Temozolomide and Other Alkylating Agents in Glioblastoma Therapy


 and
and 
Abstract
:1. Introduction
2. Alkylating Agents—Their Chemistry and Biological Uses
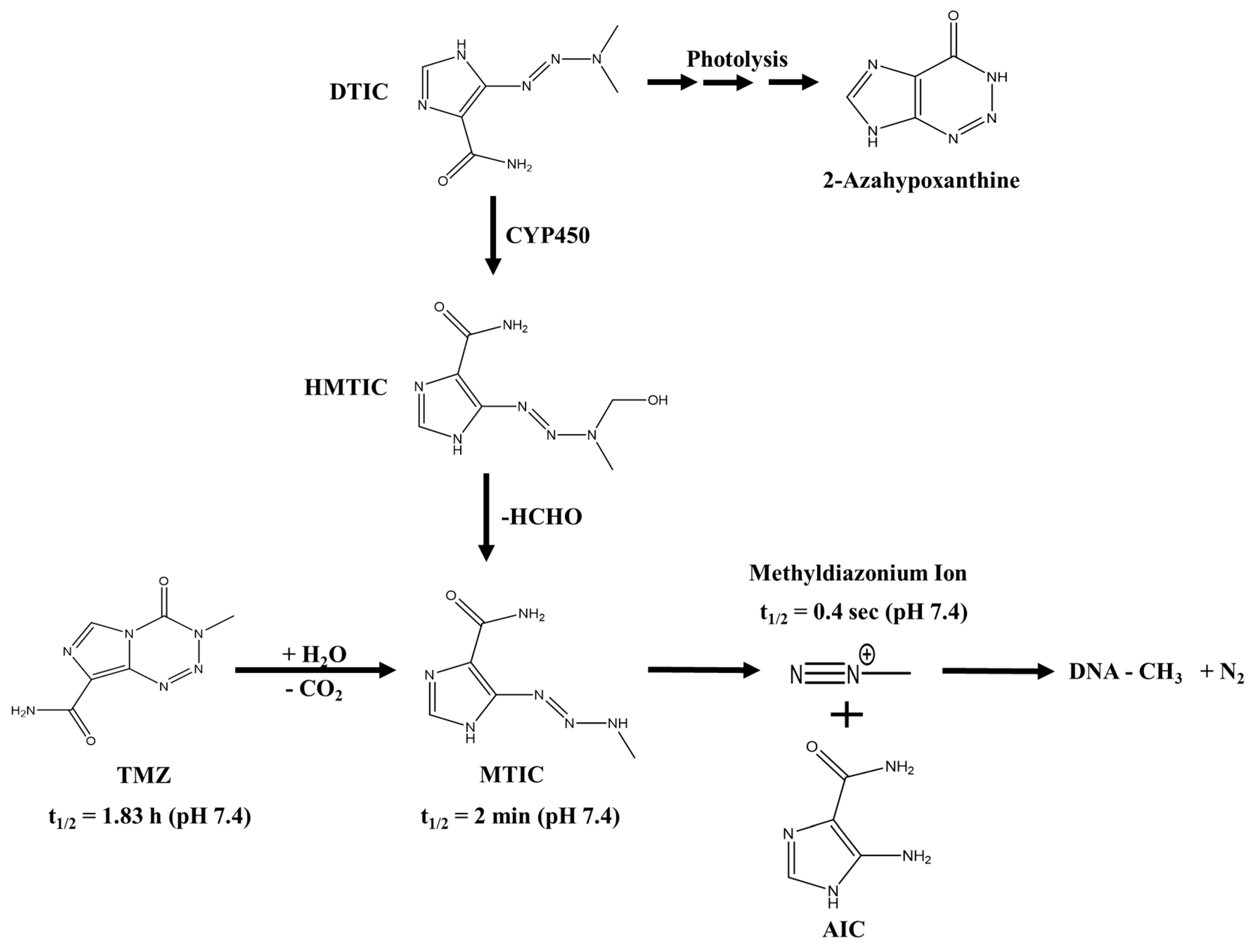
3. Triazene Compounds and Their Mode of Action
4. Additional Functions of TMZ to be Considered
5. Open Questions Regarding TMZ’s Mode of Action
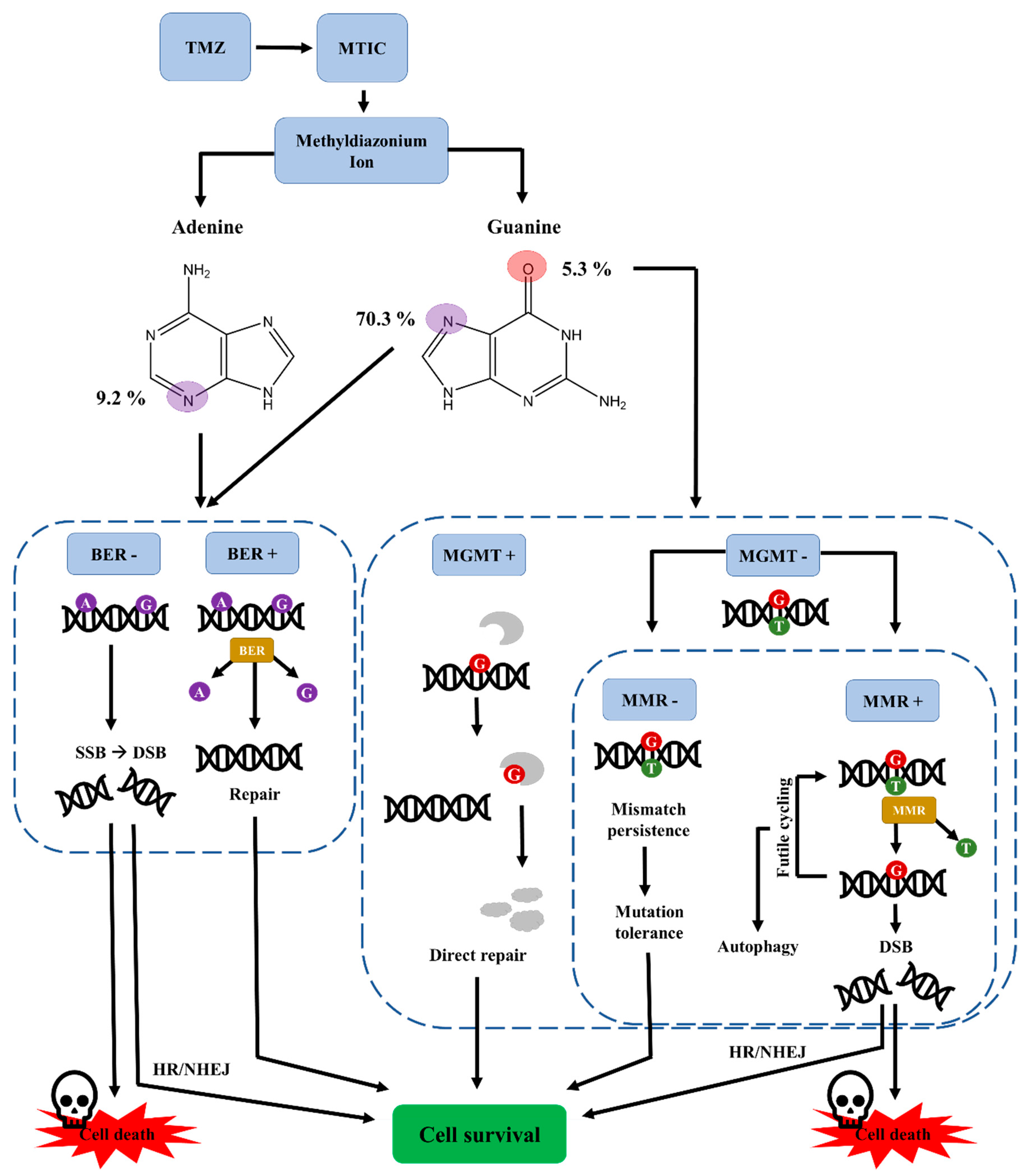
5.1. Membrane Permeability of MTIC
5.2. DNA Targets of TMZ-Mediated Methylation
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; Deimling, A.; von Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Bjerkvig, R.; Berens, M.E.; Westphal, M. Cost of migration: Invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 2003, 21, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.A.; Karajannis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Sreerama, L.; Schwab, M. (Eds.) Encyclopedia of Cancer; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1–6. [Google Scholar]
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594. [Google Scholar] [CrossRef] [PubMed]
- Drabløs, F.; Feyzi, E.; Aas, P.A.; Vaagbø, C.B.; Kavli, B.; Bratlie, M.S.; Peña-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Colvin, M.; Holland, J.F.; Frei, E.; Kufe, D.W. (Eds.) Cancer Medicine; Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Loechler, E.L. A violation of the Swain-Scott principle, and not SN1 versus SN2 reaction mechanisms, explains why carcinogenic alkylating agents can form different proportions of adducts at oxygen versus nitrogen in DNA. Chem. Res. Toxicol. 1994, 7, 277–280. [Google Scholar] [CrossRef]
- Pullman, A.; Pullman, B. Molecular electrostatic potential of the nucleic acids. Q. Rev. Biophys. 1981, 14, 289–380. [Google Scholar] [CrossRef]
- Saffhill, R.; Margison, G.; Oconnor, P. Mechanisms of carcinogenesis induced by alkylating agents. Biochim. Et Biophys. Acta (Bba) Rev. Cancer 1985, 823, 111–145. [Google Scholar] [CrossRef]
- Pearson, R.G. Acids and bases. Science (N. Y.) 1966, 151, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Hathway, D.E.; Kolar, G.F. Mechanisms of reaction between ultimate chemical carcinogens and nucleic acid. Chem. Soc. Rev. 1980, 9, 241–264. [Google Scholar] [CrossRef]
- Avendaño, C.; Menéndez, J.C. Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2015; pp. 243–271. [Google Scholar]
- Stevens, M.F.G.; Newlands, E.S. From triazines and triazenes to temozolomide. Eur. J. Cancer 1993, 29, 1045–1047. [Google Scholar] [CrossRef]
- Marchesi, F.; Turriziani, M.; Tortorelli, G.; Avvisati, G.; Torino, F.; Vecchis, L.D. Triazene compounds: Mechanism of action and related DNA repair systems. Pharmacol. Res. 2007, 56, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.A.; Barclay, R.K.; Stock, C.C.; Rondestvedt, C.S. Triazenes as inhibitors of mouse sarcoma 180. Proc. Soc. Exp. Biol. Med. 1955, 90, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Rutty, C.J.; Newell, D.R.; Vincent, R.B.; Abel, G. Abstracts for the 24th AGM of the BACR March 23rd-25th, 1983. Br. J. Cancer 1983, 48, 111–148. [Google Scholar] [Green Version]
- Stevens, M.F.; Hickman, J.A.; Langdon, S.P.; Chubb, D.; Vickers, L.; Stone, R.; Baig, G.; Goddard, C.; Gibson, N.W.; Slack, J.A.; et al. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo5,1-d-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987, 47, 5846–5852. [Google Scholar] [PubMed]
- Reid, J.M.; Kuffel, M.J.; Miller, J.K.; Rios, R.; Ames, M.M. Metabolic activation of dacarbazine by human cytochromes P450: The role of CYP1A1, CYP1A2, and CYP2E1. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 2192–2197. [Google Scholar]
- Gibson, N.W.; Hickman, J.A.; Erickson, L.C. DNA cross-linking and cytotoxicity in normal and transformed human cells treated in vitro with 8-carbamoyl-3-(2-chloroethyl)imidazo5,1-d-1,2,3,5-tetrazin-4(3H)-one. Cancer Res. 1984, 44, 1772–1775. [Google Scholar]
- Newlands, E.S.; Blackledge, G.; Slack, J.A.; Goddard, C.; Brindley, C.J.; Holden, L.; Stevens, M.F.G. Phase I clinical trial of mitozolomide. Cancer Treat. Rep. 1985, 69, 801–805. [Google Scholar] [PubMed]
- Harding, M.; Northcott, D.; Smyth, J.; Stuart, N.S.; Green, J.A.; Newlands, E. Phase II evaluation of mitozolomide in ovarian cancer. Br. J. Cancer 1988, 57, 113–114. [Google Scholar] [CrossRef] [Green Version]
- Blackledge, G.; Roberts, J.T.; Kaye, S.; Taylor, R.; Williams, J.; Stavola, B.d.; Uscinska, B. A phase II study of mitozolomide in metastatic transitional cell carcinoma of the bladder. Eur. J. Cancer Clin. Oncol. 1989, 25, 391–392. [Google Scholar] [CrossRef]
- Tatar, Z.; Thivat, E.; Planchat, E.; Gimbergues, P.; Gadea, E.; Abrial, C.; Durando, X. Temozolomide and unusual indications: Review of literature. Cancer Treat. Rev. 2013, 39, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Barciszewska, A.-M.; Gurda, D.; Głodowicz, P.; Nowak, S.; Naskręt-Barciszewska, M.Z. A New Epigenetic Mechanism of Temozolomide Action in Glioma Cells. PLoS ONE 2015, 10, e0136669. [Google Scholar] [CrossRef] [PubMed]
- Annovazzi, L.; Mellai, M.; Schiffer, D. Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers 2017, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Kirkwood, J.M. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist 2000, 5, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef] [PubMed]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Moody, C.L.; Wheelhouse, R.T. The medicinal chemistry of imidazotetrazine prodrugs. Pharmaceuticals 2014, 7, 797–838. [Google Scholar] [CrossRef] [PubMed]
- Nedelcheva, V.; Gut, I. P450 in the rat and man: Methods of investigation, substrate specificities and relevance to cancer. Xenobiotica Fate Foreign Compd. Biol. Syst. 1994, 24, 1151–1175. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, P.; Bentzen, S.M. (Eds.) Radiation Oncology Advances; Springer: New York, NY, USA; London, UK, 2008; pp. 169–185. [Google Scholar]
- Nolan, D.A.; Hong, W.K. (Eds.) Cancer Medicine; People’s Med. Publishing House: Shelton, CT, USA, 2010. [Google Scholar]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res. 2001, 61, 5843–5849. [Google Scholar] [PubMed]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar]
- Mojas, N.; Lopes, M.; Jiricny, J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007, 21, 3342–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; Tribolet, N.; de Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, X.; Zhou, B.; Zhang, L. The prognostic value of MGMT promoter methylation in Glioblastoma multiforme: A meta-analysis. Fam. Cancer 2013, 12, 449–458. [Google Scholar] [CrossRef]
- Thon, N.; Kreth, S.; Kreth, F.-W. Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. Oncotargets Ther. 2013, 6, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.N.; Almeida, K.H.; Fornsaglio, J.L.; Schamus, S.; Sobol, R.W. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005, 65, 6394–6400. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-b.; Svilar, D.; Trivedi, R.N.; Wang, X.-h.; Goellner, E.M.; Moore, B.; Hamilton, R.L.; Banze, L.A.; Brown, A.R.; Sobol, R.W. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro-Oncology 2011, 13, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Kat, A.; Thilly, W.G.; Fang, W.H.; Longley, M.J.; Li, G.M.; Modrich, P. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc. Natl. Acad. Sci. USA 1993, 90, 6424–6428. [Google Scholar] [CrossRef] [PubMed]
- Bull, V.L. Studies on the Mode of Cytotoxicity of Imidazotetrazinones. Ph.D. Thesis, Aston University, Birmingham, UK, 1988. [Google Scholar]
- Friedman, S.; Parsa, I. DNA adduct formation in rat, human and hamster pancreas treated with methylnitrosourea. Cancer Lett. 1985, 26, 269–276. [Google Scholar] [CrossRef]
- Bull, V.L.; Tisdale, M.J. Antitumor imidazotetrazines—XVI. Biochem. Pharmacol. 1987, 36, 3215–3220. [Google Scholar] [CrossRef]
- Murn, J.; Shi, Y. The winding path of protein methylation research: Milestones and new frontiers. Nat. Rev. Mol. Cell Biol. 2017, 18, 517–527. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Biggar, K.K.; Li, S.S.-C. Non-histone protein methylation as a regulator of cellular signaling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef]
- Lee, D.Y.; Teyssier, C.; Strahl, B.D.; Stallcup, M.R. Role of protein methylation in regulation of transcription. Endocr. Rev. 2005, 26, 147–170. [Google Scholar] [CrossRef]
- Ambler, R.P.; Rees, M.W. Epsilon-N-Methyl-lysine in bacterial flagellar protein. Nature 1959, 184, 56–57. [Google Scholar] [CrossRef] [PubMed]
- Paik, W.K.; Paik, D.C.; Kim, S. Historical review: The field of protein methylation. Trends Biochem. Sci. 2007, 32, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S. Protein methylation. Curr. Opin. Cell Biol. 1993, 5, 977–983. [Google Scholar] [CrossRef]
- Aletta, J.M.; Cimato, T.R.; Ettinger, M.J. Protein methylation: A signal event in post-translational modification. Trends Biochem. Sci. 1998, 23, 89–91. [Google Scholar] [CrossRef]
- Andreu-Pérez, P.; Esteve-Puig, R.; Torre-Minguela, C.; de López-Fauqued, M.; Bech-Serra, J.J.; Tenbaum, S.; García-Trevijano, E.R.; Canals, F.; Merlino, G.; Ávila, M.A.; et al. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci. Signal. 2011, 4, ra58. [Google Scholar] [CrossRef] [PubMed]
- Hein, K.; Mittler, G.; Cizelsky, W.; Kühl, M.; Ferrante, F.; Liefke, R.; Berger, I.M.; Just, S.; Sträng, J.E.; Kestler, H.A.; et al. Site-specific methylation of Notch1 controls the amplitude and duration of the Notch1 response. Sci. Signal. 2015, 8, ra30. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Moss, E.G. RNA Repair: Damage Control. Curr. Biol. 2003, 13, R482–R484. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Pickard, A.J.; Gallo, J.M. Histone Methylation by Temozolomide; A Classic DNA Methylating Anticancer Drug. Anticancer Res. 2016, 36, 3289–3299. [Google Scholar]
- Falnes, P.Ø. RNA repair—The latest addition to the toolbox for macromolecular maintenance. RNA Biol. 2005, 2, 14–16. [Google Scholar] [CrossRef]
- Andrasi, M.; Bustos, R.; Gaspar, A.; Gomez, F.A.; Klekner, A. Analysis and stability study of temozolomide using capillary electrophoresis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 1801–1808. [Google Scholar] [CrossRef]
- Newlands, E.S.; Blackledge, G.R.; Slack, J.A.; Rustin, G.J.; Smith, D.B.; Stuart, N.S.; Quarterman, C.P.; Hoffman, R.; Stevens, M.F.G.; Brampton, M.H.; et al. Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br. J. Cancer 1992, 65, 287–291. [Google Scholar] [PubMed] [Green Version]
- Newlands, E.S.; Stevens, M.F.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Baker, S.D.; Wirth, M.; Statkevich, P.; Reidenberg, P.; Alton, K.; Sartorius, S.E.; Dugan, M.; Cutler, D.; Batra, V.; Grochow, L.B.; et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 309–317. [Google Scholar]
- Patel, M.; McCully, C.; Godwin, K.; Balis, F.M. Plasma and cerebrospinal fluid pharmacokinetics of intravenous temozolomide in non-human primates. J. Neuro-Oncol. 2003, 61, 203–207. [Google Scholar] [CrossRef]
- Brock, C.S.; Matthews, J.C.; Brown, G.; Newlands, E.S. Molecular pathology of cancer. Br. J. Cancer 1997, 75, 1226–1245. [Google Scholar]
- Brown, G.D.; Luthra, S.K.; Brock, C.S.; Stevens, M.F.G.; Price, P.M.; Brady, F. Antitumor imidazotetrazines. 40. Radiosyntheses of 4-11C-carbonyl- and 3-N-11C-methyl-8-carbamoyl-3-methylimidazo-5,1-d-1,2,3,5-tetrazin-4(3H)-one (temozolomide) for positron emission tomography (PET) studies. J. Med. Chem. 2002, 45, 5448–5457. [Google Scholar] [CrossRef] [PubMed]
- Appel, E.A.; Rowland, M.J.; Loh, X.J.; Heywood, R.M.; Watts, C.; Scherman, O.A. Enhanced stability and activity of temozolomide in primary glioblastoma multiforme cells with cucurbitnuril. Chem. Commun. (Camb. Engl.) 2012, 48, 9843–9845. [Google Scholar] [CrossRef]
- Tsang, L.L.H.; Quarterman, C.P.; Gescher, A.; Slack, J.A. Comparison of the cytotoxicity in vitro of temozolomide and dacarbazine, prodrugs of 3-methyl-(triazen-1-yl)imidazole-4-carboxamide. Cancer Chemother. Pharm. 1991, 27, 342–346. [Google Scholar] [CrossRef]
- Reid, J.M.; Stevens, D.C.; Rubin, J.; Ames, M.M. Pharmacokinetics of 3-methyl-(triazen-1-yl)imidazole-4-carboximide following administration of temozolomide to patients with advanced cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 2393–2398. [Google Scholar]
- Kim, H.K.; Lin, C.C.; Parker, D.; Veals, J.; Lim, J.; Likhari, P.; Statkevich, P.; Marco, A.; Nomeir, A.A. High-performance liquid chromatographic determination and stability of 5-(3-methyltriazen-1-yl)-imidazo-4-carboximide, the biologically active product of the antitumor agent temozolomide, in human plasma. J. Chromatogr. B Biomed. Sci. Appl. 1997, 703, 225–233. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Laudicina, D.; Blumenkrantz, N.; Wirth, M.; Alton, K.B. An LC/MS/MS method for the quantitation of MTIC (5-(3-N-methyltriazen-1-yl)-imidazole-4-carboxamide), a bioconversion product of temozolomide, in rat and dog plasma. J. Pharm. Biomed. Anal. 1999, 19, 659–668. [Google Scholar] [CrossRef]
- Meer, L.; Janzer, R.C.; Kleihues, P.; Kolar, G.F. In vivo metabolism and reaction with dna of the cytostatic agent, 5-(3,3-dimethyl-1-triazeno)imidazole-4-carboxamide (DTIC). Biochem. Pharmacol. 1986, 35, 3243–3247. [Google Scholar] [CrossRef]
- Farquhar, D.; Benvenuto, J. 1-Aryl-3,3-dimethyltriazenes: Potential central nervous system active analogues of 5-(3,3-dimethyl-1-triazeno)imidazole-4-carboxamide (DTIC). J. Med. Chem. 1984, 27, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Beal, D.D.; Skibba, J.L.; Croft, W.A.; Cohen, S.M.; Bryan, G.T. Carcinogenicity of the antineoplastic agent, 5-(3,3-dimethyl-1-triazeno)-imidazole-4-carboxamide, and its metabolites in rats. J. Natl. Cancer Inst. 1975, 54, 951–957. [Google Scholar] [PubMed]
- Skibba, J.L.; Beal, D.D.; Ramirez, G.; Bryan, G.T. N-demethylation the antineoplastic agent4(5)-(3,3-dimethyl-1-triazeno)imidazole-5(4)-carboxamide by rats and man. Cancer Res. 1970, 30, 147–150. [Google Scholar] [PubMed]
- Yamagata, S.; Ohmori, S.; Suzuki, N.; Yoshino, M.; Hino, M.; Ishii, I.; Kitada, M. Metabolism of dacarbazine by rat liver microsomes contribution of CYP1A enzymes to dacarbazine N-demethylation. Drug Metab. Dispos. Biol. Fate Chem. 1998, 26, 379–382. [Google Scholar] [PubMed]
- Ferguson, C.S.; Tyndale, R.F. Cytochrome P450 enzymes in the brain: Emerging evidence of biological significance. Trends Pharmacol. Sci. 2011, 32, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.S.; Decker, R.W. Alteration of DNA by 5-(3-methyl-1- triazeno)imidazole-4-carboxamide (NSC-407347). Biochem. Pharmacol. 1976, 25, 2643–2647. [Google Scholar] [CrossRef]
- Lunn, J.M.; Harris, A.L. Cytotoxicity of 5-(3-methyl-1-triazeno)imidazole-4-carboxamide (MTIC) on Mer+, Mer+Rem- and Mer- cell lines: Differential potentiation by 3-acetamidobenzamide. Br. J. Cancer 1988, 57, 54–58. [Google Scholar] [CrossRef]
- Tsang, L.L.H.; Farmer, P.B.; Gescher, A.; Slack, J.A. Characterisation of urinary metabolites of temozolomide in humans and mice and evaluation of their cytotoxicity. Cancer Chemother. Pharm. 1990, 26, 429–436. [Google Scholar] [CrossRef]
- Pope, D.C.; Oliver, W.T. Dimethyl sulfoxide (DMSO). Can. J. Comp. Med. Vet. Sci. 1966, 30, 3–8. [Google Scholar] [PubMed]
- Santos, N.C.; Figueira-Coelho, J.; Martins-Silva, J.; Saldanha, C. Multidisciplinary utilization of dimethyl sulfoxide: Pharmacological, cellular, and molecular aspects. Biochem. Pharmacol. 2003, 65, 1035–1041. [Google Scholar] [CrossRef]
- Beal, D.D.; Skibba, J.L.; Whitnable, K.K.; Bryan, G.T. Effects of 5-(3,3-dimethyl-1-triazeno)imidazole-4-carboxamide and its metabolites on Novikoff hepatoma cells. Cancer Res. 1976, 36, 2827–2831. [Google Scholar] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1990, 231, 11–30. [Google Scholar] [CrossRef]
- Broegger, A.; LaChapelle, A.D. (Eds.) Chromosomes Today. In Proceedings of the International Chromosome Conference, Helsinki, Finland, 29–30 August 1977; Elsevier: Amsterdam, The Netherland, 1977; pp. 297–306. [Google Scholar]
- Durante, M.; Geri, C.; Bonatti, S.; Parenti, R. Non-random alkylation of DNA sequences induced in vivo by chemical mutagens. Carcinogenesis 1989, 10, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncology 2017, 19, v1–v88. [Google Scholar] [CrossRef]
- Liu, M.; Thakkar, J.P.; Garcia, C.R.; Dolecek, T.A.; Wagner, L.M.; van Dressler, E.M.; Villano, J.L. National cancer database analysis of outcomes in pediatric glioblastoma. Cancer Med. 2018, 7, 1151–1159. [Google Scholar] [CrossRef]
- Huse, J.T.; Holland, E.C. Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 2010, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, W.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network TCGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-W.; Weiss, W.A. Targeting the RTK-PI3K-mTOR axis in malignant glioma: Overcoming resistance. Curr. Top. Microbiol. Immunol. 2010, 347, 279–296. [Google Scholar] [PubMed]
- Langhans, J.; Schneele, L.; Trenkler, N.; von Bandemer, H.; Nonnenmacher, L.; Karpel-Massler, G.; Siegelin, M.D.; Zhou, S.; Halatsch, M.E.; Debatin, K.M.; et al. The effects of PI3K-mediated signaling on glioblastoma cell behaviour. Oncogenesis 2017, 6, 398. [Google Scholar] [CrossRef] [PubMed]
- Hasslacher, S.; Schneele, L.; Stroh, S.; Langhans, J.; Zeiler, K.; Kattner, P.; Karpel-Massler, G.; Siegelin, M.D.; Schneider, M.; Zhou, S.; et al. Inhibition of PI3K signaling increases the efficiency of radiotherapy in glioblastoma cells. Int. J. Oncol. 2018, 53, 1881–1896. [Google Scholar] [PubMed]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.-F.; Wang, J.; Shao, W.; Wu, C.-P.; Chen, Z.-P.; To, S.-S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef]
- Nonnenmacher, L.; Westhoff, M.-A.; Fulda, S.; Karpel-Massler, G.; Halatsch, M.-E.; Engelke, J.; Simmet, T.; Corbacioglu, S.; Debatin, K.M. RIST: A potent new combination therapy for glioblastoma. Int. J. Cancer 2015, 136, E173–E187. [Google Scholar] [CrossRef]
- Halatsch, M.E.; Kast, R.E.; Dwucet, A.; Hlavac, M.; Heiland, T.; Westhoff, M.A.; Debatin, K.M.; Wirtz, C.R.; Siegelin, M.D.; Karpel-Massler, G. Bcl-2/Bcl-xL inhibition predominantly synergistically enhances the anti-neoplastic activity of a low-dose CUSP9 repurposed drug regime against glioblastoma. Br. J. Pharm. 2019, 176, 3681–3694. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Bingham, C.A. The cell cycle and cancer chemotherapy. Am. J. Nurs. 1978, 78, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-S.; Koh, C.-G.; Li, H.-Y. Mitosis-targeted anti-cancer therapies: Where they stand. Cell Death Dis. 2012, 3, e411. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.A.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-beta-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017, 21, 650–664 e8. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.; Lyle, S. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J. Oncol. 2011, 2011, 396076. [Google Scholar] [CrossRef]
- Spies, J.; Lukas, C.; Somyajit, K.; Rask, M.-B.; Lukas, J.; Neelsen, K.J. 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 2019, 21, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, T.; Galanti, G.; Lavie, G.; Jacob-Hirsch, J.; Kventsel, I.; Zeligson, S.; Winkler, R.; Simon, A.J.; Amariglio, N.; Rechavi, G.; et al. Mechanisms operative in the antitumor activity of temozolomide in glioblastoma multiforme. Cancer J. (Sudbury Mass.) 2007, 13, 335–344. [Google Scholar] [CrossRef]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef]
- Zhang, W.-B.; Wang, Z.; Shu, F.; Jin, Y.-H.; Liu, H.-Y.; Wang, Q.-J.; Yang, Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J. Biol. Chem. 2010, 285, 40461–40471. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.-C.A.; Prestegarden, L.; Grudic, A.; Hegi, M.E.; Tysnes, B.B.; Bjerkvig, R. The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro-Oncology 2013, 15, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Hosoda, R.; Akiyama, Y.; Sebori, R.; Wanibuchi, M.; Mikami, T.; Sugino, T.; Suzuki, K.; Maruyama, M.; Tsukamoto, M.; et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J. Neuro-Oncol. 2015, 122, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xu, X.-K.; Li, J.-L.; Kong, K.-K.; Li, H.; Chen, C.; He, J.; Wang, F.; Li, P.; Ge, X.S.; et al. MALAT1 is a prognostic factor in glioblastoma multiforme and induces chemoresistance to temozolomide through suppressing miR-203 and promoting thymidylate synthase expression. Oncotarget 2017, 8, 22783–22799. [Google Scholar] [CrossRef] [PubMed]
- Rosso, L.; Brock, C.S.; Gallo, J.M.; Saleem, A.; Price, P.M.; Turkheimer, F.E.; Aboagye, E.O. A new model for prediction of drug distribution in tumor and normal tissues: Pharmacokinetics of temozolomide in glioma patients. Cancer Res. 2009, 69, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Günther, W.; Pawlak, E.; Damasceno, R.; Arnold, H.; Terzis, A.J. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br. J. Cancer 2003, 88, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| In Vitro Methylation Pattern (Cell-Free System) | Adduct Profile of Cultured Cells | Adduct Profile of Bacteria | Adduct Profile of Isolated Rat-Liver DNA | ||||
|---|---|---|---|---|---|---|---|
| Percentage of total alkylation/ binding to DNA [%] | |||||||
| Site of Alkylation | TMZ a | MNU b | MNNG b | MNU b | MNNG b | MNU b | MNNG b |
| Adenine | |||||||
| N1 | - | 0.7–1.3 | 1.0 | nd | - | 0.3 | - |
| N3 | - | 8.0–9.0 | 12.0 | 3.8–4.2 | 2.0 | 1.1–3.6 | 8.6 |
| N6 | - | nd | - | - | - | - | - |
| N7 | 9.2 | 0.8–2.0 | - | 1.5–3.1 | - | 0.8 | - |
| Cytosine | |||||||
| O2 | - | 0.1 | - | nd | - | nd | - |
| N3 | - | 0.06–0.6 | 2.0 | 0.3–0.4 | - | nd | - |
| Guanine | |||||||
| N1 | - | nd | - | - | - | - | - |
| N3 | - | 0.6–1.9 | - | 0.5–0.7 | - | 0.6–1.6 | - |
| O6 | 5.3 | 5.9–8.2 | 7.0 | 5.1–11.6 | 11.0 | 3.6–10.0 | 9.2 |
| N7 | 70.3 | 65.0–70.0 | 67.0 | 69.0–72.9 | 78.0 | 70.0–86.6 | 82.2 |
| Thymidine | |||||||
| O2 | - | 0.1–0.3 | - | nd | - | nd | - |
| N3 | - | 0.1–0.3 | - | nd | - | nd | - |
| O4 | - | 0.1–0.3 | - | 0.5 | - | 1.8 | - |
| Phospho-triesters | - | 12.0–17.0 | - | 9.0–11.7 | - | 13.4 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.-M.; Westhoff, M.-A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines7030069
Strobel H, Baisch T, Fitzel R, Schilberg K, Siegelin MD, Karpel-Massler G, Debatin K-M, Westhoff M-A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines. 2019; 7(3):69. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines7030069
Chicago/Turabian StyleStrobel, Hannah, Tim Baisch, Rahel Fitzel, Katharina Schilberg, Markus D. Siegelin, Georg Karpel-Massler, Klaus-Michael Debatin, and Mike-Andrew Westhoff. 2019. "Temozolomide and Other Alkylating Agents in Glioblastoma Therapy" Biomedicines 7, no. 3: 69. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines7030069