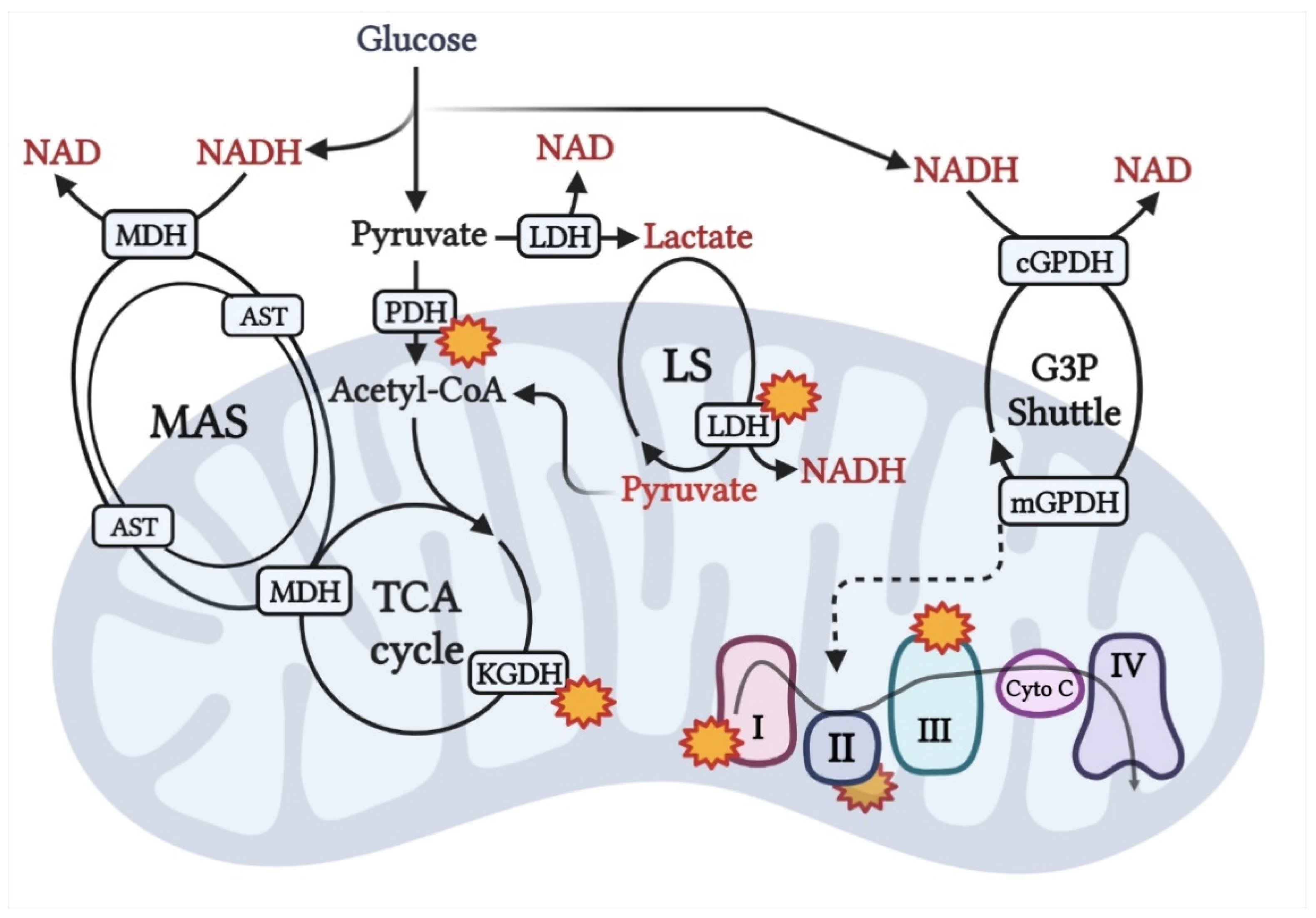{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Efficiency Increases Predisposition of African Americans to Obesity
2.1. Energy Expenditure
2.2. Mitochondrial “Substrate Preference”
3. Mitochondrial Phenotype Increases Predisposition of African Americans to Insulin Resistance
3.1. Insulin Resistance and Metabolic (In)Flexibility
3.2. Muscle Enzymatic Differences and Lower Fatty Acid Oxidation Potential
3.3. Ectopic Lipid Accumulation Is Not a Predictor of Insulin Resistance in AA
4. Oxidative Stress and Insulin Resistance
4.1. Relative Oversupply of Fatty Acids
4.2. Glycolytic Flux and ROS Production
5. Association of Mitochondrial Dynamics with Insulin Resistance in AA
6. Other Factors Influencing Obesity and Insulin Resistance in African Americans
6.1. Uncouplers
6.2. Peroxisomes
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Office of Minority Health. Available online: https://www.cdc.gov/nchs/nhis/shs/tables.htm (accessed on 27 April 2022).
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of obesity among adults and youth: United States, 2015–2016. NCHS Data Brief 2015, 288, 1–8. [Google Scholar]
- Ogden, C.; Lamb, M.; Carroll, M.; Flegal, K. Obesity and socioeconomic status in adults: United States, 2005–2008. NCHS Data Brief 2010, 50, 1–8. [Google Scholar]
- Egede, L.E.; Mueller, M.; Carrae, E.L.; Gebregziabher, M. Longitudinal differences in glycemic control by race/ethnicity among veterans with type 2 diabetes. Med. Care 2010, 48, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, A.N.; Zaslavsky, A.M.; Schneider, E.C.; Ayanian, J.Z. Relationship between quality of care and racial disparities in medicare health plans. JAMA 2006, 296, 1998–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gannon, B.; Dipietro, L.; Poehlman, E.T. Do African Americans have lower energy expenditure than Caucasians? Int. J. Obes. Relat. Metab. Disord. 2000, 24, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzmarzyk, P.T.; Most, J.; Redman, L.M.; Rood, J.; Ravussin, E. Energy expenditure and substrate oxidation in White and African American young adults without obesity. Eur. J. Clin. Nutr. 2018, 72, 920–922. [Google Scholar] [CrossRef]
- Gallagher, D.; Albu, J.; He, Q.; Heshka, S.; Boxt, L.; Krasnow, N.; Elia, M. Small organs with a high metabolic rate explain lower resting energy expenditure in African American than in white adults 2. Am. J. Clin. Nutr. 2006, 83, 1062–1067. [Google Scholar] [CrossRef] [Green Version]
- Zurlo, F.; Larson, K.; Bogardus, C.; Ravussin, E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Investig. 1990, 86, 1423–1427. [Google Scholar] [CrossRef] [Green Version]
- Browning, M.G.; Evans, R.K. The contribution of fat-free mass to resting energy expenditure: Implications for weight loss strategies in the treatment of adolescent obesity. Int. J. Adolesc. Med. Health 2015, 27, 241–246. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; Peterson, C.M.; Thomas, D.M.; Heo, M.; Schuna, J.M. Why are there race/ethnic differences in adult body mass index-adiposity relationships? A quantitative critical review. Obes. Rev. 2016, 17, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Dunham-Snary, K.J.; Ballinger, S.W. Mitochondrial genetics and obesity: Evolutionary adaptation and contemporary disease susceptibility. Free Radic. Biol. Med. 2013, 65, 1229–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, M.; Falatoonzadeh, P.; Atilano, S.; Chwa, M.; Cáceres-del-Carpio, J.; Malik, D.; Boyer, D.; Nesburn, A.; Kuppermann, B. African-origin Mitochondrial DNA Variants as a Contributing Factor to Susceptibilities for Diabetes and Age-related Diseases. Int. J. Diabetes Clin. Res. 2016, 3, 53. [Google Scholar] [CrossRef]
- Tranah, G.J.; Manini, T.M.; Lohman, K.K.; Nalls, M.A.; Kritchevsky, S.; Newman, A.B.; Harris, T.B.; Miljkovic, I.; Biffi, A.; Cummings, S.R.; et al. Mitochondrial DNA variation in human metabolic rate and energy expenditure. Mitochondrion 2011, 11, 855–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLany, J.P.; Dubé, J.J.; Standley, R.A.; Distefano, G.; Goodpaster, B.H.; Stefanovic-Racic, M.; Coen, P.M.; Toledo, F.G.S. Racial differences in peripheral insulin sensitivity and mitochondrial capacity in the absence of obesity. J. Clin. Endocrinol. Metab. 2014, 99, 4307–4314. [Google Scholar] [CrossRef]
- Toledo, F.G.S.; Dubé, J.J.; Goodpaster, B.H.; Stefanovic-Racic, M.; Coen, P.M.; DeLany, J.P. Mitochondrial respiration is associated with lower energy expenditure and lower aerobic capacity in African American women. Obesity 2018, 26, 903–909. [Google Scholar] [CrossRef] [Green Version]
- Sirikul, B.; Gower, B.A.; Hunter, G.R.; Larson-Meyer, D.E.; Newcomer, B.R.; Sirikul, B.A.; Gower, G.R.; Hunter, D.E.; Larson-Meyer, B. Relationship between insulin sensitivity and in vivo mitochondrial function in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2006, 291, 724–728. [Google Scholar] [CrossRef] [Green Version]
- Coskun, P.E.; Ruiz-Pesini, E.; Wallace, D.C. Control region mtDNA variants: Longevity, climatic adaptation, and a forensic conundrum. Proc. Natl. Acad. Sci. USA 2003, 100, 2174–2176. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Pesini, E.; Mishmar, D.; Brandon, M.; Procaccio, V.; Wallace, D.C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 2004, 303, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Gower, B.A.; Fowler, L.A. Obesity in African-Americans: The role of physiology. J. Intern. Med. 2020, 288, 295–304. [Google Scholar] [CrossRef]
- Mogensen, M.; Sahlin, K.; Fernström, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Højlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.P.; Hesselink, M.K.C.; et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berk, E.S.; Kovera, A.J.; Boozer, C.N.; Pi-Sunyer, F.X.; Albu, J.B. Metabolic inflexibility in substrate use is present in African-American but not Caucasian healthy, premenopausal, nondiabetic women. J. Clin. Endocrinol. Metab. 2006, 91, 4099–4106. [Google Scholar] [CrossRef] [PubMed]
- Hickner, R.C.; Privette, J.; McIver, K.; Barakat; Bara, H. Fatty acid oxidation in African-American and Caucasian women during physical activity. J. Appl. Physiol. 2001, 90, 2319–2324. [Google Scholar] [CrossRef]
- Cortright, R.N.; Sandhoff, K.M.; Basilio, J.L.; Berggren, J.R.; Hickner, R.C.; Hulver, M.W.; Dohm, G.L.; Houmard, J.A. Skeletal muscle fat oxidation is increased in African-American and white women after 10 days of endurance exercise training. Obesity 2006, 14, 1201–1210. [Google Scholar] [CrossRef]
- Fisher, G.; Windham, S.T.; Griffin, P.; Warren, J.L.; Gower, B.A.; Hunter, G.R. Associations of human skeletal muscle fiber type and insulin sensitivity, blood lipids, and vascular hemodynamics in a cohort of premenopausal women. Eur. J. Appl. Physiol. 2017, 117, 1413–1422. [Google Scholar] [CrossRef] [Green Version]
- Hunter, G.R.; Weinsier, R.L.; McCarthy, J.P.; Larson-Meyer, D.E.; Newcomer, B.R. Hemoglobin, muscle oxidative capacity, and VO2max in African-American and Caucasian women. Med. Sci. Sports Exerc. 2001, 33, 1739–1743. [Google Scholar] [CrossRef]
- Ama, P.F.M.; Simoneau, J.A.; Boulay, M.R.; Theriault, G.; Bouchard, C. Skeletal muscle characteristics in sedentary Black and Caucasian males. J. Appl. Physiol. 1986, 61, 1758–1761. [Google Scholar] [CrossRef]
- Swift, D.L.; Johannsen, N.M.; Lavie, C.J.; Earnest, C.P.; Johnson, W.D.; Blair, S.N.; Church, T.S.; Newton, R.L. Racial differences in the response of cardiorespiratory fitness to aerobic exercise training in Caucasian and African American postmenopausal women. J. Appl. Physiol. 2013, 114, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Tanner, C.J.; Barakat, H.A.; Dohm, G.L.; Pories, W.J.; MacDonald, K.G.; Cunningham, P.R.G.; Swanson, M.S.; Houmard, J.A. Muscle fiber type is associated with obesity and weight loss. Am. J. Physiol.-Endocrinol. Metab. 2002, 282, E1191–E1196. [Google Scholar] [CrossRef] [Green Version]
- Hasson, B.R.; Apovian, C.; Istfan, N. Racial/Ethnic Differences in Insulin Resistance and Beta Cell Function: Relationship to Racial Disparities in Type 2 Diabetes among African Americans versus Caucasians. Curr. Obes. Rep. 2015, 4, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Kodama, K.; Tojjar, D.; Yamada, S.; Toda, K.; Patel, C.J.; Butte, A.J. Ethnic differences in the relationship between insulin sensitivity and insulin response: A systematic review and meta-analysis. Diabetes Care 2013, 36, 1789–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; Yao-Borengasser, A.; Starks, T.; Tripputi, M.; Kern, P.A.; Rasouli, N. Insulin resistance in African-American and Caucasian women: Differences in lipotoxicity, adipokines, and gene expression in adipose tissue and muscle. J. Clin. Endocrinol. Metab. 2010, 95, 4441–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Privette, J.D.; Hickner, R.C.; MacDonald, K.G.; Pories, W.J.; Barakat, H.A. Fatty acid oxidation by skeletal muscle homogenates from morbidly obese black and white American women. Metab. Clin. Exp. 2003, 52, 735–738. [Google Scholar] [CrossRef]
- Kim, J.-y.; Hickner, R.C.; Cortright, R.L.; Dohm, G.L.; Houmard, J.A. Lipid oxidation is reduced in obese human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 578–591. [Google Scholar] [CrossRef] [Green Version]
- Albu, J.B.; Kovera, A.J.; Allen, L.; Wainwright, M.; Berk, E.; Raja-Khan, N.; Janumala, I.; Burkey, B.; Heshka, S.; Gallagher, D. Independent association of insulin resistance with larger amounts of intermuscular adipose tissue and a greater acute insulin response to glucose in African American than in white nondiabetic women. Am. J. Clin. Nutr. 2005, 82, 1210–1217. [Google Scholar] [CrossRef]
- Gallagher, D.; Kuznia, P.; Heshka, S.; Albu, J.; Heymsfield, S.B.; Goodpaster, B.; Visser, M.; Harris, T.B. Adipose tissue in muscle: A novel depot similar in size to visceral adipose tissue. Am. J. Clin. Nutr. 2005, 81, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, J.C.; Newcomer, B.R.; Buchthal, S.D.; Sirikul, B.; Oster, R.A.; Hunter, G.R.; Gower, B.A. Relationship of intramyocellular lipid to insulin sensitivity may differ with ethnicity in healthy girls and women. Obesity 2011, 19, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Miljkovic-Gacic, I.; Gordon, C.L.; Goodpaster, B.H.; Bunker, C.H.; Patrick, A.L.; Kuller, L.H.; Wheeler, V.W.; Evans, R.W.; Zmuda, J.M. Adipose tissue infiltration in skeletal muscle: Age patterns and association with diabetes among men of African ancestry. Am. J. Clin. Nutr. 2008, 87, 1590–1595. [Google Scholar] [CrossRef] [Green Version]
- Reed, R.M.; Nevitt, S.J.; Kemp, G.J.; Cuthbertson, D.J.; Whyte, M.B.; Goff, L.M. Ectopic fat deposition in populations of black African ancestry: A systematic review and meta-analysis. Acta Diabetol. 2021, 59, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Kitessa, S.M.; Abeywardena, M.Y. Lipid-induced insulin resistance in skeletal muscle: The chase for the culprit goes from total intramuscular fat to lipid intermediates, and finally to species of lipid intermediates. Nutrients 2016, 8, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigger, D.; Schumacher, F.; Schneider-Schaulies, S.; Kleuser, B. Sphingosine 1-phosphate metabolism and insulin signaling. Cell Signal. 2021, 82, 109959. [Google Scholar] [CrossRef] [PubMed]
- Katzmarzyk, P.T.; Bray, G.A.; Greenway, F.L.; Johnson, W.D.; Newton, R.L.; Ravussin, E.; Ryan, D.H.; Smith, S.R.; Bouchard, C. Racial differences in abdominal depot-specific adiposity in white and African American adults. Am. J. Clin. Nutr. 2010, 91, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulloch-Reid, M.K.; Hanson, R.L.; Sebring, N.G.; Reynolds, J.C.; Premkumar, A.; Genovese, D.J.; Sumner, A.E. Both subcutaneous and visceral adipose tissue correlate highly with insulin resistance in african americans. Obes. Res. 2004, 12, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Buie, J.N.J.; Hammad, S.M.; Nietert, P.J.; Magwood, G.; Adams, R.J.; Bonilha, L.; Sims-Robinson, C. Differences in plasma levels of long chain and very long chain ceramides between African Americans and whites: An observational study. PLoS ONE 2019, 14, e0216213. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.R.; Xanthakis, V.; Duncan, M.S.; Gross, S.; Friedrich, N.; Völzke, H.; Felix, S.B.; Jiang, H.; Sidhu, R.; Nauck, M.; et al. Ceramide remodeling and risk of cardiovascular events and mortality. J. Am. Heart Assoc. 2018, 7, e007931. [Google Scholar] [CrossRef]
- Hilvo, M.; Salonurmi, T.; Havulinna, A.S.; Kauhanen, D.; Pedersen, E.R.; Tell, G.S.; Meyer, K.; Teeriniemi, A.M.; Laatikainen, T.; Jousilahti, P.; et al. Ceramide stearic to palmitic acid ratio predicts incident diabetes. Diabetologia 2018, 61, 1424–1434. [Google Scholar] [CrossRef] [Green Version]
- Fisher-Wellman, K.H.; Neufer, P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 2012, 23, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.A.; Zhao, L.; Patel, R.S.; Jones, D.P.; Ahmed, Y.; Stoyanova, N.; Gibbons, G.H.; Vaccarino, V.; Din-Dzietham, R.; Quyyumi, A.A. Differences in systemic oxidative stress based on race and the metabolic syndrome: The morehouse and Emory Team up to Eliminate Health Disparities (META-Health) study. Metab. Syndr. Relat. Disord. 2012, 10, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Feairheller, D.L.; Park, J.Y.; Sturgeon, K.M.; Williamson, S.T.; Diaz, K.M.; Veerabhadrappa, P.; Brown, M.D. Racial differences in oxidative stress and inflammation: In vitro and in vivo. Clin. Transl. Sci. 2011, 4, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Watkins, S.; Kelley, D.E. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes 2001, 50, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; Hepple, R.T.; Burelle, Y. Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: Tailoring the organelle for optimal function. Am. J. Physiol. Cell Physiol. 2012, 302, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Mráček, T.; Drahota, Z.; Houštěk, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Orr, A.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Brand, M.D. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J. Biol. Chem. 2012, 287, 42921–42935. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Fisher-Wellman, K.H.; Gilliam, L.A.A.; Lin, C.T.; Cathey, B.L.; Lark, D.S.; Darrell Neufer, P. Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload. Free Radic. Biol. Med. 2013, 65, 1201–1208. [Google Scholar] [CrossRef] [Green Version]
- Brooks, G.A. Lactate as a fulcrum of metabolism. Redox Biol. 2020, 35, 101454. [Google Scholar] [CrossRef]
- Young, A.; Oldford, C.; Mailloux, R.J. Lactate dehydrogenase supports lactate oxidation in mitochondria isolated from different mouse tissues. Redox Biol. 2020, 28, 101339. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A.; Dubouchaud, H.; Brown, M.; Sicurello, J.P.; Butz, C.E. Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc. Natl. Acad. Sci. USA 1999, 96, 1129–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubé, J.J.; Collyer, M.L.; Trant, S.; Toledo, F.G.S.; Goodpaster, B.H.; Kershaw, E.E.; DeLany, J.P. Decreased mitochondrial dynamics is associated with insulin resistance, metabolic rate, and fitness in African Americans. J. Clin. Endocrinol. Metab. 2020, 105, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Bu, S.Y. Suppression of long chain acyl-CoA synthetase blocks intracellular fatty acid flux and glucose uptake in skeletal myotubes. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2020, 1865, 158678. [Google Scholar] [CrossRef]
- Trcka, F.; Durech, M.; Man, P.; Hernychova, L.; Muller, P.; Vojtesek, B. The assembly and intermolecular properties of the Hsp70-Tomm34-Hsp90 molecular chaperone complex. J. Biol. Chem. 2014, 289, 9887–9901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Gao, J.; Kosinski, P.A.; Elliman, S.J.; Hughes, T.E.; Gromada, J.; Kemp, D.M. Heat shock protein 90 (HSP90) inhibitors activate the heat shock factor 1 (HSF1) stress response pathway and improve glucose regulation in diabetic mice. Biochem. Biophys. Res. Commun. 2013, 430, 1109–1113. [Google Scholar] [CrossRef]
- Mulyani, W.R.W.; Sanjiwani, M.I.D.; Sandra; Prabawa, P.Y.; Lestari, A.A.W.; Wihandani, D.M.; Suastika, K.; Saraswati, M.R.; Bhargah, A.; Manuaba, I.B.A.P. Chaperone-based therapeutic target innovation: Heat shock protein 70 (HSP70) for type 2 diabetes mellitus. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Venojärvi, M.; Korkmaz, A.; Aunola, S.; Hällsten, K.; Virtanen, K.; Marniemi, J.; Halonen, J.P.; Hänninen, O.; Nuutila, P.; Atalay, M. Decreased thioredoxin-1 and increased HSP90 expression in skeletal muscle in subjects with type 2 diabetes or impaired glucose tolerance. BioMed Res. Int. 2014, 2014, 386351. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Nguyen, A.-K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.S.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744. [Google Scholar] [CrossRef] [Green Version]
- Hirosumi, J.; Tuncman, G.R.; Chang, L.; Rgü, C.Z.G.; Uysal, K.T.; Maeda, K.; Karin, M.; Gö; Hotamisligil, S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Vijayvargia, R.; Mann, K.; Weiss, H.R.; Pownall, H.J.; Ruan, H. JNK deficiency enhances fatty acid utilization and diverts glucose from oxidation to glycogen storage in cultured myotubes. Obesity 2010, 18, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Vernia, S.; Cavanagh-Kyros, J.; Garcia-Haro, L.; Sabio, G.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Xu, J.; Shulha, H.P.; Garber, M.; et al. The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014, 20, 512–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, S.; Than, T.A.; Fernandez-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, S.; Than, T.A.; Le, B.H.A.; García-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, S.; Than, T.A.; Kaplowitz, N. The regulation of JNK signaling pathways in cell death through the interplay with mitochondrial SAB and upstream post-translational effects. Int. J. Mol. Sci. 2018, 19, 3657. [Google Scholar] [CrossRef] [Green Version]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal kinase (JNK) in obesity and type 2 diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Argyropoulos, G.; Brown, A.M.; Willi, S.M.; Zhu, J.; He, Y.; Reitman, M.; Gevao, S.M.; Spruill, I.; Garvey, W.T. Mutation and polymorphisms in UCP3 mediate obesity and type 2 diabetes effects of mutations in the human uncoupling protein 3 gene on the respiratory quotient and fat oxidation in severe obesity and type 2 diabetes. J. Clin. Investig. 1998, 102, 1345–1351. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.K.; Luke, A.; Cooper, R.S.; Rotini, C.; Vidal-Puig, A.; Rosenbaum, M.; Chua, M.; Solanes, G.; Zheng, M.; Zhao, L.; et al. Brief Genetics Rep ort Genetic and Physiologic Analysis of the Role of Uncoupling Protein 3 in Human Energy Homeostasis. Diabetes 1999, 48, 1890–1895. [Google Scholar] [CrossRef]
- Patrick, S.; Hesselink, M. The role of uncoupling protein 3 in fatty acid metabolism: Protection against lipotoxicity? Proc. Nutr. Soc. 2004, 63, 287–292. [Google Scholar] [CrossRef]
- Astrup, A.; Toubro, S.; Dalgaard, L.T.; Urhammer, S.A.; Sùrensen, T.; Pedersen, O. Impact of the vav 55 polymorphism of the uncoupling protein 2 gene on 24-h energy expenditure and substrate oxidation. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 1030–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanovski, J.A.; Diament, A.L.; Sovik, K.N.; Nguyen, T.T.; Li, H.; Sebring, N.G.; Warden, C.H. Associations between uncoupling protein 2, body composition, and resting energy expenditure in lean and obese African American, white, and Asian children. Am. J. Clin. Nutr. 2000, 71, 1405–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.Y.; Zheng, D.; Hickner, R.C.; Brault, J.J.; Cortright, R.N. Peroxisomal gene and protein expression increase in response to a high-lipid challenge in human skeletal muscle. Metab. Clin. Exp. 2019, 98, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duve, C.D.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef]
- Lismont, C.; Nordgren, M.; Veldhoven, P.P.V.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochem. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy pathways in health and disease. J. Cell Biol. 2020, 219, e202004029. [Google Scholar] [CrossRef]
- Krako Jakovljevic, N.; Pavlovic, K.; Jotic, A.; Lalic, K.; Stoiljkovic, M.; Lukic, L.; Milicic, T.; Macesic, M.; Stanarcic Gajovic, J.; Lalic, N.M. Targeting Mitochondria in Diabetes. Int. J. Mol. Sci. 2021, 22, 6642. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jevtovic, F.; Krassovskaia, P.M.; Lopez, C.A.; Fisher-Wellman, K.H.; Cortright, R.N.; Broskey, N.T. Mitochondrial Phenotype as a Driver of the Racial Dichotomy in Obesity and Insulin Resistance. Biomedicines 2022, 10, 1456. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061456
Jevtovic F, Krassovskaia PM, Lopez CA, Fisher-Wellman KH, Cortright RN, Broskey NT. Mitochondrial Phenotype as a Driver of the Racial Dichotomy in Obesity and Insulin Resistance. Biomedicines. 2022; 10(6):1456. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061456
Chicago/Turabian StyleJevtovic, Filip, Polina M. Krassovskaia, Christian A. Lopez, Kelsey H. Fisher-Wellman, Ronald N. Cortright, and Nicholas T. Broskey. 2022. "Mitochondrial Phenotype as a Driver of the Racial Dichotomy in Obesity and Insulin Resistance" Biomedicines 10, no. 6: 1456. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10061456