
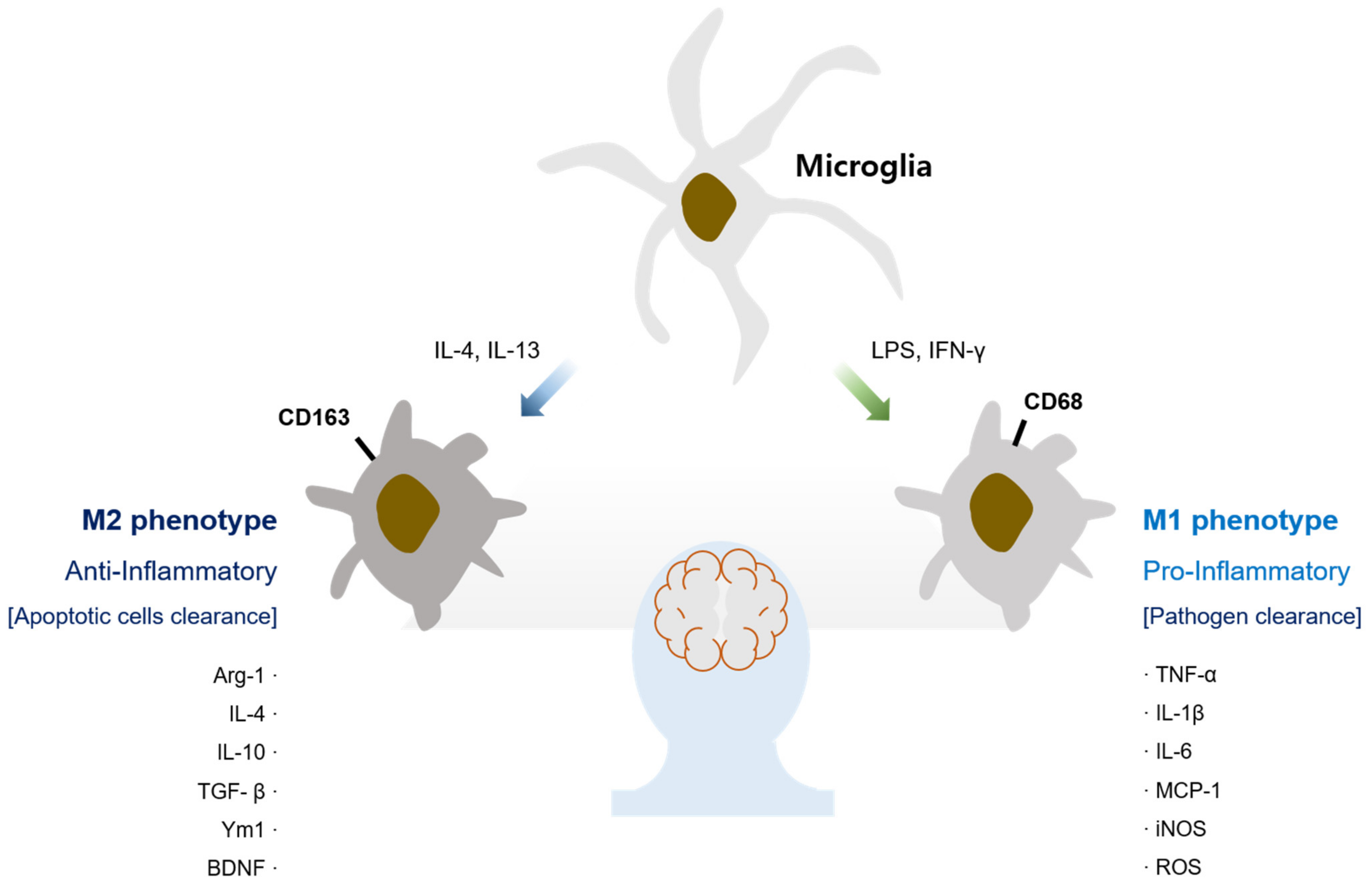
The Role of Microglia in the Development of Neurodegenerative Diseases

 , and
, and
Abstract
:1. Introduction
2. Microglia in Epilepsy
Experimental Animal Models of Epilepsy
3. Microglia in Alzheimer’s Disease (AD)
Experimental Animal Models of AD
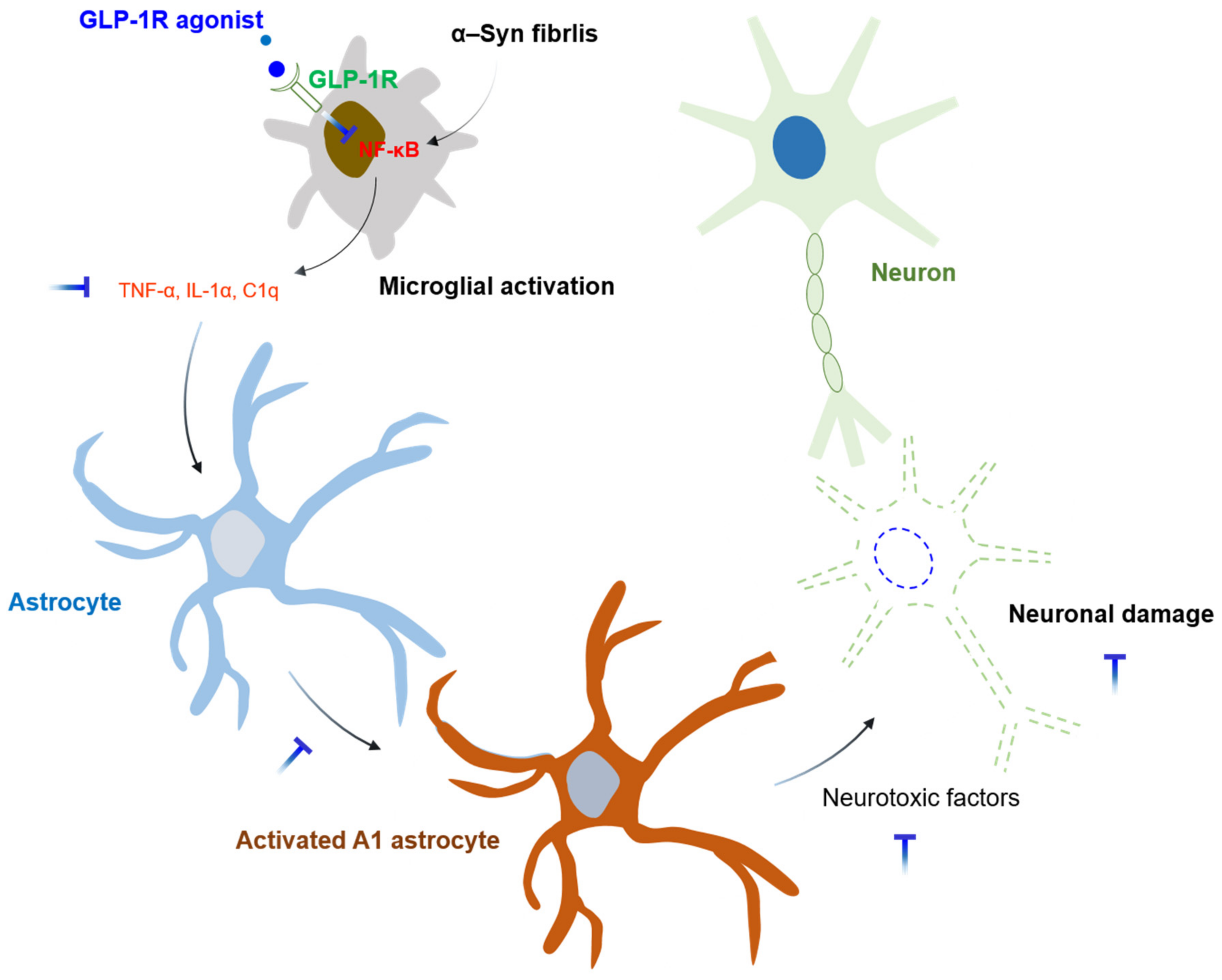
4. Microglia in Parkinson’s Disease (PD)
Experimental Animal Models of PD
5. Microglia in Huntington’s Disease (HD)
Experimental Animal Models of HD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia-Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef]
- Andoh, M.; Ikegaya, Y.; Koyama, R. Synaptic Pruning by Microglia in Epilepsy. J. Clin. Med. 2019, 8, 2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePaula-Silva, A.B.; Gorbea, C.; Doty, D.J.; Libbey, J.E.; Sanchez, J.M.S.; Hanak, T.J.; Cazalla, D.; Fujinami, R.S. Differential transcriptional profiles identify microglial- and macrophage-specific gene markers expressed during virus-induced neuroinflammation. J. Neuroinflamm. 2019, 16, 152. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Jernigan, J.; Raza, S.A.; Dammer, E.B.; Xiao, H.; Seyfried, N.T.; Levey, A.I.; Rangaraju, S. Transcriptional regulation of homeostatic and disease-associated-microglial genes by IRF1, LXRbeta, and CEBPalpha. Glia 2019, 67, 1958–1975. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Streit, W.J.; Graeber, M.B.; Kreutzberg, G.W. Functional plasticity of microglia: A review. Glia 1988, 1, 301–307. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Chiba, K. Role of microglial m1/m2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 2014, 7, 1028–1048. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflamm. 2016, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.T.; Wu, W.F.; Deng, Y.H.; Ge, J.W. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol. Med. Rep. 2020, 21, 2006–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Geladaris, A.; Hausler, D.; Weber, M.S. Microglia: The Missing Link to Decipher and Therapeutically Control MS Progression? Int. J. Mol. Sci. 2021, 22, 3461. [Google Scholar] [CrossRef]
- Fujita, Y.; Yamashita, T. Neuroprotective function of microglia in the developing brain. Neuronal Signal. 2021, 5, NS20200024. [Google Scholar] [CrossRef] [PubMed]
- Bilbo, S.; Stevens, B. Microglia: The Brain’s First Responders. Cerebrum 2017, 2017, 14–17. [Google Scholar]
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527. [Google Scholar] [CrossRef]
- Wen, Y.; Wu, K.; Xie, Y.; Dan, W.; Zhan, Y.; Shi, Q. Inhibitory effects of glucagon-like peptide-1 receptor on epilepsy. Biochem. Biophys. Res. Commun. 2019, 511, 79–86. [Google Scholar] [CrossRef]
- Liu, M.; Liu, X.; Wang, L.; Wang, Y.; Dong, F.; Wu, J.; Qu, X.; Liu, Y.; Liu, Z.; Fan, H.; et al. TRPV4 Inhibition Improved Myelination and Reduced Glia Reactivity and Inflammation in a Cuprizone-Induced Mouse Model of Demyelination. Front. Cell. Neurosci. 2018, 12, 392. [Google Scholar] [CrossRef] [Green Version]
- Ali, I.; Chugh, D.; Ekdahl, C.T. Role of fractalkine-CX3CR1 pathway in seizure-induced microglial activation, neurodegeneration, and neuroblast production in the adult rat brain. Neurobiol. Dis. 2015, 74, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Kosaraju, J.; Holsinger, R.M.D.; Guo, L.; Tam, K.Y. Linagliptin, a Dipeptidyl Peptidase-4 Inhibitor, Mitigates Cognitive Deficits and Pathology in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 6074–6084. [Google Scholar] [CrossRef]
- Wicinski, M.; Wodkiewicz, E.; Slupski, M.; Walczak, M.; Socha, M.; Malinowski, B.; Pawlak-Osinska, K. Neuroprotective Activity of Sitagliptin via Reduction of Neuroinflammation beyond the Incretin Effect: Focus on Alzheimer’s Disease. BioMed Res. Int. 2018, 2018, 6091014. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Sawmiller, D.; Habib, A.; Hou, H.; Mori, T.; Fan, A.; Tian, J.; Zeng, J.; Giunta, B.; Sanberg, P.R.; Mattson, M.P.; et al. A Novel Apolipoprotein E Antagonist Functionally Blocks Apolipoprotein E Interaction With N-terminal Amyloid Precursor Protein, Reduces beta-Amyloid-Associated Pathology, and Improves Cognition. Biol. Psychiatry 2019, 86, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Kosaraju, J.; Gali, C.C.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Holsinger, R.M.; Madhunapantula, V.S.; Nataraj, S.K.M.; Basavan, D. Saxagliptin: A dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 2013, 72, 291–300. [Google Scholar] [CrossRef]
- Moehle, M.S.; West, A.B. M1 and M2 immune activation in Parkinson’s Disease: Foe and ally? Neuroscience 2015, 302, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Nassar, N.N.; Al-Shorbagy, M.Y.; Arab, H.H.; Abdallah, D.M. Saxagliptin: A novel antiparkinsonian approach. Neuropharmacology 2015, 89, 308–317. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Erro, R.; Pappata, S.; Amboni, M.; Vicidomini, C.; Longo, K.; Santangelo, G.; Picillo, M.; Vitale, C.; Moccia, M.; Giordano, F.; et al. Anxiety is associated with striatal dopamine transporter availability in newly diagnosed untreated Parkinson’s disease patients. Parkinsonism Relat. Disord. 2012, 18, 1034–1038. [Google Scholar] [CrossRef]
- Finkelstein, D.I.; Hare, D.J.; Billings, J.L.; Sedjahtera, A.; Nurjono, M.; Arthofer, E.; George, S.; Culvenor, J.G.; Bush, A.I.; Adlard, P.A. Clioquinol Improves Cognitive, Motor Function, and Microanatomy of the Alpha-Synuclein hA53T Transgenic Mice. ACS Chem. Neurosci. 2016, 7, 119–129. [Google Scholar] [CrossRef]
- Kabel, A.M.; Omar, M.S.; Alhadhrami, A.; Alharthi, S.S.; Alrobaian, M.M. Linagliptin potentiates the effect of l-dopa on the behavioural, biochemical and immunohistochemical changes in experimentally-induced Parkinsonism: Role of toll-like receptor 4, TGF-beta1, NF-kappaB and glucagon-like peptide 1. Physiol. Behav. 2018, 188, 108–118. [Google Scholar] [CrossRef]
- Nie, S.; Ma, K.; Sun, M.; Lee, M.; Tan, Y.; Chen, G.; Zhang, Z.; Zhang, Z.; Cao, X. 7,8-Dihydroxyflavone Protects Nigrostriatal Dopaminergic Neurons from Rotenone-Induced Neurotoxicity in Rodents. Parkinsons Dis. 2019, 2019, 9193534. [Google Scholar] [CrossRef]
- Masneuf, S.; Lowery-Gionta, E.; Colacicco, G.; Pleil, K.E.; Li, C.; Crowley, N.; Flynn, S.; Holmes, A.; Kash, T. Glutamatergic mechanisms associated with stress-induced amygdala excitability and anxiety-related behavior. Neuropharmacology 2014, 85, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treiman, D.M. GABAergic mechanisms in epilepsy. Epilepsia 2001, 42 (Suppl. 3), 8–12. [Google Scholar] [CrossRef] [PubMed]
- Victor, T.R.; Tsirka, S.E. Microglial contributions to aberrant neurogenesis and pathophysiology of epilepsy. Neuroimmunol. Neuroinflamm. 2020, 7, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, L.; An, D.; Xu, W.; Wu, C.; Sha, S.; Li, Y.; Zhu, Y.; Chen, A.; Du, Y.; et al. TRPV4-induced inflammatory response is involved in neuronal death in pilocarpine model of temporal lobe epilepsy in mice. Cell Death Dis. 2019, 10, 386. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhu, C.; Huang, D. Microglial activation: An important process in the onset of epilepsy. Am. J. Transl. Res. 2018, 10, 2877–2889. [Google Scholar] [PubMed]
- Kobylarek, D.; Iwanowski, P.; Lewandowska, Z.; Limphaibool, N.; Szafranek, S.; Labrzycka, A.; Kozubski, W. Advances in the Potential Biomarkers of Epilepsy. Front. Neurol. 2019, 10, 685. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Jeon, S.J.; Cho, K.S.; Moon, E.; Sapkota, A.; Jun, H.S.; Ryu, J.H.; Choi, J.W. Activation of Glucagon-Like Peptide-1 Receptor Promotes Neuroprotection in Experimental Autoimmune Encephalomyelitis by Reducing Neuroinflammatory Responses. Mol. Neurobiol. 2018, 55, 3007–3020. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front. Cell. Neurosci. 2018, 12, 206. [Google Scholar] [CrossRef] [Green Version]
- McElroy, P.B.; Liang, L.P.; Day, B.J.; Patel, M. Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp. Neurol. 2017, 298, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Min, J.S.; Kim, B.; Chae, U.B.; Yun, J.W.; Choi, M.S.; Kong, I.K.; Chang, K.T.; Lee, D.S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-kappaB pathways. Neurosci. Lett. 2015, 584, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, J.; Chen, O.; Xue, J.; Huang, S.; Zhu, W.; Wang, Y. Neuroprotective and anti-inflammatory effects of isoliquiritigenin in kainic acid-induced epileptic rats via the TLR4/MYD88 signaling pathway. Inflammopharmacology 2019, 27, 1143–1153. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, H.; Lee, J.E.; Kang, T.C. CDDO-Me Inhibits Microglial Activation and Monocyte Infiltration by Abrogating NFkappaB- and p38 MAPK-Mediated Signaling Pathways Following Status Epilepticus. Cells 2020, 9, 1123. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [Green Version]
- Tarozzo, G.; Bortolazzi, S.; Crochemore, C.; Chen, S.C.; Lira, A.S.; Abrams, J.S.; Beltramo, M. Fractalkine protein localization and gene expression in mouse brain. J. Neurosci. Res. 2003, 73, 81–88. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bisht, K.; Tremblay, M.E. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell. Neurosci. 2014, 8, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zeng, K.; Han, Y.; Wang, L.; Chen, D.; Xi, Z.; Wang, H.; Wang, X.; Chen, G. Altered expression of CX3CL1 in patients with epilepsy and in a rat model. Am. J. Pathol. 2012, 180, 1950–1962. [Google Scholar] [CrossRef]
- Eyo, U.B.; Peng, J.; Murugan, M.; Mo, M.; Lalani, A.; Xie, P.; Xu, P.; Margolis, D.J.; Wu, L.J. Regulation of Physical Microglia-Neuron Interactions by Fractalkine Signaling after Status Epilepticus. eNeuro 2016, 3. [Google Scholar] [CrossRef]
- Varin, E.M.; Mulvihill, E.E.; Baggio, L.L.; Koehler, J.A.; Cao, X.; Seeley, R.J.; Drucker, D.J. Distinct Neural Sites of GLP-1R Expression Mediate Physiological versus Pharmacological Control of Incretin Action. Cell Rep. 2019, 27, 3371–3384. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.S.; Feng, X.B.; Zhang, B.H.; Xia, Z.Y.; Zhan, L.Y. Exendin-4 attenuates pain-induced cognitive impairment by alleviating hippocampal neuroinflammation in a rat model of spinal nerve ligation. Neural Regen. Res. 2020, 15, 1333–1339. [Google Scholar] [CrossRef]
- White, J.P.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiragi, T.; Ikegaya, Y.; Koyama, R. Microglia after Seizures and in Epilepsy. Cells 2018, 7, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Shan, W.; Yang, H.; Liu, R.; Wu, J.; Wang, Q. The Role of Neuroinflammation in Post-traumatic Epilepsy. Front. Neurol. 2021, 12, 646152. [Google Scholar] [CrossRef]
- Shi, M.; Du, F.; Liu, Y.; Li, L.; Cai, J.; Zhang, G.F.; Xu, X.F.; Lin, T.; Cheng, H.R.; Liu, X.D.; et al. Glial cell-expressed mechanosensitive channel TRPV4 mediates infrasound-induced neuronal impairment. Acta Neuropathol. 2013, 126, 725–739. [Google Scholar] [CrossRef]
- Byun, J.S.; Lee, J.W.; Kim, S.Y.; Kwon, K.J.; Sohn, J.H.; Lee, K.; Oh, D.; Kim, S.S.; Chun, W.; Lee, H.J. Neuroprotective effects of stanniocalcin 2 following kainic acid-induced hippocampal degeneration in ICR mice. Peptides 2010, 31, 2094–2099. [Google Scholar] [CrossRef]
- Alves, M.; Kenny, A.; de Leo, G.; Beamer, E.H.; Engel, T. Tau Phosphorylation in a Mouse Model of Temporal Lobe Epilepsy. Front. Aging Neurosci. 2019, 11, 308. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Jimenez-Mateos, E.M.; Matsushima, S.; Taki, W.; Henshall, D.C. Hippocampal damage after intra-amygdala kainic acid-induced status epilepticus and seizure preconditioning-mediated neuroprotection in SJL mice. Epilepsy Res. 2010, 88, 151–161. [Google Scholar] [CrossRef]
- Ortiz-Perez, A.; Limon-Morales, O.; Rojas-Castaneda, J.C.; Cerbon, M.; Picazo, O. Prolactin prevents the kainic acid-induced neuronal loss in the rat hippocampus by inducing prolactin receptor and putatively increasing the VGLUT1 overexpression. Neurosci. Lett. 2019, 694, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, C.; Shi, Y.; Guan, L.; Li, H.; Li, S.; Li, Y.; Zhang, Y.; Lin, J. Abnormal neuronal damage and inflammation in the hippocampus of kainic acid-induced epilepsy mice. Cell Biochem. Funct. 2021. [Google Scholar] [CrossRef]
- Sabilallah, M.; Fontanaud, P.; Linck, N.; Boussadia, B.; Peyroutou, R.; Lasgouzes, T.; Rassendren, F.A.; Marchi, N.; Hirbec, H.E. Evidence for Status Epilepticus and Pro-Inflammatory Changes after Intranasal Kainic Acid Administration in Mice. PLoS ONE 2016, 11, e0150793. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, M.; Liu, H.; Wang, J. Signaling by growth/differentiation factor 5 through the bone morphogenetic protein receptor type IB protects neurons against kainic acid-induced neurodegeneration. Neurosci. Lett 2017, 651, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Rong, S.; Wan, D.; Fan, Y.; Liu, S.; Sun, K.; Huo, J.; Zhang, P.; Li, X.; Xie, X.; Wang, F.; et al. Amentoflavone Affects Epileptogenesis and Exerts Neuroprotective Effects by Inhibiting NLRP3 Inflammasome. Front. Pharmacol. 2019, 10, 856. [Google Scholar] [CrossRef] [Green Version]
- Loscher, W. Animal Models of Seizures and Epilepsy: Past, Present, and Future Role for the Discovery of Antiseizure Drugs. Neurochem. Res. 2017, 42, 1873–1888. [Google Scholar] [CrossRef]
- Zhen, J.; Qu, Z.; Fang, H.; Fu, L.; Wu, Y.; Wang, H.; Zang, H.; Wang, W. Effects of grape seed proanthocyanidin extract on pentylenetetrazole-induced kindling and associated cognitive impairment in rats. Int. J. Mol. Med. 2014, 34, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Guan, Q.; Wang, Y.; Shen, H.; Zhai, L.; Lu, X.; Jin, Y. Anticonvulsant and anti-apoptosis effects of salvianolic acid B on pentylenetetrazole-kindled rats via AKT/CREB/BDNF signaling. Epilepsy Res. 2019, 154, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Bertoglio, D.; Amhaoul, H.; Van Eetveldt, A.; Houbrechts, R.; Van De Vijver, S.; Ali, I.; Dedeurwaerdere, S. Kainic Acid-Induced Post-Status Epilepticus Models of Temporal Lobe Epilepsy with Diverging Seizure Phenotype and Neuropathology. Front. Neurol. 2017, 8, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.S.; Zhu, L. MiR-134 expression and changes in inflammatory cytokines of rats with epileptic seizures. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3479–3484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Mong, M.C.; Yang, Y.C.; Yin, M.C. Asiatic acid and maslinic acid attenuated kainic acid-induced seizure through decreasing hippocampal inflammatory and oxidative stress. Epilepsy Res. 2018, 139, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Chun-Yan, H.; Hua, P.; Bin-Bin, Y.; Xiaoping, T. Phyllathin From Phyllanthus Amarus Ameliorates Epileptic Convulsion and Kindling Associated Post-Ictal Depression in Mice via Inhibition of NF-kappaB/TLR-4 Pathway. Dose Response 2020, 18. [Google Scholar] [CrossRef]
- Huang, X.Y.; Hu, Q.P.; Shi, H.Y.; Zheng, Y.Y.; Hu, R.R.; Guo, Q. Everolimus inhibits PI3K/Akt/mTOR and NF-kB/IL-6 signaling and protects seizure-induced brain injury in rats. J. Chem. Neuroanat. 2021, 114, 101960. [Google Scholar] [CrossRef]
- Cao, J.; Tang, C.; Gao, M.; Rui, Y.; Zhang, J.; Wang, L.; Wang, Y.; Xu, B.; Yan, B.C. Hyperoside alleviates epilepsy-induced neuronal damage by enhancing antioxidant levels and reducing autophagy. J. Ethnopharmacol. 2020, 257, 112884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, Y.; Zhao, Y.; Li, Q.; Jin, H.; Qin, J. Inhibition of TRIB3 Protects Against Neurotoxic Injury Induced by Kainic Acid in Rats. Front. Pharmacol. 2019, 10, 585. [Google Scholar] [CrossRef]
- Rusina, E.; Bernard, C.; Williamson, A. The Kainic Acid Models of Temporal Lobe Epilepsy. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Hussein, A.M.; Eldosoky, M.; El-Shafey, M.; El-Mesery, M.; Abbas, K.M.; Ali, A.N.; Helal, G.M.; Abulseoud, O.A. Effects of GLP-1 Receptor Activation on a Pentylenetetrazole-Kindling Rat Model. Brain Sci. 2019, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Serrano, J.R.; Hoyle, R.; Holtzman, D.M. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Tan, Y.; Zheng, Y.; Xu, D.; Sun, Z.; Yang, H.; Yin, Q. Galectin-3: A key player in microglia-mediated neuroinflammation and Alzheimer’s disease. Cell Biosci. 2021, 11, 78. [Google Scholar] [CrossRef]
- Wang, Z.H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates delta-Secretase by Upregulating C/EBPbeta in Alzheimer’s Disease. Cell Rep. 2019, 28, 655–669.e5. [Google Scholar] [CrossRef] [Green Version]
- Numakawa, T.; Odaka, H. Brain-Derived Neurotrophic Factor Signaling in the Pathophysiology of Alzheimer’s Disease: Beneficial Effects of Flavonoids for Neuroprotection. Int. J. Mol. Sci. 2021, 22, 5719. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.P.; de Castro, A.A.; Soares, F.V.; da Cunha, E.F.F.; Ramalho, T.C. Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis. Molecules 2019, 24, 4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elangovan, S.; Holsinger, R.M.D. Cyclical amyloid beta-astrocyte activity induces oxidative stress in Alzheimer’s disease. Biochimie 2020, 171–172, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.T.; Wang, A.; To, E.; Gao, J.; Cao, S.; Cui, J.Z.; Matsubara, J.A. Vinpocetine inhibits amyloid-beta induced activation of NF-kappaB, NLRP3 inflammasome and cytokine production in retinal pigment epithelial cells. Exp. Eye Res. 2014, 127, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Grimaldi, A.; Pediconi, N.; Oieni, F.; Pizzarelli, R.; Rosito, M.; Giubettini, M.; Santini, T.; Limatola, C.; Ruocco, G.; Ragozzino, D.; et al. Neuroinflammatory Processes, A1 Astrocyte Activation and Protein Aggregation in the Retina of Alzheimer’s Disease Patients, Possible Biomarkers for Early Diagnosis. Front. Neurosci. 2019, 13, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Lian, H.; Zheng, H. Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J. Neurochem. 2016, 136, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Bolos, M.; Perea, J.R.; Avila, J. Alzheimer’s disease as an inflammatory disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. beta-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J. Neuroinflamm. 2020, 17, 280. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Lempriere, S. NLRP3 inflammasome activation implicated in tau pathology. Nat. Rev. Neurol. 2020, 16, 4. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 2006, 26, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Weisgraber, K.H.; Mucke, L.; Mahley, R.W. Apolipoprotein E: Diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 189–204. [Google Scholar] [CrossRef]
- Kockx, M.; Traini, M.; Kritharides, L. Cell-specific production, secretion, and function of apolipoprotein E. J. Mol. Med. 2018, 96, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Saura, J.; Petegnief, V.; Wu, X.; Liang, Y.; Paul, S.M. Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: Opposite effects of lipopolysaccharide. J. Neurochem. 2003, 85, 1455–1467. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017, 168, 427–441.e421. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.; Borchelt, D.R.; Chakrabarty, P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 8. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, C.M.; Fitz, N.F.; Nam, K.N.; Lefterov, I.; Koldamova, R. The Role of APOE and TREM2 in Alzheimer’s Disease—Current Understanding and Perspectives. Int. J. Mol. Sci. 2018, 20, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Fitz, N.F.; Nam, K.N.; Wolfe, C.M.; Letronne, F.; Playso, B.E.; Iordanova, B.E.; Kozai, T.D.Y.; Biedrzycki, R.J.; Kagan, V.E.; Tyurina, Y.Y.; et al. Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer’s disease. Nat. Commun. 2021, 12, 3416. [Google Scholar] [CrossRef] [PubMed]
- Ruganzu, J.B.; Zheng, Q.; Wu, X.; He, Y.; Peng, X.; Jin, H.; Zhou, J.; Ma, R.; Ji, S.; Ma, Y.; et al. TREM2 overexpression rescues cognitive deficits in APP/PS1 transgenic mice by reducing neuroinflammation via the JAK/STAT/SOCS signaling pathway. Exp. Neurol. 2021, 336, 113506. [Google Scholar] [CrossRef] [PubMed]
- Franzmeier, N.; Suarez-Calvet, M.; Frontzkowski, L.; Moore, A.; Hohman, T.J.; Morenas-Rodriguez, E.; Nuscher, B.; Shaw, L.; Trojanowski, J.Q.; Dichgans, M.; et al. Higher CSF sTREM2 attenuates ApoE4-related risk for cognitive decline and neurodegeneration. Mol. Neurodegener. 2020, 15, 57. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, Y.D.; Gao, Q.; Ou, Z.; Gong, P.Y.; Shi, J.Q.; Wu, L.; Zhou, J.S. TREM2 Ameliorates Neuronal Tau Pathology Through Suppression of Microglial Inflammatory Response. Inflammation 2018, 41, 811–823. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Tweedie, D.; Rachmany, L.; Rubovitch, V.; Li, Y.; Holloway, H.W.; Lehrmann, E.; Zhang, Y.; Becker, K.G.; Perez, E.; Hoffer, B.J.; et al. Blast traumatic brain injury-induced cognitive deficits are attenuated by preinjury or postinjury treatment with the glucagon-like peptide-1 receptor agonist, exendin-4. Alzheimers Dement. 2016, 12, 34–48. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Kam, T.I.; Lee, S.; Park, H.; Oh, Y.; Kwon, S.H.; Song, J.J.; Kim, D.; Kim, H.; Jhaldiyal, A.; et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 78. [Google Scholar] [CrossRef]
- Takeda, M.; Martinez, R.; Kudo, T.; Tanaka, T.; Okochi, M.; Tagami, S.; Morihara, T.; Hashimoto, R.; Cacabelos, R. Apolipoprotein E and central nervous system disorders: Reviews of clinical findings. Psychiatry Clin. Neurosci. 2010, 64, 592–607. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Holtzman, D.M. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011, 10, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Tai, L.M.; Youmans, K.L.; Jungbauer, L.; Yu, C.; Ladu, M.J. Introducing Human APOE into Abeta Transgenic Mouse Models. Int. J. Alzheimers Dis. 2011, 2011, 810981. [Google Scholar] [CrossRef] [Green Version]
- Yossef, R.R.; Al-Yamany, M.F.; Saad, M.A.; El-Sahar, A.E. Neuroprotective effects of vildagliptin on drug induced Alzheimer’s disease in rats with metabolic syndrome: Role of hippocampal klotho and AKT signaling pathways. Eur. J. Pharmacol. 2020, 889, 173612. [Google Scholar] [CrossRef]
- Wang, S.; Yao, H.; Xu, Y.; Hao, R.; Zhang, W.; Liu, H.; Huang, Y.; Guo, W.; Lu, B. Therapeutic potential of a TrkB agonistic antibody for Alzheimer’s disease. Theranostics 2020, 10, 6854–6874. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2012, 37, 434–444. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 578–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatziagapiou, K.; Kakouri, E.; Lambrou, G.I.; Bethanis, K.; Tarantilis, P.A. Antioxidant Properties of Crocus Sativus L. and Its Constituents and Relevance to Neurodegenerative Diseases; Focus on Alzheimer’s and Parkinson’s Disease. Curr. Neuropharmacol. 2019, 17, 377–402. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Shin, M.; Park, Y.; Choi, B.; Jang, S.; Lim, C.; Yun, H.S.; Lee, I.S.; Won, S.Y.; Cho, K.S. Linalool Alleviates Abeta42-Induced Neurodegeneration via Suppressing ROS Production and Inflammation in Fly and Rat Models of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2021, 2021, 8887716. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Lv, L.; Mao, S.; Dong, H.; Liu, B. Cognition Deficits in Parkinson’s Disease: Mechanisms and Treatment. Parkinsons Dis. 2020, 2020, 2076942. [Google Scholar] [CrossRef]
- Boecker, H.; Ceballos-Baumann, A.O.; Volk, D.; Conrad, B.; Forstl, H.; Haussermann, P. Metabolic alterations in patients with Parkinson disease and visual hallucinations. Arch. Neurol. 2007, 64, 984–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvatore, M.F.; McInnis, T.R.; Cantu, M.A.; Apple, D.M.; Pruett, B.S. Tyrosine Hydroxylase Inhibition in Substantia Nigra Decreases Movement Frequency. Mol. Neurobiol. 2019, 56, 2728–2740. [Google Scholar] [CrossRef]
- Palermo, G.; Ceravolo, R. Molecular Imaging of the Dopamine Transporter. Cells 2019, 8, 872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaarmann, A.; Gandhi, S.; Abramov, A.Y. Dopamine induces Ca2+ signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. J. Biol. Chem. 2010, 285, 25018–25023. [Google Scholar] [CrossRef] [Green Version]
- Zaichick, S.V.; McGrath, K.M.; Caraveo, G. The role of Ca(2+) signaling in Parkinson’s disease. Dis. Model. Mech. 2017, 10, 519–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaheer, A.; Zaheer, S.; Sahu, S.K.; Knight, S.; Khosravi, H.; Mathur, S.N.; Lim, R. A novel role of glia maturation factor: Induction of granulocyte-macrophage colony-stimulating factor and pro-inflammatory cytokines. J. Neurochem. 2007, 101, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, A.; Zaheer, S.; Sahu, S.K.; Yang, B.; Lim, R. Reduced severity of experimental autoimmune encephalomyelitis in GMF-deficient mice. Neurochem. Res. 2007, 32, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Fong, T.; Chen, X.; Chen, C.; Luo, P.; Xie, H. Glia maturation factor-beta: A potential therapeutic target in neurodegeneration and neuroinflammation. Neuropsychiatr. Dis. Treat. 2018, 14, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, H.; Thangavel, R.; Selvakumar, G.P.; Dubova, I.; Schwartz, N.; Ahmed, M.E.; Zaheer, S.; Kempuraj, D.; Iyer, S.; Zaheer, A.; et al. NLRP3 inflammasome and glia maturation factor coordinately regulate neuroinflammation and neuronal loss in MPTP mouse model of Parkinson’s disease. Int. Immunopharmacol. 2020, 83, 106441. [Google Scholar] [CrossRef]
- Joers, V.; Tansey, M.G.; Mulas, G.; Carta, A.R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 2017, 155, 57–75. [Google Scholar] [CrossRef]
- Ghosh, A.; Roy, A.; Liu, X.; Kordower, J.H.; Mufson, E.J.; Hartley, D.M.; Ghosh, S.; Mosley, R.L.; Gendelman, H.E.; Pahan, K. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18754–18759. [Google Scholar] [CrossRef] [Green Version]
- Ho, M.S. Microglia in Parkinson’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Fyfe, I. APOE(*)epsilon4 promotes synucleinopathy. Nat. Rev. Neurol. 2020, 16, 185. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Inman, C.E.; Wargel, Z.M.; Dube, U.; Freeberg, B.M.; Galluppi, A.; Haines, J.N.; Dhavale, D.D.; Miller, R.; Choudhury, F.A.; et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, S.; Nie, K.; Li, Y.; Gao, Y.; Gan, R.; Wang, L.; Li, B.; Sun, X.; Wang, L.; et al. TREM2 modulates microglia phenotypes in the neuroinflammation of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2018, 499, 797–802. [Google Scholar] [CrossRef]
- Jeong, S.H.; Chung, S.J.; Yoo, H.S.; Hong, N.; Jung, J.H.; Baik, K.; Lee, Y.H.; Sohn, Y.H.; Lee, P.H. Beneficial effects of dipeptidyl peptidase-4 inhibitors in diabetic Parkinson’s disease. Brain 2021, 144, 1127–1137. [Google Scholar] [CrossRef]
- Jamali-Raeufy, N.; Mojarrab, Z.; Baluchnejadmojarad, T.; Roghani, M.; Fahanik-Babaei, J.; Goudarzi, M. The effects simultaneous inhibition of dipeptidyl peptidase-4 and P2X7 purinoceptors in an in vivo Parkinson’s disease model. Metab. Brain. Dis. 2020, 35, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, D.; Newberg, A.B.; Cary, M.S.; Siderowf, A.D.; Moberg, P.J.; Kleiner-Fisman, G.; Duda, J.E.; Stern, M.B.; Mozley, D.; Katz, I.R. Striatal dopamine transporter imaging correlates with anxiety and depression symptoms in Parkinson’s disease. J. Nucl. Med. 2005, 46, 227–232. [Google Scholar]
- Martin, B.; Golden, E.; Carlson, O.D.; Pistell, P.; Zhou, J.; Kim, W.; Frank, B.P.; Thomas, S.; Chadwick, W.A.; Greig, N.H.; et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes 2009, 58, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Wang, V.; Kuo, T.T.; Huang, E.Y.; Ma, K.H.; Chou, Y.C.; Fu, Z.Y.; Lai, L.W.; Jung, J.; Choi, H.I.; Choi, D.S.; et al. Sustained Release GLP-1 Agonist PT320 Delays Disease Progression in a Mouse Model of Parkinson’s Disease. ACS Pharmacol. Transl. Sci. 2021, 4, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.H.; Chen, Y.; Li, F.F.; Wang, S.; Zhang, X.L.; Yuan, Y.H.; Chen, N.H. TLR4 deficiency has a protective effect in the MPTP/probenecid mouse model of Parkinson’s disease. Acta Pharmacol. Sin. 2019, 40, 1503–1512. [Google Scholar] [CrossRef]
- Zhang, Y.; Qin, L.; Xie, J.; Li, J.; Wang, C. Eupatilin prevents behavioral deficits and dopaminergic neuron degeneration in a Parkinson’s disease mouse model. Life Sci. 2020, 253, 117745. [Google Scholar] [CrossRef]
- Song, S.Y.; Kim, I.S.; Koppula, S.; Park, J.Y.; Kim, B.W.; Yoon, S.H.; Choi, D.K. 2-Hydroxy-4-Methylbenzoic Anhydride Inhibits Neuroinflammation in Cellular and Experimental Animal Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8195. [Google Scholar] [CrossRef]
- George, S.; Mok, S.S.; Nurjono, M.; Ayton, S.; Finkelstein, D.I.; Masters, C.L.; Li, Q.X.; Culvenor, J.G. alpha-Synuclein transgenic mice reveal compensatory increases in Parkinson’s disease-associated proteins DJ-1 and parkin and have enhanced alpha-synuclein and PINK1 levels after rotenone treatment. J. Mol. Neurosci. 2010, 42, 243–254. [Google Scholar] [CrossRef]
- Jin, W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson’s Disease. J. Clin. Med. 2020, 9, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, D.; Shi, Y.; Wang, J.; Lin, Q.; Sun, Y.; Ye, K.; Yan, Q.; Zhang, H. 7,8-dihydroxyflavone protects 6-OHDA and MPTP induced dopaminergic neurons degeneration through activation of TrkB in rodents. Neurosci. Lett. 2016, 620, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Ajitkumar, A.; De Jesus, O. Huntington Disease; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Bano, D.; Zanetti, F.; Mende, Y.; Nicotera, P. Neurodegenerative processes in Huntington’s disease. Cell Death Dis. 2011, 2, e228. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.H.; Fathy, N.; Kortam, M.A.; Rabie, M.A.; Mohamed, A.F.; Kamel, A.S. Vildagliptin Attenuates Huntington’s Disease through Activation of GLP-1 Receptor/PI3K/Akt/BDNF Pathway in 3-Nitropropionic Acid Rat Model. Neurotherapeutics 2020, 17, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Faideau, M.; Kim, J.; Cormier, K.; Gilmore, R.; Welch, M.; Auregan, G.; Dufour, N.; Guillermier, M.; Brouillet, E.; Hantraye, P.; et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: A correlation with Huntington’s disease subjects. Hum. Mol. Genet. 2010, 19, 3053–3067. [Google Scholar] [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [Green Version]
- Bjorkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [Green Version]
- Connolly, C.; Magnusson-Lind, A.; Lu, G.; Wagner, P.K.; Southwell, A.L.; Hayden, M.R.; Bjorkqvist, M.; Leavitt, B.R. Enhanced immune response to MMP3 stimulation in microglia expressing mutant huntingtin. Neuroscience 2016, 325, 74–88. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Garcia-Martinez, V.; Poejo, J.; Garcia-Lopez, V.; Salazar, J.; Gutierrez-Merino, C. Early Reactive A1 Astrocytes Induction by the Neurotoxin 3-Nitropropionic Acid in Rat Brain. Int. J. Mol. Sci. 2020, 21, 3609. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; St-Pierre, M.K.; Carrier, M.; El Hajj, H.; Novak, S.W.; Sanchez, M.G.; Cicchetti, F.; Tremblay, M.E. Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J. Neuroinflamm. 2020, 17, 98. [Google Scholar] [CrossRef] [PubMed]
- Crapser, J.D.; Ochaba, J.; Soni, N.; Reidling, J.C.; Thompson, L.M.; Green, K.N. Microglial depletion prevents extracellular matrix changes and striatal volume reduction in a model of Huntington’s disease. Brain 2020, 143, 266–288. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Chen, W.Y.; Chen, S.D. The Emerging Role of GLP-1 Receptors in DNA Repair: Implications in Neurological Disorders. Int. J. Mol. Sci. 2017, 18, 1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.; Kwatra, M.; Gawali, B.; Panda, S.R.; Naidu, V.G.M. Potential role of TrkB agonist in neuronal survival by promoting CREB/BDNF and PI3K/Akt signaling in vitro and in vivo model of 3-nitropropionic acid (3-NP)-induced neuronal death. Apoptosis 2021, 26, 52–70. [Google Scholar] [CrossRef]
- Yang, X.; Chu, S.F.; Wang, Z.Z.; Li, F.F.; Yuan, Y.H.; Chen, N.H. Ginsenoside Rg1 exerts neuroprotective effects in 3-nitropronpionic acid-induced mouse model of Huntington’s disease via suppressing MAPKs and NF-kappaB pathways in the striatum. Acta Pharmacol. Sin. 2021, 42, 1409–1421. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Diseases | Targeting | Agent | Reference | |
|---|---|---|---|---|
| Epilepsy | GLP-1R | Agonist | Liraglutide | [19] |
| TRPV4 | Antagonist | HC-067047 | [20] | |
| CX3CR1 | Antibody | Anti-CX3CR1 antibody | [21] | |
| AD | DPP-4 | Inhibitor | Saxagliptin | [22] |
| Linagliptin | [23] | |||
| GLP-1R | Agonist | Exendin-4 | [24] | |
| NLY01 | [25] | |||
| TREM2 | Agonist | AL002c | [26] | |
| TrкB | Agonist | AS86 | [27] | |
| 7,8-dihydroxyflavone | [28] | |||
| PD | DPP-4 | Inhibitor | Saxagliptin | [29,30] |
| Linagliptin | [31] | |||
| GLP-1R | Agonist | NLY01 | [32] | |
| PT320 | [33] | |||
| TrкB | Agonist | 7,8-dihydroxyflavone | [34,35] | |
| HD | DPP-4 | Inhibitor | Vildagliptin | [36] |
| GLP-1R | Agonist | Exendin-4 | [30] | |
| TrкB | Agonist | 7,8-dihydroxyflavone | [36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-W.; Chun, W.; Lee, H.J.; Kim, S.-M.; Min, J.-H.; Kim, D.-Y.; Kim, M.-O.; Ryu, H.W.; Lee, S.U. The Role of Microglia in the Development of Neurodegenerative Diseases. Biomedicines 2021, 9, 1449. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101449
Lee J-W, Chun W, Lee HJ, Kim S-M, Min J-H, Kim D-Y, Kim M-O, Ryu HW, Lee SU. The Role of Microglia in the Development of Neurodegenerative Diseases. Biomedicines. 2021; 9(10):1449. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101449
Chicago/Turabian StyleLee, Jae-Won, Wanjoo Chun, Hee Jae Lee, Seong-Man Kim, Jae-Hong Min, Doo-Young Kim, Mun-Ock Kim, Hyung Won Ryu, and Su Ui Lee. 2021. "The Role of Microglia in the Development of Neurodegenerative Diseases" Biomedicines 9, no. 10: 1449. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9101449