Targeting Alternative Splicing as a Potential Therapy for Episodic Ataxia Type 2

 ,
,
Abstract
:
1. Introduction
2. Alternative Splicing
3. Splicing Modulating Therapies
3.1. Antisense Oligonucleotides (AONs)
3.1.1. AON-Mediated Exon Inclusion
3.1.2. AON-Mediated Exon Skipping
3.1.3. AON-Mediated Masking of Cryptic Splice Sites
3.2. Spliceosome-Mediated RNA Trans-Splicing (SMaRT)
3.3. CRISPR-Based Approaches
4. Conclusions: Potential Advantages of Splicing Therapies for EA2
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2′MOE | 2′-O-methoxyethyl |
| 2′OMePS | 2’-O-methyl-phosphorothioate RNA |
| AMO | Antisense morpholino oligonucleotide |
| AON | Antisense oligonucleotide |
| AS | Alternative splicing |
| BMD | Becker muscular dystrophy |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DMD | Duchenne muscular dystrophy |
| DSB | Double strand break |
| EA2 | Episodic ataxia type 2 |
| ESE/S | Exonic splicing enhancer/silencer |
| FDA | Food and Drug Administration |
| FTDP-17 | Frontotemporal dementia with parkinsonism linked to chromosome 17 |
| gRNA | Guide RNA |
| HDR | Homology-directed repair |
| hnRNP | Heterogeneous nuclear ribonucleoprotein particle |
| ISE/S | Intronic splicing enhancer/silencer |
| NHEJ | Non-homologous end joining |
| PMO | Phosphorodiamidate antisense morpholino oligonucleotide |
| PS | Phosphorothioate |
| PTM | Pre-trans-splicing RNA molecule |
| SMA | Spinal muscular atrophy |
| SMaRT | Spliceosome-Mediated RNA Trans-Splicing |
| SMN | Survival of motor neuron |
| tcDNA | tricyclo-DNA |
| VGCC | Voltage-gated calcium channel |
References
- Jen, J.; Kim, G.W.; Baloh, R.W. Clinical spectrum of episodic ataxia type 2. Neurology 2004, 62, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Strupp, M.; Zwergal, A.; Brandt, T. Episodic ataxia type 2. Neurother. J. Am. Soc. Exp. Neurother. 2007, 4, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbrici, P.; Eunson, L.H.; Graves, T.D.; Bhatia, K.P.; Wadia, N.H.; Kullmann, D.M.; Hanna, M.G. Late-onset episodic ataxia type 2 due to an in-frame insertion in CACNA1A. Neurology 2005, 65, 944–946. [Google Scholar] [CrossRef] [PubMed]
- Mantuano, E.; Romano, S.; Veneziano, L.; Gellera, C.; Castellotti, B.; Caimi, S.; Testa, D.; Estienne, M.; Zorzi, G.; Bugiani, M.; et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J. Neurol. Sci. 2010, 291, 30–36. [Google Scholar] [CrossRef]
- Jen, J.C.; Graves, T.D.; Hess, E.J.; Hanna, M.G.; Griggs, R.C.; Baloh, R.W.; The CINCH Investigators. Primary episodic ataxias: Diagnosis, pathogenesis and treatment. Brain J. Neurol. 2007, 130, 2484–2493. [Google Scholar] [CrossRef] [Green Version]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Thalhammer, A.; Jaudon, F.; Cingolani, L.A. Emerging Roles of Activity-Dependent Alternative Splicing in Homeostatic Plasticity. Front. Cell Neurosci. 2020, 14, 104. [Google Scholar] [CrossRef]
- Cao, Y.Q.; Piedras-Renteria, E.S.; Smith, G.B.; Chen, G.; Harata, N.C.; Tsien, R.W. Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron 2004, 43, 387–400. [Google Scholar] [CrossRef] [Green Version]
- Mintz, I.M.; Sabatini, B.L.; Regehr, W.G. Calcium control of transmitter release at a cerebellar synapse. Neuron 1995, 15, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.G.; Westenbroek, R.E.; Borst, J.G.; Catterall, W.A.; Sakmann, B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J. Neurosci. 1999, 19, 726–736. [Google Scholar] [CrossRef]
- Lipscombe, D.; Allen, S.E.; Toro, C.P. Control of neuronal voltage-gated calcium ion channels from RNA to protein. Trends Neurosci. 2013, 36, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Sintas, C.; Carreno, O.; Fernandez-Castillo, N.; Corominas, R.; Vila-Pueyo, M.; Toma, C.; Cuenca-Leon, E.; Barroeta, I.; Roig, C.; Volpini, V.; et al. Mutation Spectrum in the CACNA1A Gene in 49 Patients with Episodic Ataxia. Sci. Rep. 2017, 7, 2514. [Google Scholar] [CrossRef] [PubMed]
- Denier, C.; Ducros, A.; Vahedi, K.; Joutel, A.; Thierry, P.; Ritz, A.; Castelnovo, G.; Deonna, T.; Gerard, P.; Devoize, J.L.; et al. High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology 1999, 52, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Eunson, L.H.; Graves, T.D.; Hanna, M.G. New calcium channel mutations predict aberrant RNA splicing in episodic ataxia. Neurology 2005, 65, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Subramony, S.H.; Schott, K.; Raike, R.S.; Callahan, J.; Langford, L.R.; Christova, P.S.; Anderson, J.H.; Gomez, C.M. Novel CACNA1A mutation causes febrile episodic ataxia with interictal cerebellar deficits. Ann. Neurol. 2003, 54, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Damaj, L.; Lupien-Meilleur, A.; Lortie, A.; Riou, E.; Ospina, L.H.; Gagnon, L.; Vanasse, C.; Rossignol, E. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet. 2015, 23, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Nachbauer, W.; Nocker, M.; Karner, E.; Stankovic, I.; Unterberger, I.; Eigentler, A.; Schneider, R.; Poewe, W.; Delazer, M.; Boesch, S. Episodic ataxia type 2: Phenotype characteristics of a novel CACNA1A mutation and review of the literature. J. Neurol. 2014, 261, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Carr, J.R.; Baloh, R.W.; Jen, J.C. Nonconsensus intronic mutations cause episodic ataxia. Ann. Neurol. 2005, 57, 131–135. [Google Scholar] [CrossRef]
- Kaunisto, M.A.; Harno, H.; Kallela, M.; Somer, H.; Sallinen, R.; Hamalainen, E.; Miettinen, P.J.; Vesa, J.; Orpana, A.; Palotie, A.; et al. Novel splice site CACNA1A mutation causing episodic ataxia type 2. Neurogenetics 2004, 5, 69–73. [Google Scholar] [CrossRef]
- Bourinet, E.; Soong, T.W.; Sutton, K.; Slaymaker, S.; Mathews, E.; Monteil, A.; Zamponi, G.W.; Nargeot, J.; Snutch, T.P. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat. Neurosci. 1999, 2, 407–415. [Google Scholar] [CrossRef]
- Chaudhuri, D.; Chang, S.Y.; DeMaria, C.D.; Alvania, R.S.; Soong, T.W.; Yue, D.T. Alternative splicing as a molecular switch for Ca2+/calmodulin-dependent facilitation of P/Q-type Ca2+ channels. J. Neurosci. 2004, 24, 6334–6342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soong, T.W.; DeMaria, C.D.; Alvania, R.S.; Zweifel, L.S.; Liang, M.C.; Mittman, S.; Agnew, W.S.; Yue, D.T. Systematic identification of splice variants in human P/Q-type channel alpha1(2.1) subunits: Implications for current density and Ca2+-dependent inactivation. J. Neurosci. 2002, 22, 10142–10152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalhammer, A.; Contestabile, A.; Ermolyuk, Y.S.; Ng, T.; Volynski, K.E.; Soong, T.W.; Goda, Y.; Cingolani, L.A. Alternative Splicing of P/Q-Type Ca2+ Channels Shapes Presynaptic Plasticity. Cell Rep. 2017, 20, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Thalhammer, A.; Jaudon, F.; Cingolani, L.A. Combining Optogenetics with Artificial microRNAs to Characterize the Effects of Gene Knockdown on Presynaptic Function within Intact Neuronal Circuits. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vigues, S.; Gastaldi, M.; Massacrier, A.; Cau, P.; Valmier, J. The alpha(1A) subunits of rat brain calcium channels are developmentally regulated by alternative RNA splicing. Neuroscience 2002, 113, 509–517. [Google Scholar] [CrossRef]
- Graves, T.D.; Imbrici, P.; Kors, E.E.; Terwindt, G.M.; Eunson, L.H.; Frants, R.R.; Haan, J.; Ferrari, M.D.; Goadsby, P.J.; Hanna, M.G.; et al. Premature stop codons in a facilitating EF-hand splice variant of CaV2.1 cause episodic ataxia type 2. Neurobiol. Dis. 2008, 32, 10–15. [Google Scholar] [CrossRef]
- Imbrici, P.; Jaffe, S.L.; Eunson, L.H.; Davies, N.P.; Herd, C.; Robertson, R.; Kullmann, D.M.; Hanna, M.G. Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain J. Neurol. 2004, 127, 2682–2692. [Google Scholar] [CrossRef] [Green Version]
- Maksemous, N.; Roy, B.; Smith, R.A.; Griffiths, L.R. Next-generation sequencing identifies novel CACNA1A gene mutations in episodic ataxia type 2. Mol. Genet. Genom. Med. 2016, 4, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Soden, S.E.; Saunders, C.J.; Willig, L.K.; Farrow, E.G.; Smith, L.D.; Petrikin, J.E.; LePichon, J.B.; Miller, N.A.; Thiffault, I.; Dinwiddie, D.L.; et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 2014, 6, 265ra168. [Google Scholar] [CrossRef] [Green Version]
- Indelicato, E.; Nachbauer, W.; Karner, E.; Eigentler, A.; Wagner, M.; Unterberger, I.; Poewe, W.; Delazer, M.; Boesch, S. The neuropsychiatric phenotype in CACNA1A mutations: A retrospective single center study and review of the literature. Eur. J. Neurol. 2019, 26, 66–67. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.D.; Kim, J.S.; Kim, H.J.; Jung, I.; Jeong, S.H.; Lee, S.H.; Kim, D.U.; Kim, S.H.; Choi, S.Y.; Shin, J.H.; et al. Genetic Variants Associated with Episodic Ataxia in Korea. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zafeiriou, D.I.; Lehmann-Horn, F.; Vargiami, E.; Teflioudi, E.; Ververi, A.; Jurkat-Rott, K. Episodic ataxia type 2 showing ictal hyperhidrosis with hypothermia and interictal chronic diarrhea due to a novel CACNA1A mutation. Eur. J. Paediatr. Neurol. 2009, 13, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Van den Maagdenberg, A.M.; Kors, E.E.; Brunt, E.R.; van Paesschen, W.; Pascual, J.; Ravine, D.; Keeling, S.; Vanmolkot, K.R.; Vermeulen, F.L.; Terwindt, G.M.; et al. Episodic ataxia type 2. Three novel truncating mutations and one novel missense mutation in the CACNA1A gene. J. Neurol. 2002, 249, 1515–1519. [Google Scholar] [CrossRef]
- Mantuano, E.; Veneziano, L.; Spadaro, M.; Giunti, P.; Guida, S.; Leggio, M.G.; Verriello, L.; Wood, N.; Jodice, C.; Frontali, M. Clusters of non-truncating mutations of P/Q type Ca2+ channel subunit Ca(v)2.1 causing episodic ataxia 2. J. Med. Genet. 2004, 41, e82. [Google Scholar] [CrossRef] [Green Version]
- Yue, Q.; Jen, J.C.; Nelson, S.F.; Baloh, R.W. Progressive ataxia due to a missense mutation in a calcium-channel gene. Am. J. Hum. Genet. 1997, 61, 1078–1087. [Google Scholar] [CrossRef] [Green Version]
- Tantsis, E.M.; Gill, D.; Griffiths, L.; Gupta, S.; Lawson, J.; Maksemous, N.; Ouvrier, R.; Riant, F.; Smith, R.; Troedson, C.; et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev. Med. Child Neurol. 2016, 58, 639–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikaido, K.; Tachi, N.; Ohya, K.; Wada, T.; Tsutsumi, H. New mutation of CACNA1A gene in episodic ataxia type 2. Pediatr. Int. 2011, 53, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Cricchi, F.; Di Lorenzo, C.; Grieco, G.S.; Rengo, C.; Cardinale, A.; Racaniello, M.; Santorelli, F.M.; Nappi, G.; Pierelli, F.; Casali, C. Early-onset progressive ataxia associated with the first CACNA1A mutation identified within the I-II loop. J. Neurol. Sci. 2007, 254, 69–71. [Google Scholar] [CrossRef]
- Isaacs, D.A.; Bradshaw, M.J.; Brown, K.; Hedera, P. Case report of novel CACNA1A gene mutation causing episodic ataxia type 2. SAGE Open Med. Case Rep. 2017, 5, 2050313X17706044. [Google Scholar] [CrossRef]
- Scoggan, K.A.; Friedman, J.H.; Bulman, D.E. CACNA1A mutation in a EA-2 patient responsive to acetazolamide and valproic acid. Can. J. Neurol. Sci. 2006, 33, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Rajakulendran, S.; Graves, T.D.; Labrum, R.W.; Kotzadimitriou, D.; Eunson, L.; Davis, M.B.; Davies, R.; Wood, N.W.; Kullmann, D.M.; Hanna, M.G.; et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J. Physiol. 2010, 588, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Leon, E.; Banchs, I.; Serra, S.A.; Latorre, P.; Fernandez-Castillo, N.; Corominas, R.; Valverde, M.A.; Volpini, V.; Fernandez-Fernandez, J.M.; Macaya, A.; et al. Late-onset episodic ataxia type 2 associated with a novel loss-of-function mutation in the CACNA1A gene. J. Neurol. Sci. 2009, 280, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Guerin, A.A.; Feigenbaum, A.; Donner, E.J.; Yoon, G. Stepwise developmental regression associated with novel CACNA1A mutation. Pediatr. Neurol. 2008, 39, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Roubertie, A.; Echenne, B.; Leydet, J.; Soete, S.; Krams, B.; Rivier, F.; Riant, F.; Tournier-Lasserve, E. Benign paroxysmal tonic upgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family. J. Neurol. 2008, 255, 1600–1602. [Google Scholar] [CrossRef] [PubMed]
- Bertholon, P.; Chabrier, S.; Riant, F.; Tournier-Lasserve, E.; Peyron, R. Episodic ataxia type 2: Unusual aspects in clinical and genetic presentation. Special emphasis in childhood. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1289–1292. [Google Scholar] [CrossRef]
- Scoggan, K.A.; Chandra, T.; Nelson, R.; Hahn, A.F.; Bulman, D.E. Identification of two novel mutations in the CACNA1A gene responsible for episodic ataxia type 2. J. Med. Genet. 2001, 38, 249–253. [Google Scholar] [CrossRef]
- Blumkin, L.; Michelson, M.; Leshinsky-Silver, E.; Kivity, S.; Lev, D.; Lerman-Sagie, T. Congenital ataxia, mental retardation, and dyskinesia associated with a novel CACNA1A mutation. J. Child Neurol. 2010, 25, 892–897. [Google Scholar] [CrossRef]
- Jen, J.; Wan, J.; Graves, M.; Yu, H.; Mock, A.F.; Coulin, C.J.; Kim, G.; Yue, Q.; Papazian, D.M.; Baloh, R.W. Loss-of-function EA2 mutations are associated with impaired neuromuscular transmission. Neurology 2001, 57, 1843–1848. [Google Scholar] [CrossRef]
- Pietrobon, D. CaV2.1 channelopathies. Pflug. Arch. Eur. J. Physiol. 2010, 460, 375–393. [Google Scholar] [CrossRef]
- Guida, S.; Trettel, F.; Pagnutti, S.; Mantuano, E.; Tottene, A.; Veneziano, L.; Fellin, T.; Spadaro, M.; Stauderman, K.; Williams, M.; et al. Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am. J. Hum. Genet. 2001, 68, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Jen, J.; Yue, Q.; Nelson, S.F.; Yu, H.; Litt, M.; Nutt, J.; Baloh, R.W. A novel nonsense mutation in CACNA1A causes episodic ataxia and hemiplegia. Neurology 1999, 53, 34–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spacey, S.D.; Materek, L.A.; Szczygielski, B.I.; Bird, T.D. Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch. Neurol. 2005, 62, 314–316. [Google Scholar] [CrossRef]
- Friend, K.L.; Crimmins, D.; Phan, T.G.; Sue, C.M.; Colley, A.; Fung, V.S.; Morris, J.G.; Sutherland, G.R.; Richards, R.I. Detection of a novel missense mutation and second recurrent mutation in the CACNA1A gene in individuals with EA-2 and FHM. Hum. Genet. 1999, 105, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, A.; D’Angelo, M.G.; Salati, R.; Villa, L.; Germinasi, C.; Frattini, T.; Meola, G.; Turconi, A.C.; Bresolin, N.; Bassi, M.T. Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J. Neurol. Sci. 2006, 241, 13–17. [Google Scholar] [CrossRef]
- Spacey, S.D.; Hildebrand, M.E.; Materek, L.A.; Bird, T.D.; Snutch, T.P. Functional implications of a novel EA2 mutation in the P/Q-type calcium channel. Ann. Neurol. 2004, 56, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Denier, C.; Ducros, A.; Durr, A.; Eymard, B.; Chassande, B.; Tournier-Lasserve, E. Missense CACNA1A mutation causing episodic ataxia type 2. Arch. Neurol. 2001, 58, 292–295. [Google Scholar] [CrossRef]
- Ohba, C.; Osaka, H.; Iai, M.; Yamashita, S.; Suzuki, Y.; Aida, N.; Shimozawa, N.; Takamura, A.; Doi, H.; Tomita-Katsumoto, A.; et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 2013, 14, 225–232. [Google Scholar] [CrossRef]
- Jouvenceau, A.; Eunson, L.H.; Spauschus, A.; Ramesh, V.; Zuberi, S.M.; Kullmann, D.M.; Hanna, M.G. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet 2001, 358, 801–807. [Google Scholar] [CrossRef]
- Melzer, N.; Classen, J.; Reiners, K.; Buttmann, M. Fluctuating neuromuscular transmission defects and inverse acetazolamide response in episodic ataxia type 2 associated with the novel CaV2.1 single amino acid substitution R2090Q. J. Neurol. Sci. 2010, 296, 104–106. [Google Scholar] [CrossRef]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Valcarcel, J. Building specificity with nonspecific RNA-binding proteins. Nat. Struct. Mol. Biol. 2005, 12, 645–653. [Google Scholar] [CrossRef]
- Smith, C.W.; Valcarcel, J. Alternative pre-mRNA splicing: The logic of combinatorial control. Trends Biochem. Sci. 2000, 25, 381–388. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhou, T. Alternative-splicing-mediated gene expression. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2014, 89, 012713. [Google Scholar] [CrossRef]
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266. [Google Scholar] [CrossRef]
- Montes, M.; Sanford, B.L.; Comiskey, D.F.; Chandler, D.S. RNA Splicing and Disease: Animal Models to Therapies. Trends Genet. TIG 2019, 35, 68–87. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef]
- DeVos, S.L.; Miller, T.M. Antisense oligonucleotides: Treating neurodegeneration at the level of RNA. Neurother. J. Am. Soc. Exp. Neurother. 2013, 10, 486–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozaffari-Jovin, S.; Wandersleben, T.; Santos, K.F.; Will, C.L.; Luhrmann, R.; Wahl, M.C. Inhibition of RNA helicase Brr2 by the C-terminal tail of the spliceosomal protein Prp8. Science 2013, 341, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef] [Green Version]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [Green Version]
- Dominski, Z.; Kole, R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1993, 90, 8673–8677. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.; Palma, E.; Sazani, P.; Orum, H.; Cho, M.; Kole, R. Efficient and persistent splice switching by systemically delivered LNA oligonucleotides in mice. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 471–475. [Google Scholar] [CrossRef]
- Sazani, P.; Gemignani, F.; Kang, S.H.; Maier, M.A.; Manoharan, M.; Persmark, M.; Bortner, D.; Kole, R. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol. 2002, 20, 1228–1233. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Brzustowicz, L.M.; Lehner, T.; Castilla, L.H.; Penchaszadeh, G.K.; Wilhelmsen, K.C.; Daniels, R.; Davies, K.E.; Leppert, M.; Ziter, F.; Wood, D.; et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature 1990, 344, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Farrar, M.A.; Kiernan, M.C. The Genetics of Spinal Muscular Atrophy: Progress and Challenges. Neurother. J. Am. Soc. Exp. Neurother. 2015, 12, 290–302. [Google Scholar] [CrossRef]
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef]
- Melki, J.; Abdelhak, S.; Sheth, P.; Bachelot, M.F.; Burlet, P.; Marcadet, A.; Aicardi, J.; Barois, A.; Carriere, J.P.; Fardeau, M.; et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature 1990, 344, 767–768. [Google Scholar] [CrossRef]
- Feldkotter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Wirth, B.; Brichta, L.; Schrank, B.; Lochmuller, H.; Blick, S.; Baasner, A.; Heller, R. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum. Genet. 2006, 119, 422–428. [Google Scholar] [CrossRef]
- Cartegni, L.; Hastings, M.L.; Calarco, J.A.; de Stanchina, E.; Krainer, A.R. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am. J. Hum. Genet. 2006, 78, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, P.W.; Wee, K.B.; Yee, W.C.; Pramono, Z.A. Dual masking of specific negative splicing regulatory elements resulted in maximal exon 7 inclusion of SMN2 gene. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darras, B.T.; Farrar, M.A.; Mercuri, E.; Finkel, R.S.; Foster, R.; Hughes, S.G.; Bhan, I.; Farwell, W.; Gheuens, S. An Integrated Safety Analysis of Infants and Children with Symptomatic Spinal Muscular Atrophy (SMA) Treated with Nusinersen in Seven Clinical Trials. CNS Drugs 2019, 33, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Hache, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J. Child Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. NMD 2018, 28, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for spinal muscular atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 1756285618754459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ydewalle, C.; Ramos, D.M.; Pyles, N.J.; Ng, S.Y.; Gorz, M.; Pilato, C.M.; Ling, K.; Kong, L.; Ward, A.J.; Rubin, L.L.; et al. The Antisense Transcript SMN-AS1 Regulates SMN Expression and is a Novel Therapeutic Target for Spinal Muscular Atrophy. Neuron 2017, 93, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; de Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.P.; Dressman, D. Molecular pathophysiology and targeted therapeutics for muscular dystrophy. Trends Pharmacol. Sci. 2001, 22, 465–470. [Google Scholar] [CrossRef]
- Nowak, K.J.; Davies, K.E. Duchenne muscular dystrophy and dystrophin: Pathogenesis and opportunities for treatment. EMBO Rep. 2004, 5, 872–876. [Google Scholar] [CrossRef]
- Helderman-van den Enden, A.T.; Straathof, C.S.; Aartsma-Rus, A.; den Dunnen, J.T.; Verbist, B.M.; Bakker, E.; Verschuuren, J.J.; Ginjaar, H.B. Becker muscular dystrophy patients with deletions around exon 51; a promising outlook for exon skipping therapy in Duchenne patients. Neuromuscul. Disord. NMD 2010, 20, 251–254. [Google Scholar] [CrossRef]
- Arechavala-Gomeza, V.; Graham, I.R.; Popplewell, L.J.; Adams, A.M.; Aartsma-Rus, A.; Kinali, M.; Morgan, J.E.; van Deutekom, J.C.; Wilton, S.D.; Dickson, G.; et al. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum. Gene Ther. 2007, 18, 798–810. [Google Scholar] [CrossRef] [Green Version]
- Van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Janson, A.A.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.J.; van Deutekom, J.C. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Alter, J.; Lou, F.; Rabinowitz, A.; Yin, H.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Lu, Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kole, R.; Krieg, A.M. Exon skipping therapy for Duchenne muscular dystrophy. Adv. Drug Deliv. Rev. 2015, 87, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for Duchenne muscular dystrophy. Hum. Mol. Genet. 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Relizani, K.; Griffith, G.; Echevarria, L.; Zarrouki, F.; Facchinetti, P.; Vaillend, C.; Leumann, C.; Garcia, L.; Goyenvalle, A. Efficacy and Safety Profile of Tricyclo-DNA Antisense Oligonucleotides in Duchenne Muscular Dystrophy Mouse Model. Mol. Ther. Nucleic Acids 2017, 8, 144–157. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef]
- Perlman, S.; Becker-Catania, S.; Gatti, R.A. Ataxia-telangiectasia: Diagnosis and treatment. Semin. Pediatr. Neurol. 2003, 10, 173–182. [Google Scholar] [CrossRef]
- Cavalieri, S.; Pozzi, E.; Gatti, R.A.; Brusco, A. Deep-intronic ATM mutation detected by genomic resequencing and corrected in vitro by antisense morpholino oligonucleotide (AMO). Eur. J. Hum. Genet. EJHG 2013, 21, 774–778. [Google Scholar] [CrossRef]
- Nakamura, K.; Du, L.; Tunuguntla, R.; Fike, F.; Cavalieri, S.; Morio, T.; Mizutani, S.; Brusco, A.; Gatti, R.A. Functional characterization and targeted correction of ATM mutations identified in Japanese patients with ataxia-telangiectasia. Hum. Mutat. 2012, 33, 198–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teraoka, S.N.; Telatar, M.; Becker-Catania, S.; Liang, T.; Onengut, S.; Tolun, A.; Chessa, L.; Sanal, O.; Bernatowska, E.; Gatti, R.A.; et al. Splicing defects in the ataxia-telangiectasia gene, ATM: Underlying mutations and consequences. Am. J. Hum. Genet. 1999, 64, 1617–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Pollard, J.M.; Gatti, R.A. Correction of prototypic ATM splicing mutations and aberrant ATM function with antisense morpholino oligonucleotides. Proc. Natl. Acad. Sci. USA 2007, 104, 6007–6012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Kayali, R.; Bertoni, C.; Fike, F.; Hu, H.; Iversen, P.L.; Gatti, R.A. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011, 20, 3151–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, N.J.; Gordon, L.B. Hutchinson-Gilford progeria syndrome. Handb. Clin. Neurol. 2015, 132, 249–264. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, I.C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef]
- Berger, A.; Maire, S.; Gaillard, M.C.; Sahel, J.A.; Hantraye, P.; Bemelmans, A.P. mRNA trans-splicing in gene therapy for genetic diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, S.G.; Chao, H.; Walsh, C.E. RNA repair using spliceosome-mediated RNA trans-splicing. Trends Mol. Med. 2004, 10, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Murauer, E.M.; Bauer, J.W. Spliceosome-mediated trans-splicing: The therapeutic cut and paste. J. Investig. Dermatol. 2012, 132, 1959–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, Q.; Mansfield, S.G.; Puttaraju, M.; Zhang, Y.; Zhou, W.; Cohn, J.A.; Garcia-Blanco, M.A.; Mitchell, L.G.; Engelhardt, J.F. Partial correction of endogenous DeltaF508 CFTR in human cystic fibrosis airway epithelia by spliceosome-mediated RNA trans-splicing. Nat. Biotechnol. 2002, 20, 47–52. [Google Scholar] [CrossRef]
- Andreadis, A.; Brown, W.M.; Kosik, K.S. Structure and novel exons of the human tau gene. Biochemistry 1992, 31, 10626–10633. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- D’Souza, I.; Schellenberg, G.D. Regulation of tau isoform expression and dementia. Biochim. Biophys. Acta 2005, 1739, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Kar, A.; Kuo, D.; He, R.; Zhou, J.; Wu, J.Y. Tau alternative splicing and frontotemporal dementia. Alzheimer Dis. Assoc. Disord. 2005, 19 (Suppl. 1), 29–36. [Google Scholar] [CrossRef]
- Qian, W.; Liu, F. Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 2014, 30, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avale, M.E.; Rodriguez-Martin, T.; Gallo, J.M. Trans-splicing correction of tau isoform imbalance in a mouse model of tau mis-splicing. Hum. Mol. Genet. 2013, 22, 2603–2611. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Martin, T.; Anthony, K.; Garcia-Blanco, M.A.; Mansfield, S.G.; Anderton, B.H.; Gallo, J.M. Correction of tau mis-splicing caused by FTDP-17 MAPT mutations by spliceosome-mediated RNA trans-splicing. Hum. Mol. Genet. 2009, 18, 3266–3273. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martin, T.; Garcia-Blanco, M.A.; Mansfield, S.G.; Grover, A.C.; Hutton, M.; Yu, Q.; Zhou, J.; Anderton, B.H.; Gallo, J.M. Reprogramming of tau alternative splicing by spliceosome-mediated RNA trans-splicing: Implications for tauopathies. Proc. Natl. Acad. Sci. USA 2005, 102, 15659–15664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coady, T.H.; Baughan, T.D.; Shababi, M.; Passini, M.A.; Lorson, C.L. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS ONE 2008, 3, e3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coady, T.H.; Lorson, C.L. Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J. Neurosci. 2010, 30, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Coady, T.H.; Shababi, M.; Tullis, G.E.; Lorson, C.L. Restoration of SMN function: Delivery of a trans-splicing RNA re-directs SMN2 pre-mRNA splicing. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1471–1478. [Google Scholar] [CrossRef]
- Shababi, M.; Glascock, J.; Lorson, C.L. Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum. Gene Ther. 2011, 22, 135–144. [Google Scholar] [CrossRef]
- Li, B.; Niu, Y.; Ji, W.; Dong, Y. Strategies for the CRISPR-Based Therapeutics. Trends Pharmacol. Sci. 2020, 41, 55–65. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, F.; Gao, G. CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell 2020, 181, 136–150. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin-Jing, L.; Xiang, L.; Cheng, T.; Ying-Qian, L.; Xinde, H.; Erwei, Z.; He, L.; Wenqin, Y.; Yidi, S.; Lu-Lu, L.; et al. Disruption of splicing-regulatory elements using CRISPR/Cas9 to rescue spinal muscular atrophy in human iPSCs and mice. Natl. Sci. Rev. 2020, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In Vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In Vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Yuan, J.; Ma, Y.; Huang, T.; Chen, Y.; Peng, Y.; Li, B.; Li, J.; Zhang, Y.; Song, B.; Sun, X.; et al. Genetic Modulation of RNA Splicing with a CRISPR-Guided Cytidine Deaminase. Mol. Cell 2018, 72, 380–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Jun, K.; Piedras-Renteria, E.S.; Smith, S.M.; Wheeler, D.B.; Lee, S.B.; Lee, T.G.; Chin, H.; Adams, M.E.; Scheller, R.H.; Tsien, R.W.; et al. Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc. Natl. Acad. Sci. USA 1999, 96, 15245–15250. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Noebels, J.L. Presynaptic Ca2+ influx at a mouse central synapse with Ca2+ channel subunit mutations. J. Neurosci. 2000, 20, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Inchauspe, C.G.; Forsythe, I.D.; Uchitel, O.D. Changes in synaptic transmission properties due to the expression of N-type calcium channels at the calyx of Held synapse of mice lacking P/Q-type calcium channels. J. Physiol. 2007, 584, 835–851. [Google Scholar] [CrossRef]
- Inchauspe, C.G.; Martini, F.J.; Forsythe, I.D.; Uchitel, O.D. Functional compensation of P/Q by N-type channels blocks short-term plasticity at the calyx of Held presynaptic terminal. J. Neurosci. 2004, 24, 10379–10383. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Kaneko, M.; Shin, H.S.; Takahashi, T. Presynaptic N-type and P/Q-type Ca2+ channels mediating synaptic transmission at the calyx of Held of mice. J. Physiol. 2005, 568, 199–209. [Google Scholar] [CrossRef]
- Maejima, T.; Wollenweber, P.; Teusner, L.U.; Noebels, J.L.; Herlitze, S.; Mark, M.D. Postnatal loss of P/Q-type channels confined to rhombic-lip-derived neurons alters synaptic transmission at the parallel fiber to purkinje cell synapse and replicates genomic Cacna1a mutation phenotype of ataxia and seizures in mice. J. Neurosci. 2013, 33, 5162–5174. [Google Scholar] [CrossRef]
- Mark, M.D.; Maejima, T.; Kuckelsberg, D.; Yoo, J.W.; Hyde, R.A.; Shah, V.; Gutierrez, D.; Moreno, R.L.; Kruse, W.; Noebels, J.L.; et al. Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J. Neurosci. 2011, 31, 4311–4326. [Google Scholar] [CrossRef] [PubMed]
- Gray, A.C.; Raingo, J.; Lipscombe, D. Neuronal calcium channels: Splicing for optimal performance. Cell Calcium 2007, 42, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altier, C.; Dale, C.S.; Kisilevsky, A.E.; Chapman, K.; Castiglioni, A.J.; Matthews, E.A.; Evans, R.M.; Dickenson, A.H.; Lipscombe, D.; Vergnolle, N.; et al. Differential role of N-type calcium channel splice isoforms in pain. J. Neurosci. 2007, 27, 6363–6373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, A.; Denome, S.; Jiang, Y.Q.; Marangoudakis, S.; Lipscombe, D. Opioid inhibition of N-type Ca2+ channels and spinal analgesia couple to alternative splicing. Nat. Neurosci. 2010, 13, 1249–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, T.J.; Thaler, C.; Castiglioni, A.J.; Helton, T.D.; Lipscombe, D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 2004, 41, 127–138. [Google Scholar] [CrossRef] [Green Version]



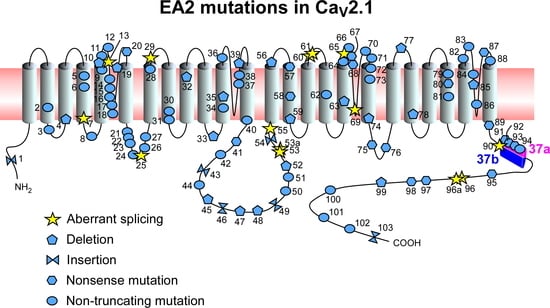
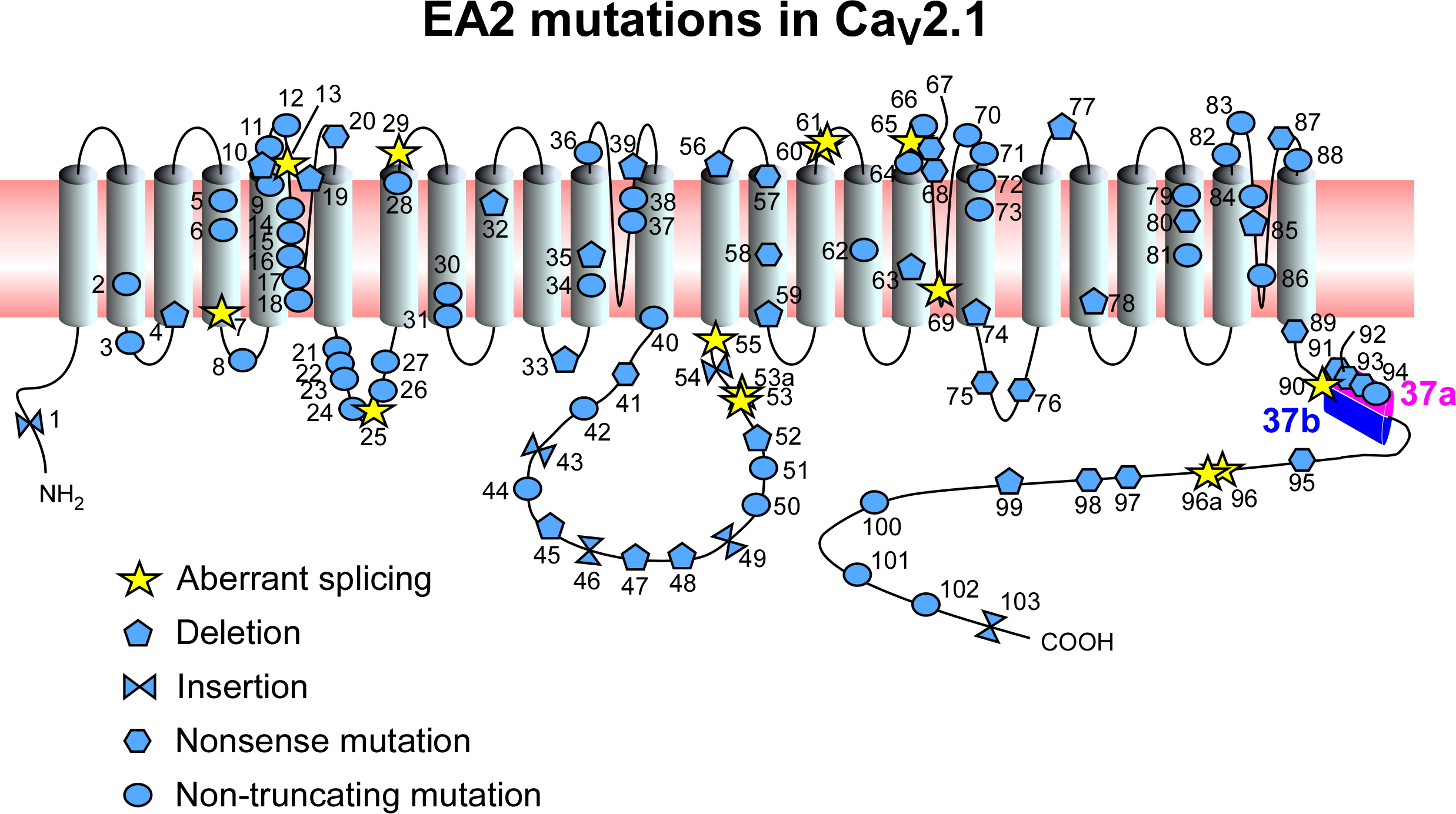{kind=link}
{kind=link}
{kind=link}
| No. | Amino Acid | DNA Mutation | References |
|---|---|---|---|
| 1 | p.(Ala56Serfs*20) | c.165dupA | [4] |
| 2 | p.Glu147Lys | c.439G>A | [27] |
| 3 | p.Gly162Val | c.485G>T | [28] |
| 4 | p.(Trp168Glyfs*10) | c.504delC | [4] |
| 5 | p.Arg192Trp | c.574C>T | [29] |
| 6 | p.Arg198Gln | c.593G>A | [30] |
| 7 | c.868+5G>A; possible aberrant splicing | [16] | |
| 8 | p.Ser218Leu | c.653C>T | [30] |
| 9 | p.Tyr248Asn/Cys | c.742T>A/c.743A>G | [31,32] |
| 10 | p.Gly250Glufs*60 | c.749delG | [12] |
| 11 | p.His253Tyr | c.1032C >T | [33] |
| 12 | p.(Cys256Arg) | c.1041T>C | [34] |
| 13 | c.983-1G>A; aberrant splicing | [4] | |
| 14 | p.Arg279Cys | c.835C>T | [28] |
| 15 | p.Cys287Tyr | c.1096G>A | [1] |
| 16 | p.Gly293Arg | c.1152G>A | [35] |
| 17 | p.Gly297Arg | c.889G>A | [36] |
| 18 | p.Asp302Asn | c.904G>A | [28] |
| 19 | p.Thr310 fs*5 | c.928_931delACTG | [28] |
| 20 | p.Trp320* | c.959G>A | [12] |
| 21 | p.Arg387Gly | c.1159C>G | [28] |
| 22 | p.Glu388Lys | c.1161G>A | [37] |
| 23 | p.(Leu389Phe) | c.1165C>T | [4] |
| 24 | p.Gly411Trp | c.1231G>T | [28] |
| 25 | c.1253+1G>A; probable aberrant splicing | [15] | |
| 26 | p.Ala454Thr | c.1360G>A | [38] |
| 27 | p.Arg455Gln | c.1364G>A | [39] |
| 28 | p.(Thr501Met) | c.1502C>T | [4] |
| 29 | c.1557+1G>A; aberrant splicing | [13] | |
| 30 | p.Glu533Lys | c.1597G>A | [40] |
| 31 | p.Gly540Arg | c.1618G>A | [41] |
| 32 | p.Val558Serfs*13 | c.1672-1_1675delGGTTA | [28] |
| 33 | p.Leu600 fs*41 | c.1799_1800delTC | [28] |
| 34 | p.Leu621Arg | c.2144T>G | [41] |
| 35 | p.Leu624Phe | c.1870-1873del | [13] |
| 36 | p.Gly638Asp | c.1913G>A | [42] |
| 37 | p.Trp670Cys | c.2010G>C | [30] |
| 38 | p.Gly677Glu | c.2030G>A | [31] |
| 39 | p.(Gln681Argfs*100) | c.2042_2043del | [33] |
| 40 | p.Ile712Val | c.2134A>G | [43] |
| 41 | p.Gln736* | c.2206C>T | [44] |
| 42 | p.(Met798Thr) | c.2393T>C | [4] |
| 43 | p.(Arg822Profs*246) | c.2464dupC | [4] |
| 44 | p.(Pro897Arg) | c.2690C>G | [4] |
| 45 | p.Gly939* | c.2816delG | [45] |
| 46 | p.Ser943Gln | c.2825+1insG | [46] |
| 47 | p.(Ala952Serfs*115) | c.2852_2861del | [4] |
| 48 | p.(Arg957Aspfs*113) | c.2867_2869del | [16] |
| 49 | p.Glu1004Argfs*66 | c.3244+1insG | [1] |
| 50 | p.Glu998Gln | c.2992G>C | [31] |
| 51 | p.Gln1154* | c.3460C>T | [45] |
| 52 | p.Cys1178Pro | c.3531delC | [13] |
| 53 | c.3089+2T; possible aberrant splicing | [17] | |
| 53a | c.3102+2T; possible aberrant splicing | [30] | |
| 54 | c.3603dupC | [30] | |
| 55 | c.3977+1G>A; aberrant splicing | [14] | |
| 56 | p.Pro1267Leu | c.4073delC | [6] |
| 57 | p.(Arg1278*) | c.3832C<T | [16] |
| 58 | p.Arg1281* | c.4077C>T | [1] |
| 59 | p.Glu1294Del | c.3871_3873delGAG | [31] |
| 60 | c.4270+1G>A; aberrant splicing | [6] | |
| 61 | c.4001+3insT; cryptic splice donor site in exon 24 | [18] | |
| 62 | p.Arg1350Gln | c.4049G>A | [47] |
| 63 | p.Phe1394Leu | c.4182delC | [33] |
| 64 | p.Phe1404Cys | c.4486T>G | [48] |
| 65 | c.4261+1G>A; aberrant splicing | [13] | |
| 66 | p.Arg1433Gln | c.4298G>A | [49] |
| 67 | p.Tyr1443* | c.4331C>G | [13] |
| 68 | p.Trp1451* | c.4588G>A | [1] |
| 69 | c.4636+1G>T; aberrant splicing | [1] | |
| 70 | p.(Gly1483Arg) | c.4722G>A | [34] |
| 71 | p.Met1488_Ser1489del | c.4739_4744del | [34] |
| 72 | p.Phe1491Ser | c.4747T>C | [50] |
| 73 | p.(Val1494Ile) | c.4755G>A | [34] |
| 74 | p.Phe1503Del | c.4509-11delCTT | [13] |
| 75 | p.Arg1549* | c.4645C>T | [51] |
| 76 | p.Gln1561* | c.4963C>T | [52] |
| 77 | p.Tyr1594Del | c.4778-80delCTT | [13] |
| 78 | p.Val1620Ser | c.4854delG | [46] |
| 79 | p.Arg1666His/Gln | c.5260G>A/c.4991G>A | [53,54] |
| 80 | p.Arg1669* | c.5005C>T | [31] |
| 81 | p.(Arg1680Cys) | c.5038C>T | [4] |
| 82 | p.His1737Leu | c.5211A>T | [55] |
| 83 | p.Leu1749Pro | c.5246T>C | [28] |
| 84 | p.Arg1751Trp | c.5251C>T | [45] |
| 85 | p.Ser1753Cysfs*2 | c.5253-2259_5403+1135del | [12] |
| 86 | p.Glu1757Lys | c.5271G>A | [56] |
| 87 | p.Arg1785* | c.5589C>T | [1] |
| 88 | p.Ser1799Leu | c.5396C>T | [57] |
| 89 | p.Arg1824* | c.5733C>T | [58] |
| 90 | c.IVS36+2T>C (c.4582+2T>C); possible aberrant splicing | [19] | |
| 91 | p.Tyr1849* | c.5547T>A | [12] |
| 92 | p.Tyr1854* | c.5562C>G | [26] |
| 93 | p.Arg1858* | c.5571C>T | [26] |
| 94 | p.(Cys1870Arg) | c.5608T>C | [4] |
| 95 | p.(Glu1927*) | c.5779G>T | [4] |
| 96 | c.IVS41+(3–6)delGAGT (c.6068+(3-6)delGAGT); aberrant splicing | [18] | |
| 96a | c.6335+4delAGTG; aberrant splicing | [14] | |
| 97 | p.Gln1986* | c.5956C>T | [31] |
| 98 | p.Gln2039* | c.6351C>T | [1] |
| 99 | p.Pro2058Leufs*69 | c.6404delC | [1] |
| 100 | p.Arg2090Gln | c.6269G>A | [59] |
| 101 | p.(Arg2136Cys) | c.6681C>T | [34] |
| 102 | p.Pro2222Leu | c.6665C>T | [12] |
| 103 | p.2319Gln[n] | c.7191CAG[n] | [60] |
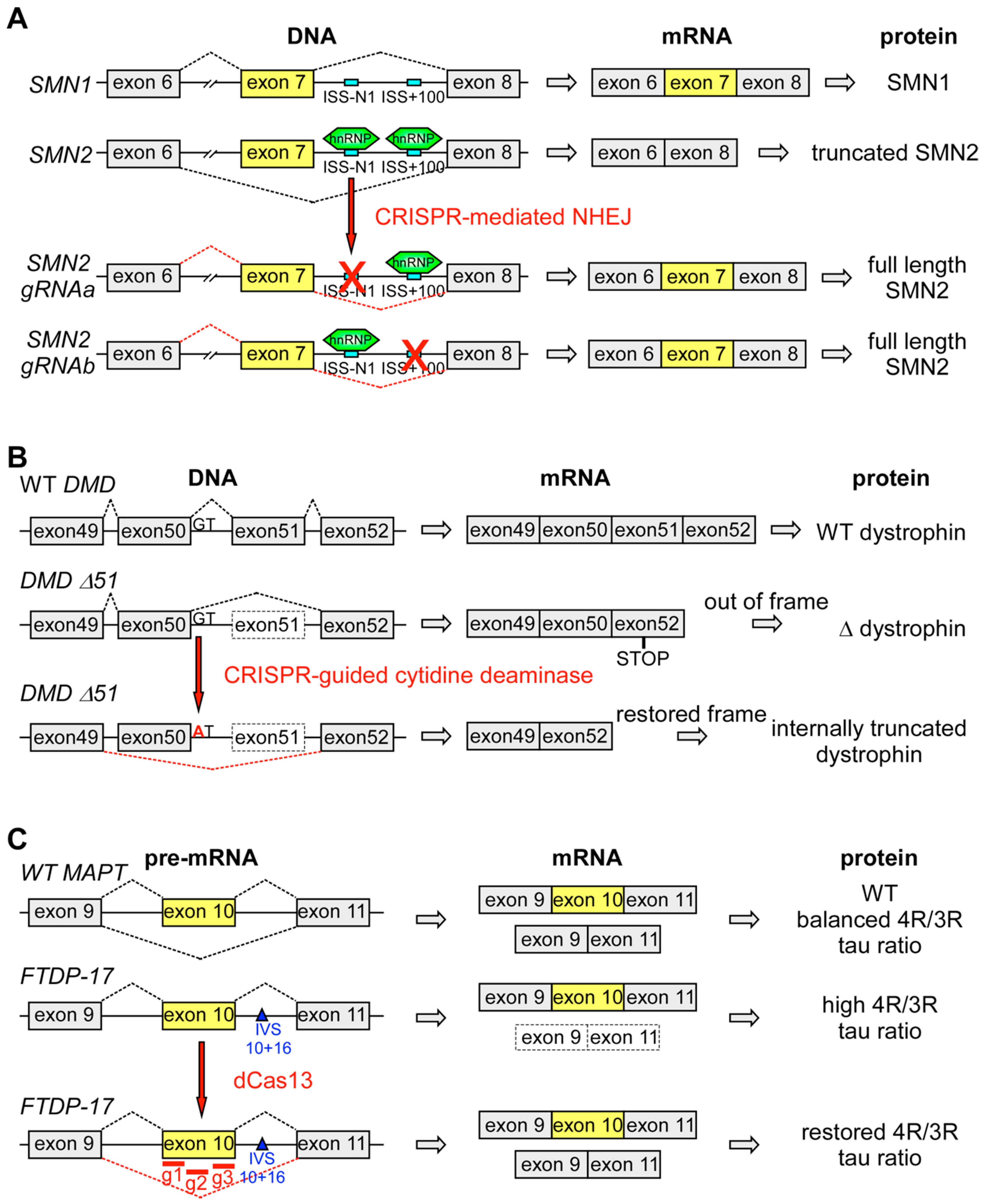
| Disease | Gene | Aberrant Splicing | Therapy | References |
|---|---|---|---|---|
| SMA | Mutated: SMN1 Therapy: SMN2 | Skipping of exon 7 due to C > T mutation at position 6 of exon 7 | 2′OMePS AONs targeting ISS-N1 to promote exon 7 inclusion in human fibroblasts and mouse brain | [92,93,94] |
| Intrathecal injection of nusinersen to increase exon 7 inclusion in patients | [95,96,97,98,99,100] | |||
| Splice switching and lncRNA-targeting AONs to promote inclusion of exon 7 in patient-derived cells and mouse brain | [102] | |||
| Trans-splicing-mediated insertion of exon 7 in fibroblasts and SMAΔ7 mice | [145,146,147,148] | |||
| CRISPR/Cas9-mediated disruption of intronic splicing silencers to enhance exon 7 inclusion in iPSC-derived motor neurons and mice | [153] | |||
| DMD | DMD | Formation of nonfunctional truncated dystrophin due to point mutations and frame-shifting deletions | 2′OMePS- or PMO-mediated skipping of exon 51 in patients | [109,110,111,112,113,114,115] |
| tcDNA-mediated exon skipping in mice | [116,117] | |||
| Restoration of open reading frame by dual Cas9-mediated exon skipping in mdx mice | [155,156,157] | |||
| dCas9-cytidine deaminase-mediated base editing to induce skipping of exon 50 and restore reading frame in iPSCs | [161] | |||
| FTDP-17 | MAPT | Increased exon 10 inclusion | Trans-splicing to reverse aberrant exon 10 inclusion in mice | [142,143,144] |
| dCas13 targeted to exon 10 splice sites on pre-mRNA to mediate exon exclusion in iPSCs-derived cortical neurons | [164] | |||
| Ataxia telangiectasia | ATM | Activation of cryptic splice sites | AMOs to mask cryptic splice sites and restore correct splicing in cell lines | [123,124] |
| Hutchinson-Gilford progeria syndrome | LMNA | Activation of an exonic cryptic donor splice site in exon 11 | AMOs to block the recognition of cryptic splice sites in fibroblasts and mice | [128,129] |
| CF | CFTR | Three base-pair deletion in exon 10 causing deletion of F508 | Correction of exon 10 by trans-splicing in epithelial cells and a xenograft model | [131,136] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaudon, F.; Baldassari, S.; Musante, I.; Thalhammer, A.; Zara, F.; Cingolani, L.A. Targeting Alternative Splicing as a Potential Therapy for Episodic Ataxia Type 2. Biomedicines 2020, 8, 332. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8090332
Jaudon F, Baldassari S, Musante I, Thalhammer A, Zara F, Cingolani LA. Targeting Alternative Splicing as a Potential Therapy for Episodic Ataxia Type 2. Biomedicines. 2020; 8(9):332. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8090332
Chicago/Turabian StyleJaudon, Fanny, Simona Baldassari, Ilaria Musante, Agnes Thalhammer, Federico Zara, and Lorenzo A. Cingolani. 2020. "Targeting Alternative Splicing as a Potential Therapy for Episodic Ataxia Type 2" Biomedicines 8, no. 9: 332. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines8090332