
CX-4945 and siRNA-Mediated Knockdown of CK2 Improves Cisplatin Response in HPV(+) and HPV(−) HNSCC Cell Lines

 ,
, 
Abstract
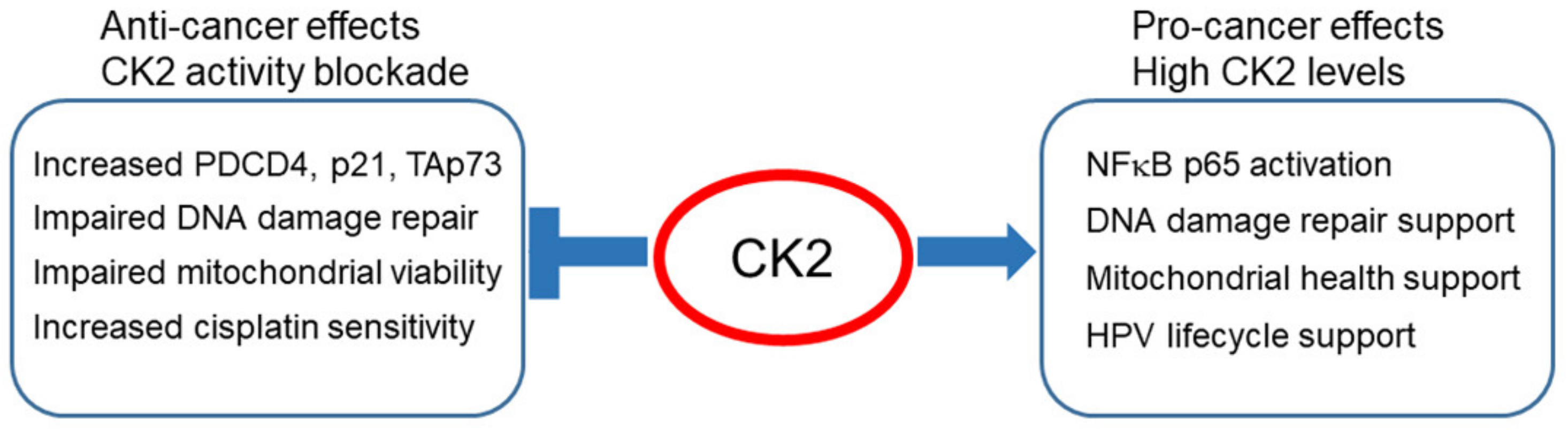
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture, and Drugs
2.2. siRNA Transfections
2.3. Combination Treatments and Viability Assays
2.4. Cell Treatments and Immunoblot Analysis
2.5. Statistical Analysis
3. Results
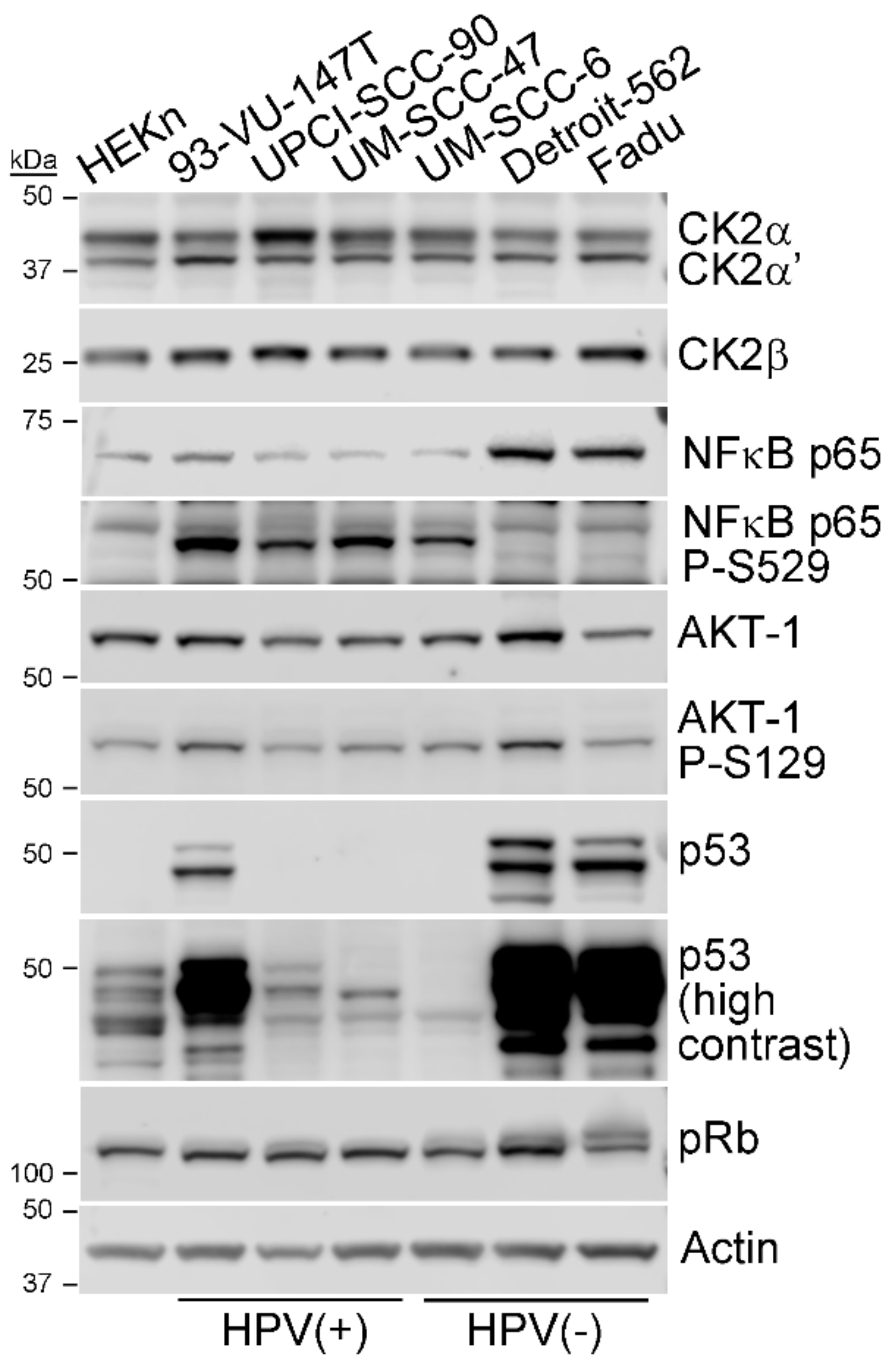
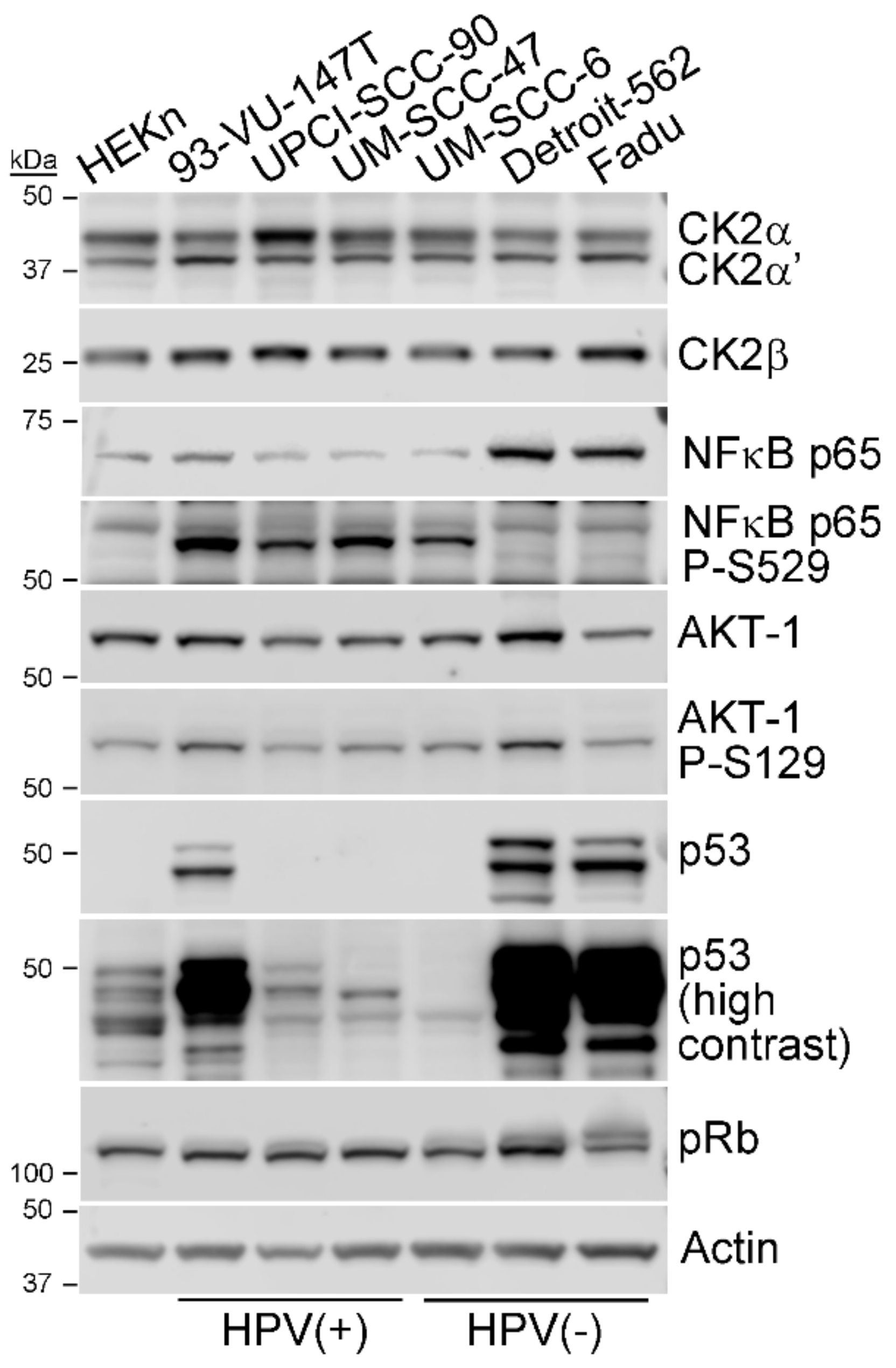
3.1. CK2 Expression and Activity in HPV+ and HPV- HNSCC Cell Lines
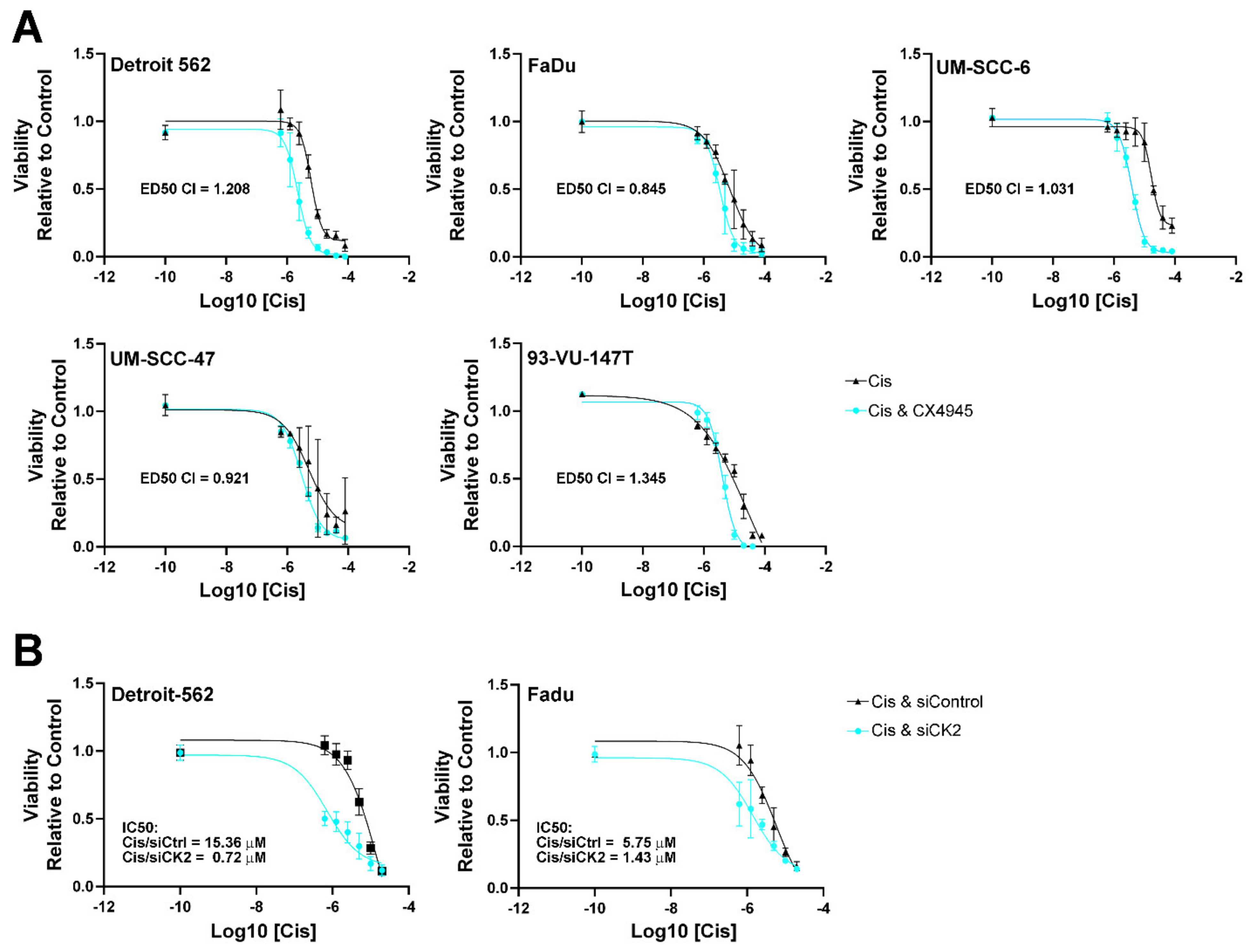
3.2. HNSCC Cell Viability Following Cisplatin Treatment and Reduced CK2 Activity or Expression
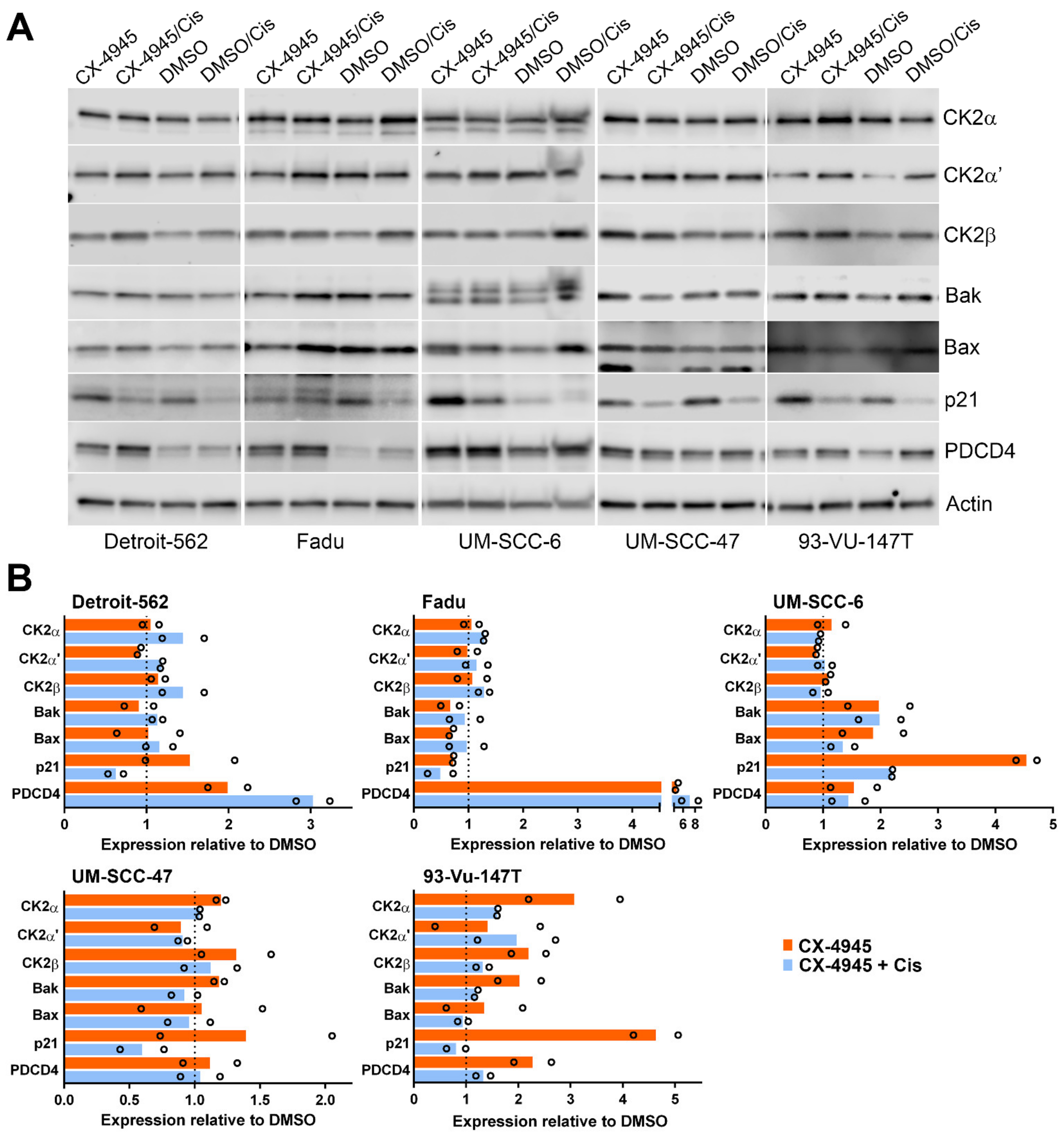
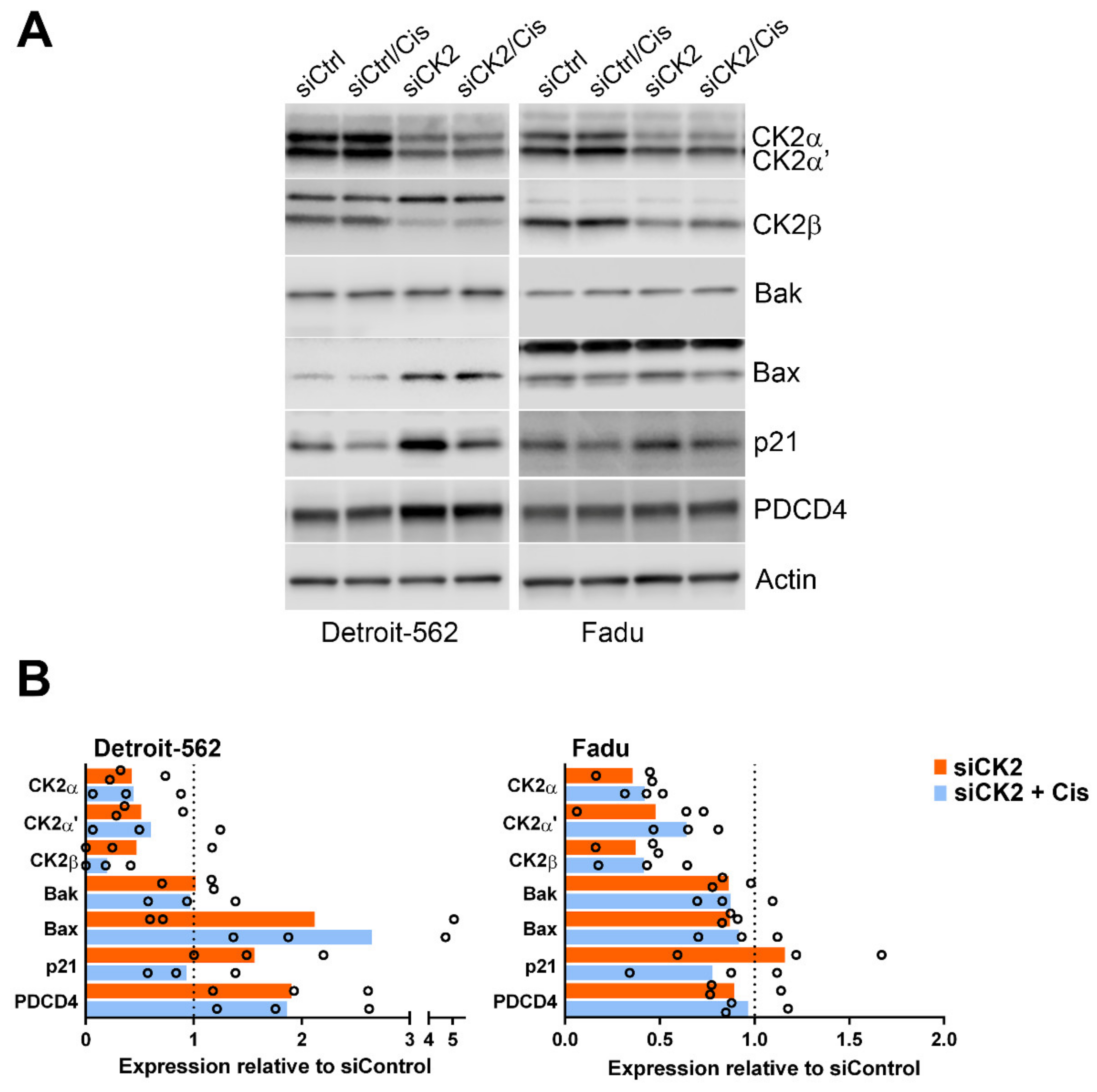
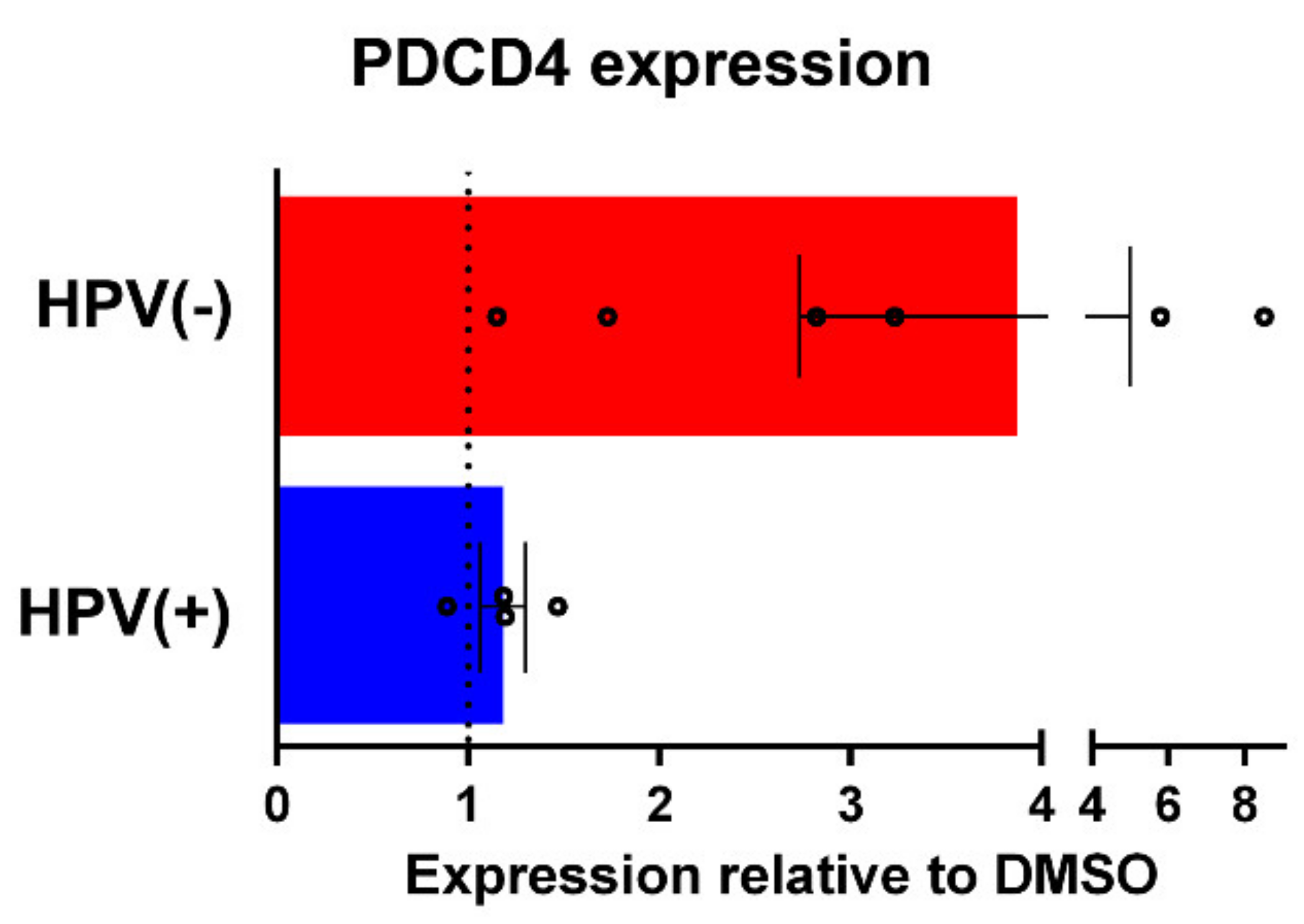
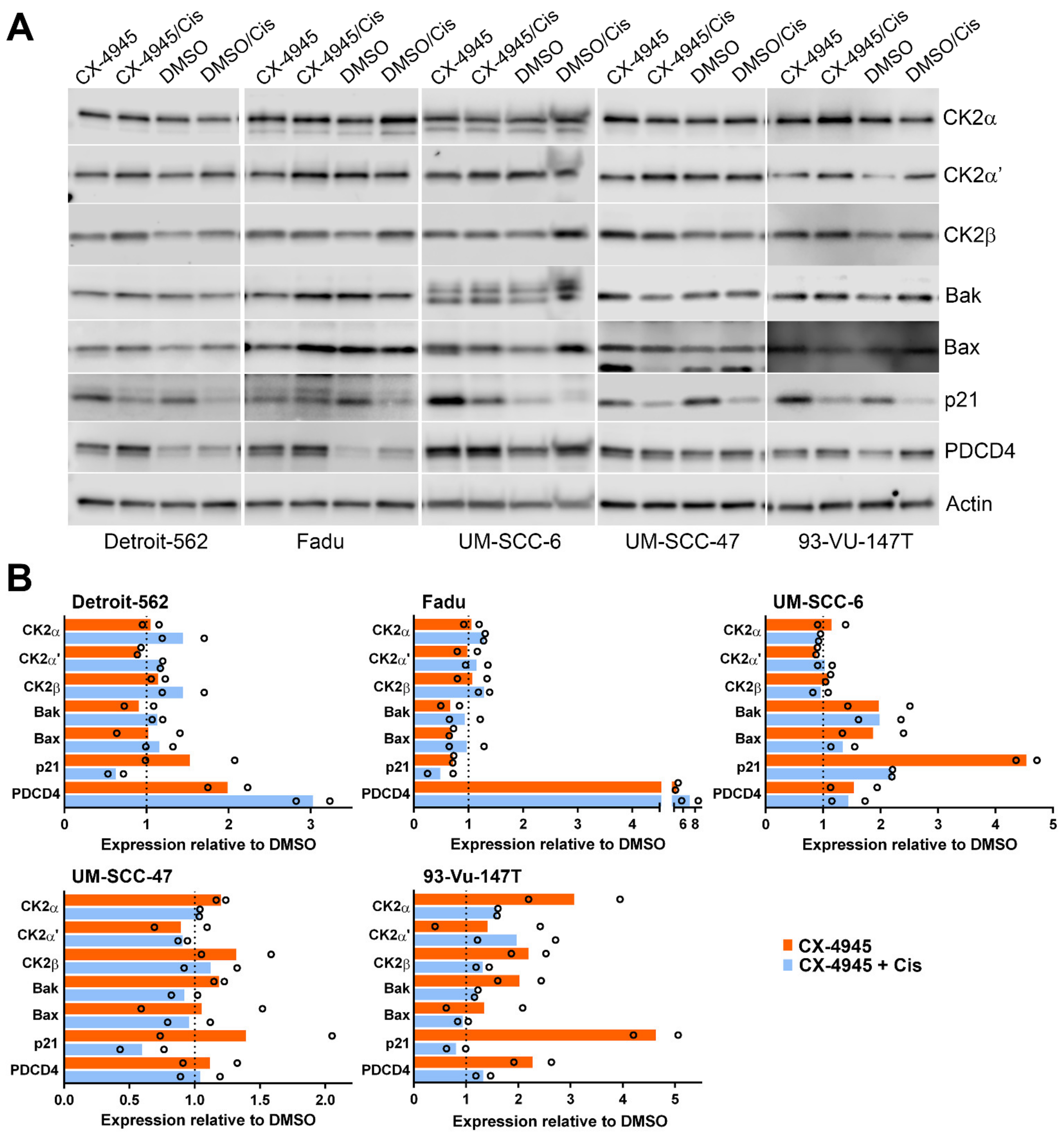
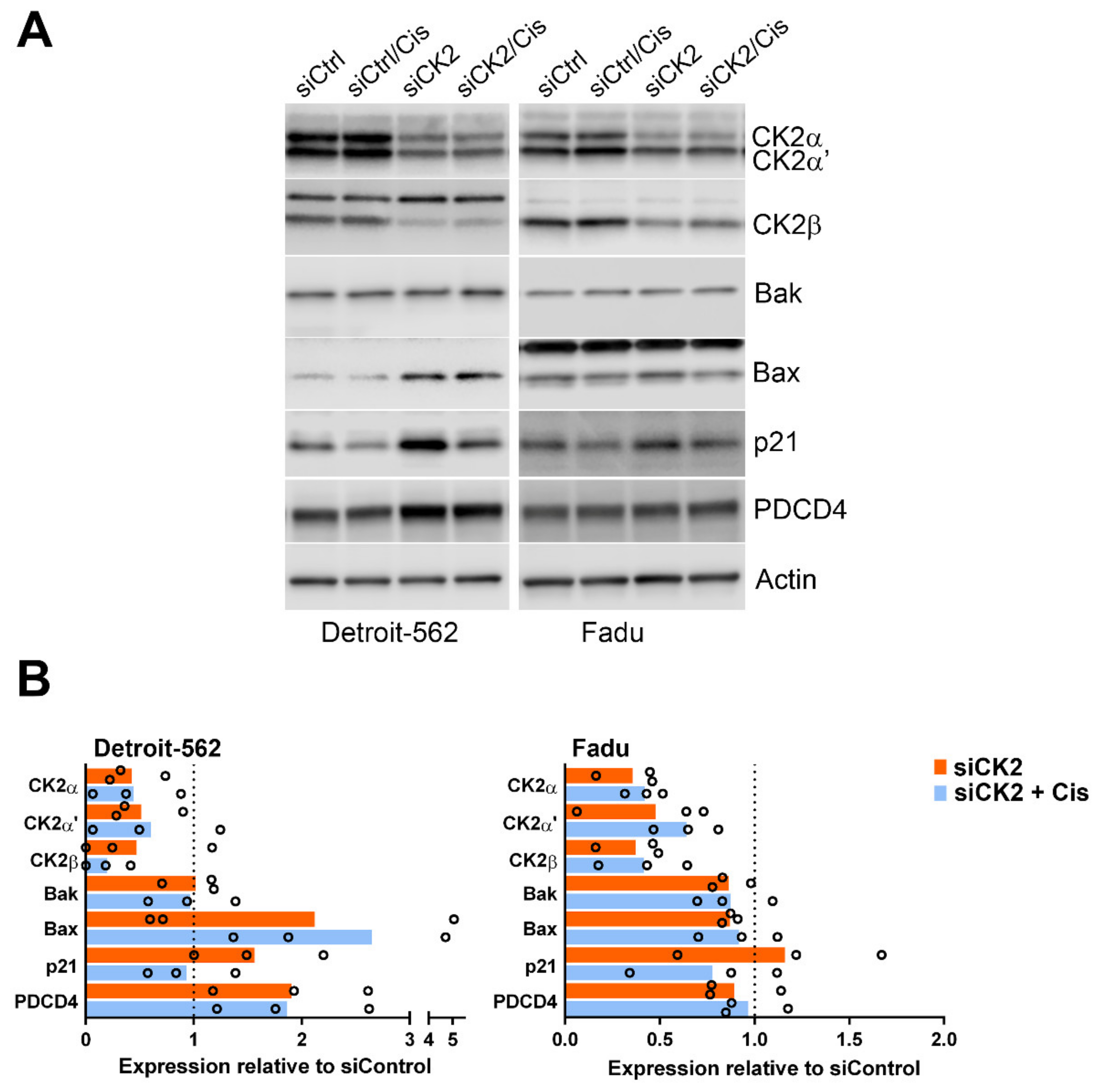
3.3. Signaling Response of HNSCC cells to CK2 Targeting Using CX-4945 or siRNA and Cisplatin Treatment
4. Discussion
Future Directions and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Silva-Pavez, E.; Tapia, J.C. Protein Kinase CK2 in Cancer Energetics. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, C.; Salizzato, V.; Borgo, C.; Cesaro, L.; Pinna, L.A.; Salvi, M. A Journey through the Cytoskeleton with Protein Kinase CK2. Curr. Protein Pept. Sci. 2019, 20, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Castello, J.; Ragnauth, A.; Friedman, E.; Rebholz, H. CK2-An Emerging Target for Neurological and Psychiatric Disorders. Pharmaceuticals 2017, 10, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuñez de Villavicencio-Diaz, T.; Rabalski, A.J.; Litchfield, D.W. Protein Kinase CK2: Intricate Relationships within Regulatory Cellular Networks. Pharmaceuticals 2017, 10, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Faust, M.; Montenarh, M. Subcellular localization of protein kinase CK2. A key to its function? Cell Tissue Res. 2000, 301, 329–340. [Google Scholar] [CrossRef]
- Ahmed, K.; Gerber, D.A.; Cochet, C. Joining the cell survival squad: An emerging role for protein kinase CK2. Trends Cell Biol. 2002, 12, 226–230. [Google Scholar] [CrossRef]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. CK2: A key player in cancer biology. Cell Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- Chua, M.M.J.; Lee, M.; Dominguez, I. Cancer-type dependent expression of CK2 transcripts. PLoS ONE 2017, 12, e0188854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gapany, M.; Faust, R.A.; Tawfic, S.; Davis, A.; Adams, G.L.; Ahmed, K. Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol. Med. 1995, 1, 659–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, R.A.; Gapany, M.; Tristani, P.; Davis, A.; Adams, G.L.; Ahmed, K. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: Association with malignant transformation. Cancer Lett. 1996, 101, 31–35. [Google Scholar] [CrossRef]
- Pfister, D.G.; Fury, M.G. New chapter in our understanding of human papillomavirus-related head and neck cancer. J. Clin. Oncol. 2014, 32, 3349–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhry, C.; Zhang, Q.; Nguyen-Tan, P.F.; Rosenthal, D.; El-Naggar, A.; Garden, A.S.; Soulieres, D.; Trotti, A.; Avizonis, V.; Ridge, J.A.; et al. Human Papillomavirus and Overall Survival After Progression of Oropharyngeal Squamous Cell Carcinoma. J. Clin. Oncol. 2014, 32, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Cesaro, L.; Sarno, S.; Pinna, L.A.; Ruzzene, M.; Salvi, M. How can a traffic light properly work if it is always green? The paradox of CK2 signaling. Crit. Rev. Biochem. Mol. Biol. 2021, 1–39. [Google Scholar] [CrossRef]
- Firzlaff, J.M.; Galloway, D.A.; Eisenman, R.N.; Luscher, B. The E7 protein of human papillomavirus type 16 is phosphorylated by casein kinase II. New Biol. 1989, 1, 44–53. [Google Scholar]
- Basukala, O.; Mittal, S.; Massimi, P.; Bestagno, M.; Banks, L. The HPV-18 E7 CKII phospho acceptor site is required for maintaining the transformed phenotype of cervical tumour-derived cells. PLoS Pathog. 2019, 15, e1007769. [Google Scholar] [CrossRef]
- Zine El Abidine, A.; Tomaić, V.; Bel Haj Rhouma, R.; Massimi, P.; Guizani, I.; Boubaker, S.; Ennaifer, E.; Banks, L. A naturally occurring variant of HPV-16 E7 exerts increased transforming activity through acquisition of an additional phospho-acceptor site. Virology 2017, 500, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Piirsoo, A.; Piirsoo, M.; Kala, M.; Sankovski, E.; Lototskaja, E.; Levin, V.; Salvi, M.; Ustav, M. Activity of CK2α protein kinase is required for efficient replication of some HPV types. PLoS Pathog. 2019, 15, e1007788. [Google Scholar] [CrossRef]
- Rampias, T.; Sasaki, C.; Weinberger, P.; Psyrri, A. E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. J. Natl. Cancer Inst. 2009, 101, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Y.; Nin, D.S.; Lee, A.Y.; Simanski, S.; Kodadek, T.; Chiang, C.M. BRD4 Phosphorylation Regulates HPV E2-Mediated Viral Transcription, Origin Replication, and Cellular MMP-9 Expression. Cell Rep. 2016, 16, 1733–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Y.; Lee, A.Y.; Lai, H.T.; Zhang, H.; Chiang, C.M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trembley, J.H.; Unger, G.M.; Korman, V.L.; Tobolt, D.K.; Kazimierczuk, Z.; Pinna, L.A.; Kren, B.T.; Ahmed, K. Nanoencapsulated anti-CK2 small molecule drug or siRNA specifically targets malignant cancer but not benign cells. Cancer Lett. 2012, 315, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, K.; Unger, G.M.; Kren, B.T.; Trembley, J.H. Targeting CK2 for cancer therapy using a nanomedicine approach. In Protein Kinase CK2 in Cellular Function in Normal and Disease States; Ahmed, K., Issinger, O.-G., Szyszka, R., Dhalla, N.S., Eds.; Advances in Biochemistry in Health and Disease; Springer: Cham, Switzerland, 2015; Volume 12, pp. 299–315. [Google Scholar]
- Qaiser, F.; Trembley, J.H.; Kren, B.T.; Wu, J.J.; Naveed, A.K.; Ahmed, K. Protein Kinase CK2 Inhibition Induces Cell Death via Early Impact on Mitochondrial Function. J. Cell Biochem. 2014, 115, 2103–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trembley, J.H.; Unger, G.M.; Tobolt, D.K.; Korman, V.L.; Wang, G.; Ahmad, K.A.; Slaton, J.W.; Kren, B.T.; Ahmed, K. Systemic administration of antisense oligonucleotides simultaneously targeting CK2alpha and alpha’ subunits reduces orthotopic xenograft prostate tumors in mice. Mol. Cell Biochem. 2011, 356, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Preclinical versus clinical drug combination studies. Leuk Lymphoma 2008, 49, 2059–2080. [Google Scholar] [CrossRef]
- Ponce, D.P.; Maturana, J.L.; Cabello, P.; Yefi, R.; Niechi, I.; Silva, E.; Armisen, R.; Galindo, M.; Antonelli, M.; Tapia, J.C. Phosphorylation of AKT/PKB by CK2 is necessary for the AKT-dependent up-regulation of beta-catenin transcriptional activity. J. Cell. Physiol. 2011, 226, 1953–1959. [Google Scholar] [CrossRef]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S. Tumor necrosis factor α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Trotti, A.M.; Harris, J.; Eisbruch, A.; Harari, P.M.; Adelstein, D.J.; Jordan, R.C.K.; Zhao, W.; Sturgis, E.M.; Burtness, B.; et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): A randomised, multicentre, non-inferiority trial. Lancet 2019, 393, 40–50. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Eckhardt, M.; Zhang, W.; Gross, A.M.; Von Dollen, J.; Johnson, J.R.; Franks-Skiba, K.E.; Swaney, D.L.; Johnson, T.L.; Jang, G.M.; Shah, P.S.; et al. Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus-Host Protein Network. Cancer Discov. 2018, 8, 1474–1489. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, M.O.; Hosek, T.; Calcada, E.O.; Castiglia, F.; Massimi, P.; Banks, L.; Felli, I.C.; Pierattelli, R. Monitoring HPV-16 E7 phosphorylation events. Virology 2017, 503, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.M.; Parker, J.N.; Schmidt-Grimminger, D.C.; Broker, T.R.; Chow, L.T. Casein kinase II phosphorylation of the human papillomavirus-18 E7 protein is critical for promoting S-phase entry. Cell Growth Differ. 2000, 11, 425–435. [Google Scholar]
- Genovese, N.J.; Banerjee, N.S.; Broker, T.R.; Chow, L.T. Casein kinase II motif-dependent phosphorylation of human papillomavirus E7 protein promotes p130 degradation and S-phase induction in differentiated human keratinocytes. J. Virol. 2008, 82, 4862–4873. [Google Scholar] [CrossRef] [Green Version]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e619. [Google Scholar] [CrossRef]
- Sarduy, M.R.; Garcia, I.; Coca, M.A.; Perera, A.; Torres, L.A.; Valenzuela, C.M.; Baladron, I.; Solares, M.; Reyes, V.; Hernandez, I.; et al. Optimizing CIGB-300 intralesional delivery in locally advanced cervical cancer. Br. J. Cancer 2015, 112, 1636–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solares, A.M.; Santana, A.; Baladron, I.; Valenzuela, C.; Gonzalez, C.A.; Diaz, A.; Castillo, D.; Ramos, T.; Gomez, R.; Alonso, D.F.; et al. Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer 2009, 9, 146. [Google Scholar] [CrossRef] [Green Version]
- Perea, S.E.; Reyes, O.; Puchades, Y.; Mendoza, O.; Vispo, N.S.; Torrens, I.; Santos, A.; Silva, R.; Acevedo, B.; Lopez, E.; et al. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2). Cancer Res. 2004, 64, 7127–7129. [Google Scholar] [CrossRef] [Green Version]
- Perera, Y.; Toro, N.D.; Gorovaya, L.; Fernandez, D.E.C.J.; Farina, H.G.; Perea, S.E. Synergistic interactions of the anti-casein kinase 2 CIGB-300 peptide and chemotherapeutic agents in lung and cervical preclinical cancer models. Mol. Clin. Oncol. 2014, 2, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Sancho-Martinez, S.M.; Prieto-Garcia, L.; Prieto, M.; Lopez-Novoa, J.M.; Lopez-Hernandez, F.J. Subcellular targets of cisplatin cytotoxicity: An integrated view. Pharmacol. Ther. 2012, 136, 35–55. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- Afzal, M.; Kren, B.T.; Naveed, A.K.; Trembley, J.H.; Ahmed, K. Protein kinase CK2 impact on intracellular calcium homeostasis in prostate cancer. Mol. Cell. Biochem. 2020, 470, 131–143. [Google Scholar] [CrossRef]
- Yata, K.; Lloyd, J.; Maslen, S.; Bleuyard, J.Y.; Skehel, M.; Smerdon, S.J.; Esashi, F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell 2012, 45, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Wang, J.; Hu, Y.; Zhao, H.; Hou, W.; Zhao, H.; Wang, H.; Liao, J.; Xu, X. Modulation of LSD1 phosphorylation by CK2/WIP1 regulates RNF168-dependent 53BP1 recruitment in response to DNA damage. Nucleic Acids Res. 2015, 43, 5936–5947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herhaus, L.; Perez-Oliva, A.B.; Cozza, G.; Gourlay, R.; Weidlich, S.; Campbell, D.G.; Pinna, L.A.; Sapkota, G.P. Casein kinase 2 (CK2) phosphorylates the deubiquitylase OTUB1 at Ser16 to trigger its nuclear localization. Sci. Signal. 2015, 8, ra35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.S.; Diallo, O.T.; Hu, M.; Ehsanian, R.; Yang, X.; Arun, P.; Lu, H.; Korman, V.; Unger, G.; Ahmed, K.; et al. CK2 Modulation of NF-κB, TP53, and the Malignant Phenotype in Head and Neck Cancer by Anti-CK2 Oligonucleotides In vitro or In vivo via Sub–50-nm Nanocapsules. Clin. Cancer Res. 2010, 16, 2295–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui-Jain, A.; Bliesath, J.; Macalino, D.; Omori, M.; Huser, N.; Streiner, N.; Ho, C.B.; Anderes, K.; Proffitt, C.; O’Brien, S.E.; et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: Mechanistic rationale for drug combination therapy. Mol. Cancer Ther. 2012, 11, 994–1005. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Yang, B.; Shi, S.; Jiang, X. RNA interference (RNAi) mediated stable knockdown of protein casein kinase 2-alpha (CK2α) inhibits migration and invasion and enhances cisplatin-induced apoptosis in HEp-2 laryngeal carcinoma cells. Acta Histochem. 2014, 116, 1000–1006. [Google Scholar] [CrossRef]
- So, K.S.; Rho, J.K.; Choi, Y.J.; Kim, S.Y.; Choi, C.M.; Chun, Y.J.; Lee, J.C. AKT/mTOR Down-regulation by CX-4945, a CK2 Inhibitor, Promotes Apoptosis in Chemorefractory Non-small Cell Lung Cancer Cells. Anticancer Res. 2015, 35, 1537–1542. [Google Scholar]
- Dutta, A.; Eckelmann, B.; Adhikari, S.; Ahmed, K.M.; Sengupta, S.; Pandey, A.; Hegde, P.M.; Tsai, M.-S.; Tainer, J.A.; Weinfeld, M.; et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Q.; Johnson, C.L.; Kumar, A.; Molkentine, D.P.; Molkentine, J.M.; Rabin, T.; Mason, K.A.; Milas, L.; Raju, U. Inhibition of P-TEFb by DRB Suppresses SIRT1/CK2alpha Pathway and Enhances Radiosensitivity of Human Cancer Cells. Anticancer Res. 2014, 34, 6981–6989. [Google Scholar]
- Olsen, B.B.; Wang, S.Y.; Svenstrup, T.H.; Chen, B.P.; Guerra, B. Protein kinase CK2 localizes to sites of DNA double-strand break regulating the cellular response to DNA damage. BMC Mol. Biol. 2012, 13, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.L.; Dianova, I.I.; Finch, D.; Tait, P.S.; Strom, C.E.; Helleday, T.; Dianov, G.L. XRCC1 phosphorylation by CK2 is required for its stability and efficient DNA repair. DNA Repair 2010. [Google Scholar] [CrossRef]
- Kang, H.; Jung, J.W.; Kim, M.K.; Chung, J.H. CK2 Is the Regulator of SIRT1 Substrate-Binding Affinity, Deacetylase Activity and Cellular Response to DNA-Damage. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Yamane, K.; Kinsella, T.J. CK2 inhibits apoptosis and changes its cellular localization following ionizing radiation. Cancer Res. 2005, 65, 4362–4367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montenarh, M. Protein kinase CK2 in DNA damage and repair. Transl. Cancer Res. 2016, 5, 49–63. [Google Scholar] [CrossRef]
- Jin, C.; Song, P.; Pang, J. The CK2 inhibitor CX4945 reverses cisplatin resistance in the A549/DDP human lung adenocarcinoma cell line. Oncol. Lett. 2019, 18, 3845–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Yao, J.; Li, B.; Shao, G.; Cui, Y. Inhibition of protein kinase CK2 sensitizes non-small cell lung cancer cells to cisplatin via upregulation of PML. Mol. Cell Biochem. 2017, 436, 87–97. [Google Scholar] [CrossRef]
- Xu, W.; Chen, Q.; Wang, Q.; Sun, Y.; Wang, S.; Li, A.; Xu, S.; Roe, O.D.; Wang, M.; Zhang, R.; et al. JWA reverses cisplatin resistance via the CK2-XRCC1 pathway in human gastric cancer cells. Cell Death Dis. 2014, 5, e1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Pei, S.; Wang, X.; Zhu, Q.; Gou, S. Emerging JWA-targeted Pt(IV) prodrugs conjugated with CX-4945 to overcome chemo-immune-resistance. Biochem. Biophys. Res. Commun. 2020, 521, 753–761. [Google Scholar] [CrossRef]
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; van Waes, C.; Ahmed, K. Emergence of protein kinase CK2 as a key target in cancer therapy. BioFactors 2010, 36, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Sewell, A.; Brown, B.; Biktasova, A.; Mills, G.B.; Lu, Y.; Tyson, D.R.; Issaeva, N.; Yarbrough, W.G. Reverse-phase protein array profiling of oropharyngeal cancer and significance of PIK3CA mutations in HPV-associated head and neck cancer. Clin. Cancer Res. 2014, 20, 2300–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, I.; Sonenshein, G.E.; Seldin, D.C. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: Linking development and cancer. Cell. Mol. Life Sci. 2009, 66, 1850–1857. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, H.; Chen, J.; Wang, R.; Saleh, A.; Si, H.; Lee, S.; Guven-Maiorov, E.; Keskin, O.; Gursoy, A.; et al. Head and Neck Cancers Promote an Inflammatory Transcriptome through Coactivation of Classic and Alternative NF-κB Pathways. Cancer Immunol. Res. 2019, 7, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yan, C.; Quan, X.X.; Yang, X.; Zhang, J.; Bian, Y.; Chen, Z.; van Waes, C. CK2 Phosphorylates and Inhibits TAp73 Tumor Suppressor Function to Promote Expression of Cancer Stem Cell Genes and Phenotype in Head and Neck Cancer. Neoplasia 2014, 16, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cmarik, J.L.; Min, H.; Hegamyer, G.; Zhan, S.; Kulesz-Martin, M.; Yoshinaga, H.; Matsuhashi, S.; Colburn, N.H. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc. Natl. Acad. Sci. USA 1999, 96, 14037–14042. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yang, H.S. The role of Pdcd4 in tumour suppression and protein translation. Biol. Cell 2018. [Google Scholar] [CrossRef]
- Matsuhashi, S.; Manirujjaman, M.; Hamajima, H.; Ozaki, I. Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions. Int. J. Mol. Sci. 2019, 20, 2304. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gee, H.; Rose, B.; Lee, C.S.; Clark, J.; Elliott, M.; Gamble, J.R.; Cairns, M.J.; Harris, A.; Khoury, S.; et al. Regulation of the tumour suppressor PDCD4 by miR-499 and miR-21 in oropharyngeal cancers. BMC Cancer 2016, 16, 86. [Google Scholar] [CrossRef]
- Sun, Z.; Li, S.; Kaufmann, A.M.; Albers, A.E. miR-21 increases the programmed cell death 4 gene-regulated cell proliferation in head and neck squamous carcinoma cell lines. Oncol. Rep. 2014, 32, 2283–2289. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Wang, X.; Gao, L.; Li, S.; Yan, X.; Zhang, J.; Huang, C.; Zhang, Y.; Zhi, K. MiR-21 modulates chemosensitivity of tongue squamous cell carcinoma cells to cisplatin by targeting PDCD4. Mol. Cell Biochem. 2014, 390, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Ye, M.; Wang, C.; Cheng, K.; Song, C.; Dong, M.; Pan, Y.; Qin, H.; Zou, H. Global screening of CK2 kinase substrates by an integrated phosphoproteomics workflow. Sci. Rep. 2013, 3, 3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, M.K.; Shibata, T.; Suzuki, T.; Ogura, M.; Kozuka-Hata, H.; Oyama, M.; Homma, Y. Role for Protein Kinase CK2 on Cell Proliferation: Assessing CK2 Complex Components in the Nucleus During the Cell Cycle Progression. In Protein Kinase CK2 Cellular Function in Normal and Disease States; Ahmed, K., Issinger, O.-G., Szyszka, R., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 197–226. [Google Scholar]
- Parveen, A.; Akash, M.S.; Rehman, K.; Kyunn, W.W. Dual Role of p21 in the Progression of Cancer and Its Treatment. Crit. Rev. Eukaryot Gene Expr. 2016, 26, 49–62. [Google Scholar] [CrossRef]
- Robinson, A.M.; Rathore, R.; Redlich, N.J.; Adkins, D.R.; VanArsdale, T.; van Tine, B.A.; Michel, L.S. Cisplatin exposure causes c-Myc-dependent resistance to CDK4/6 inhibition in HPV-negative head and neck squamous cell carcinoma. Cell Death Dis. 2019, 10, 867. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [Green Version]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef]
- Ahmed, K.; Kren, B.T.; Abedin, M.J.; Vogel, R.I.; Shaughnessy, D.P.; Nacusi, L.; Korman, V.L.; Li, Y.; Dehm, S.M.; Zimmerman, C.L.; et al. CK2 targeted RNAi therapeutic delivered via malignant cell-directed tenfibgen nanocapsule: Dose and molecular mechanisms of response in xenograft prostate tumors. Oncotarget 2016, 7, 61789–61805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, C.M.; Trembley, J.H.; Kren, B.T.; Unger, G.M.; O’Sullivan, M.G.; Cornax, I.; Modiano, J.F.; Ahmed, K. Therapeutic Targeting of Protein Kinase CK2 Gene Expression in Feline Oral Squamous Cell Carcinoma: A Naturally Occurring Large-Animal Model of Head and Neck Cancer. Hum. Gene Ther. Clin. Dev. 2017, 28, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Kren, B.; Unger, G.; Abedin, M.; Vogel, R.; Henzler, C.; Ahmed, K.; Trembley, J. Preclinical evaluation of cyclin dependent kinase 11 and casein kinase 2 survival kinases as RNA interference targets for triple negative breast cancer therapy. Breast Cancer Res. 2015, 17, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, G.M.; Kren, B.T.; Korman, V.L.; Kimbrough, T.G.; Vogel, R.I.; Ondrey, F.G.; Trembley, J.H.; Ahmed, K. Mechanism and efficacy of sub-50-nm tenfibgen nanocapsules for cancer cell-directed delivery of anti-CK2 RNAi to primary and metastatic squamous cell carcinoma. Mol. Cancer Ther. 2014, 13, 2018–2029. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Setoguchi, T.; Tsuru, A.; Saitoh, Y.; Nagano, S.; Ishidou, Y.; Maeda, S.; Furukawa, T.; Komiya, S. Inhibition of casein kinase 2 prevents growth of human osteosarcoma. Oncol. Rep. 2017, 37, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.B.; Svenstrup, T.H.; Guerra, B. Downregulation of protein kinase CK2 induces autophagic cell death through modulation of the mTOR and MAPK signaling pathways in human glioblastoma cells. Int. J. Oncol. 2012, 41, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Di Maira, G.; Gentilini, A.; Pastore, M.; Caligiuri, A.; Piombanti, B.; Raggi, C.; Rovida, E.; Lewinska, M.; Andersen, J.B.; Borgo, C.; et al. The protein kinase CK2 contributes to the malignant phenotype of cholangiocarcinoma cells. Oncogenesis 2019, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Ham, S.; Yang, K.; Kim, K. Protein kinase CK2 activation is required for transforming growth factor β-induced epithelial-mesenchymal transition. Mol. Oncol. 2018, 12, 1811–1826. [Google Scholar] [CrossRef] [Green Version]
- Sass, G.; Klinger, N.; Sirma, H.; Hashemolhosseini, S.; Hellerbrand, C.; Neureiter, D.; Wege, H.; Ocker, M.; Tiegs, G. Inhibition of experimental HCC growth in mice by use of the kinase inhibitor DMAT. Int. J. Oncol 2011, 39, 433–442. [Google Scholar] [CrossRef]
- Wu, D.; Sui, C.; Meng, F.; Tian, X.; Fu, L.; Li, Y.; Qi, X.; Cui, H.; Liu, Y.; Jiang, Y. Stable knockdown of protein kinase CK2-alpha (CK2α) inhibits migration and invasion and induces inactivation of hedgehog signaling pathway in hepatocellular carcinoma Hep G2 cells. Acta Histochem. 2014, 116, 1501–1508. [Google Scholar] [CrossRef]
- Zhang, H.X.; Jiang, S.S.; Zhang, X.F.; Zhou, Z.Q.; Pan, Q.Z.; Chen, C.L.; Zhao, J.J.; Tang, Y.; Xia, J.C.; Weng, D.S. Protein kinase CK2α catalytic subunit is overexpressed and serves as an unfavorable prognostic marker in primary hepatocellular carcinoma. Oncotarget 2015, 6, 34800–34817. [Google Scholar] [CrossRef] [Green Version]
- Licciardello, M.P.; Workman, P. A New Chemical Probe Challenges the Broad Cancer Essentiality of CK2. Trends Pharmacol. Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.I.; Drewry, D.H.; Pickett, J.E.; Tjaden, A.; Krämer, A.; Müller, S.; Gyenis, L.; Menyhart, D.; Litchfield, D.W.; Knapp, S.; et al. Development of a potent and selective chemical probe for the pleiotropic kinase CK2. Cell Chem. Biol. 2021, 28, 546–558.e10. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Tissue Origin | Age (yr) | Sex | HPV Status | CDKN2A Status 1 | TP53 Status 1 |
|---|---|---|---|---|---|---|
| HEKn 2 | Foreskin | <1 | Male | - | ND 3 | ND 3 |
| Detroit 562 | Pharynx (metastatic pleural effusion) | ND 3 | Female | - | Homozygous mutant | Homozygous mutant |
| Fadu | Hypopharynx | 56 | Male | - | Homozygous mutant | Heterozygous mutant, both alleles |
| UM-SCC-6 | Base of tongue | 32 | Male | - | Homozygous deletion | Wild-type |
| UM-SCC-47 | Lateral tongue | 53 | Male | + | Wild-type | Wild-type |
| UPCI-SCC-90 | Base of tongue | 46 | Male | + | Wild-type | Wild-type |
| VU-SCC-147T (93-Vu-147T) | Floor of mouth | 58 | Male | + | Wild-type | Mutant |
| Status | CK2α | CK2α’ | CK2β | NFκB p65 total | NFκB p65 P-S529 | p65 P-S529/Total | AKT-1 Total | AKT-1 P-S129 | AKT-1 P-S129/Total |
|---|---|---|---|---|---|---|---|---|---|
| HPV+ | 1.00 ± 0.49 | 0.62 ± 0.14 | 2.92 ± 0.86 | 0.02 ± 0.01 | 2.18 ± 0.59 | 145.97 ± 44.79 | 0.56 ± 0.04 | 1.12 ±0.23 | 2.02 ± 0.31 |
| HPV- | 0.55 ± 0.10 | 0.423 ± 0.02 | 1.96 ± 0.23 | 0.03 ± 0.02 | 0.33 ± 0.51 | 35.52 ± 67.14 | 0.51 ± 0.19 | 1.10 ± 0.56 | 2.07 ± 0.29 |
| HPV+/HPV- | 1.82 | 1.450 | 1.49 | 0.48 | 6.65 | 3.69 | 1.08 | 1.02 | 0.97 |
| IC50 (µM) | IC50 (µM) | |||
|---|---|---|---|---|
| Cisplatin Anchored Analysis | CX-4945 Anchored Analysis | |||
| Cell Line | Cisplatin | Cisplatin with CX-4945 | CX-4945 | CX-4945 with Cisplatin |
| Detroit 562 | 8.03 | 2.42 | 1.92 | 1.46 |
| (5.49, 11.97) | (1.72, 3.46) | (1.51, 2.45) | (1.03, 2.09) | |
| Fadu | 7.35 | 3.41 | 4.36 | 2.13 |
| (5.06, 10.82) | (2.51, 4.67) | (2.88, 6.78) | (1.54, 2.98) | |
| UM-SCC-6 | 31.37 | 3.95 | 5.58 | 4.94 |
| (18.99, 55.56) | (3.00, 5.24) | (4.24, 7.40) | (3.75, 6.56) | |
| UM-SCC-47 | 5.12 | 2.94 | 4.77 | 3.67 |
| (2.20, 12.19) | (2.39, 3.63) | (3.82, 5.99) | (2.98, 4.53) | |
| 93-Vu-147T | 9.52 | 4.05 | 5.7 | 5.07 |
| (6.37, 14.23) | (3.18, 5.19) | (4.43, 7.37) | (3.98, 6.48) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trembley, J.H.; Li, B.; Kren, B.T.; Gravely, A.A.; Caicedo-Granados, E.; Klein, M.A.; Ahmed, K. CX-4945 and siRNA-Mediated Knockdown of CK2 Improves Cisplatin Response in HPV(+) and HPV(−) HNSCC Cell Lines. Biomedicines 2021, 9, 571. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050571
Trembley JH, Li B, Kren BT, Gravely AA, Caicedo-Granados E, Klein MA, Ahmed K. CX-4945 and siRNA-Mediated Knockdown of CK2 Improves Cisplatin Response in HPV(+) and HPV(−) HNSCC Cell Lines. Biomedicines. 2021; 9(5):571. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050571
Chicago/Turabian StyleTrembley, Janeen H., Bin Li, Betsy T. Kren, Amy A. Gravely, Emiro Caicedo-Granados, Mark A. Klein, and Khalil Ahmed. 2021. "CX-4945 and siRNA-Mediated Knockdown of CK2 Improves Cisplatin Response in HPV(+) and HPV(−) HNSCC Cell Lines" Biomedicines 9, no. 5: 571. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050571