
Systemic Actions of SGLT2 Inhibition on Chronic mTOR Activation as a Shared Pathogenic Mechanism between Alzheimer’s Disease and Diabetes

 ,
,  and
and
Abstract
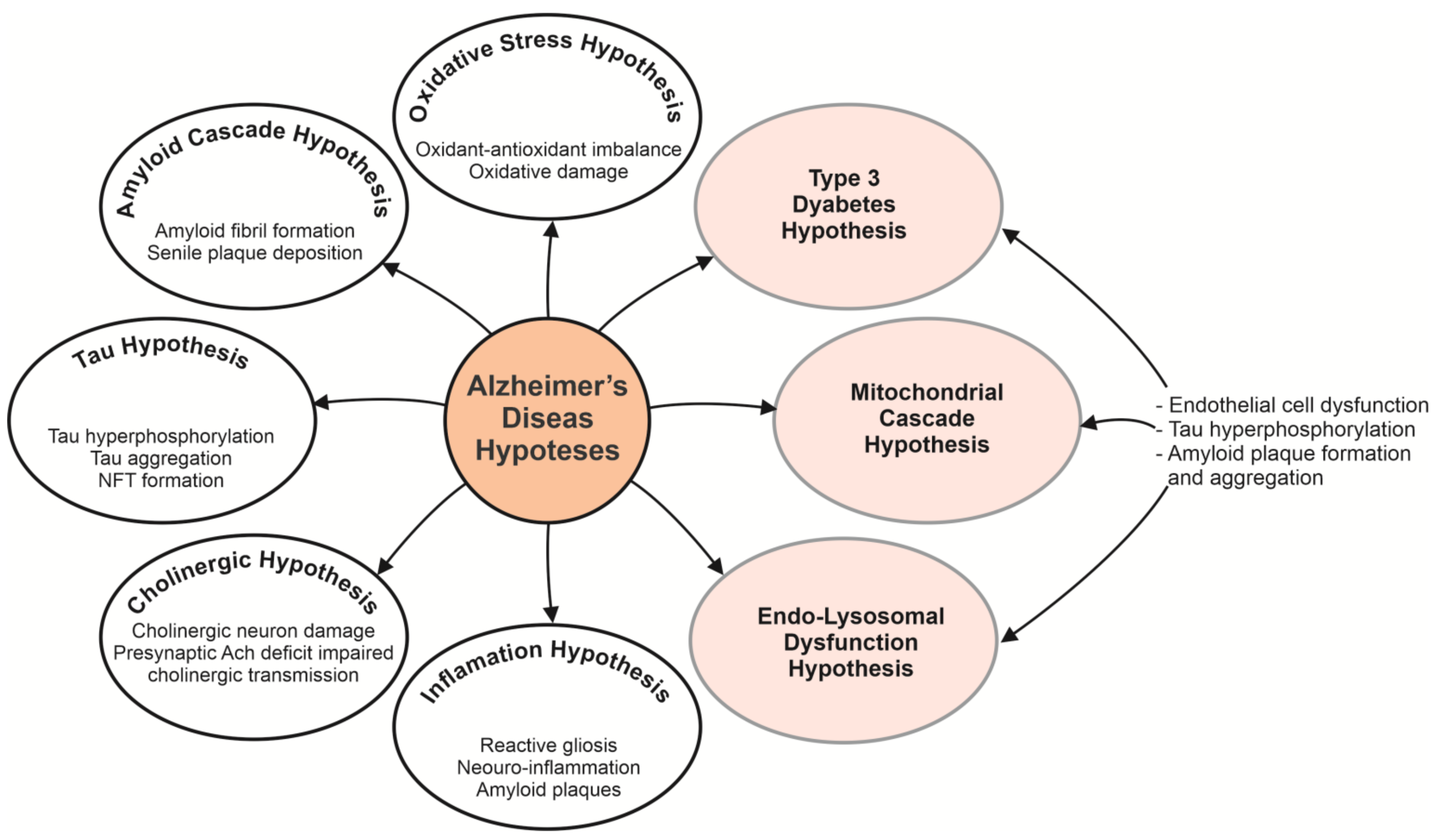
:1. Introduction
2. Pharmacological Approaches Able of Impacting Alzheimer’s Disease and Its Progression
3. Implications of Restoring Metabolic Health in the Therapy of Alzheimer’s Disease
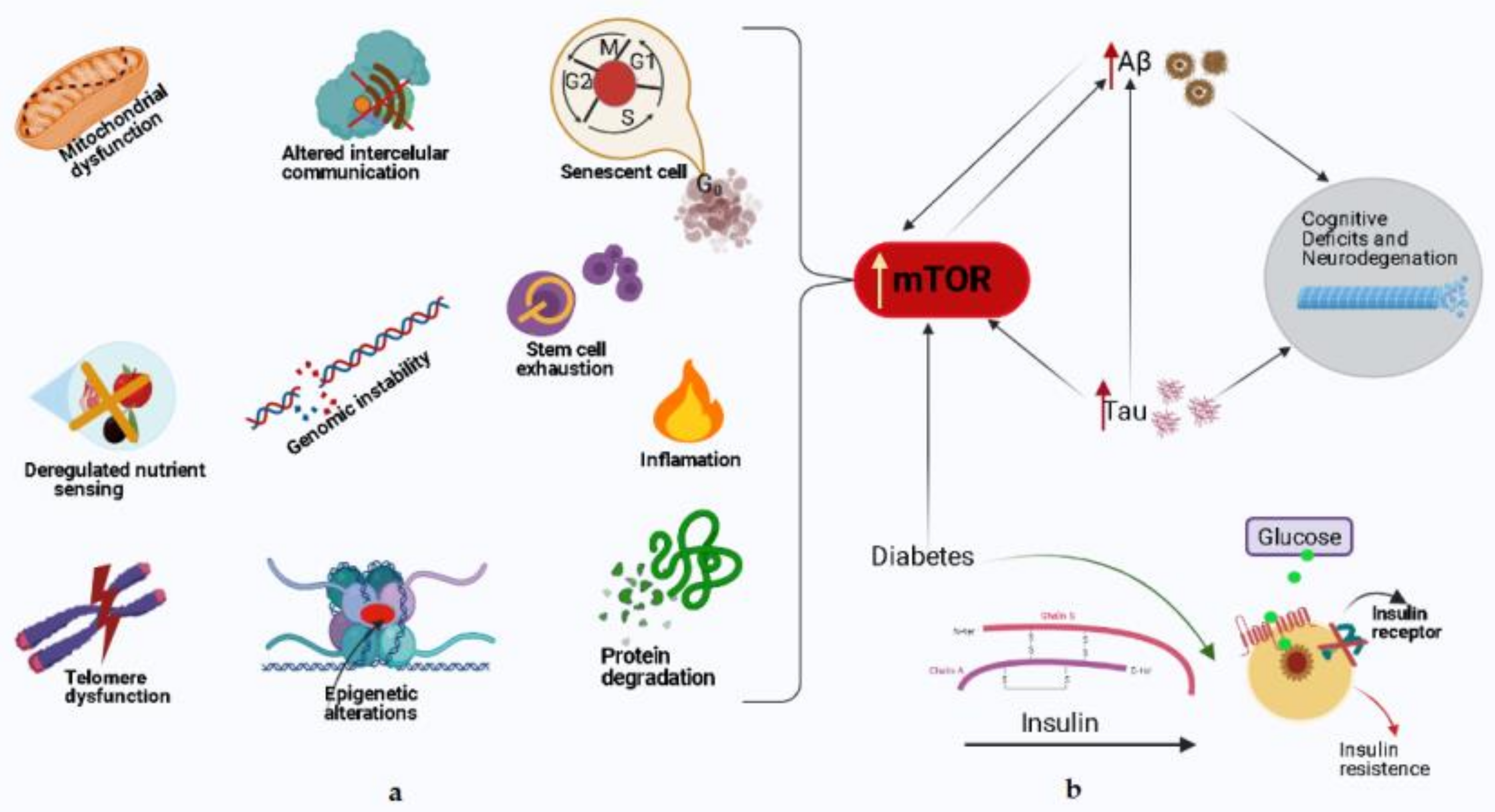
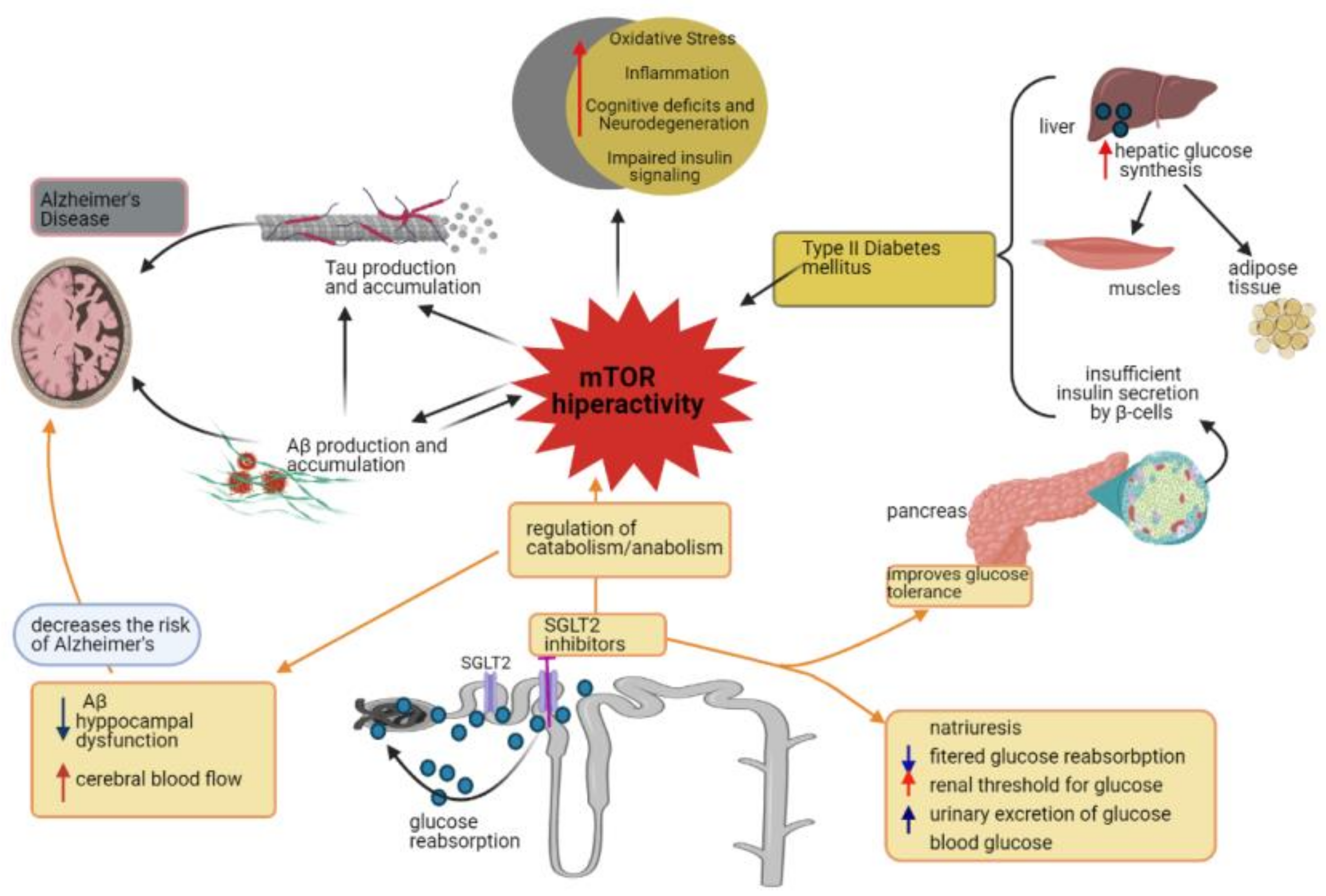
4. Impact of SGLT2 Inhibition on Chronic mTOR Activation: Is the Brain a Target?
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | amyloid β |
| mTOR | mechanistic target of rapamycin kinase |
| SGLT2 | sodium glucose cotransporter 2 |
| APP/PS1 mice | double transgenic mouse model of Alzheimer’s disease over expressing amyloid precursor protein, encoding the Swedish mutations at amino acids 595/596 and an exon-9-deleted human PS1 |
| Tg2576 mice | transgenic mouse model, which express a 695-aa residue splice form of human amyloid precursor protein modified by the Swedish Familial AD double mutation K670N-M671L |
| 3xTg-AD mice | triple-transgenic mouse model harboring PS1M146V, APPSwe, and tauP301L transgenes |
| ATP | adenosine triphosphate |
| MAPT | microtubule associated protein tau |
| ROS | reactive oxygen species |
| IDE | insulin-degrading enzymes |
| APP | amyloid precursor protein |
| TZDs | thiazolidinediones |
| GLP1 | glucagon-like peptide-1 |
| DPP-4 | dipeptidyl peptidase-4 |
| IGF-1 | insulin-like growth factor-1 |
| PS1 | presenilin 1 |
| PS 2 | presenilin 2 |
| APOE | Apolipoprotein E |
| PPARs | peroxisome proliferator-activated receptors |
| AChE | acetylcholinesterase |
| AMPK | adenosine 5′mmonophosphate-activated protein kinase |
| PPARs | peroxisome proliferator-activated receptors |
| PI3K | phosphatidylinositol-3-OH kinase |
| IRS-2 | insulin receptor-2 |
| NLRP3 | nod-like receptor pyrin domain containing 3 |
References
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 5, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanciu, G.D.; Bild, V.; Ababei, D.C.; Rusu, R.N.; Cobzaru, A.; Paduraru, L.; Bulea, D. Link between diabetes and Alzheimer’s disease due to the shared amyloid aggregation and deposition involving both neurodegenerative changes and neurovascular damages. J. Clin. Med. 2020, 9, 1713. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Vieira, T.; Guimaraes, I.; Silva, F.; Ribeiro, F. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimer’s Dement. 2020. [Google Scholar] [CrossRef]
- Wallin, K.; Boström, G.; Kivipelto, M.; Gustafson, Y. Risk factors for incident dementia in the very old. Int. Psychogeriatr. 2013, 25, 1135–1143. [Google Scholar] [CrossRef] [Green Version]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Chiriac, S.I.B.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules 2020, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The early events that initiate β-amyloid aggregation in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 1–13. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Chen, X.Q.; Mobley, W.C. Exploring the pathogenesis of Alzheimer disease in basal forebrain cholinergic neurons:Converging insights from alternative hypotheses. Front. Neurosci. 2019, 13, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippens, G.; Sillen, A.; Landrieu, I.; Amniai, L.; Sibille, N.; Barbier, P.; Leroy, A.; Hanoulle, X.; Wieruszeski, J.M. Tau aggregation in Alzheimer’s disease: What role for phosphorylation? Prion 2007, 1, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Dexheimer, T.; Sui, D.; Hovde, S.; Deng, X.; Kwok, R.; Bochar, D.A.; Kuo, M.H. Hyperphosphorylated tau aggregation and cytotoxicity modulators screen identified prescription drugs linked to Alzheimer’s disease and cognitive functions. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.P.; Castro, A.A.D.; Soares, F.V.; Cunha, E.F.F.D.; Ramalho, T.C. Future therapeutic perspectives into the Alzheimer’s disease targeting the oxidative stress hypothesis. Molecules 2019, 24, 4410. [Google Scholar] [CrossRef] [Green Version]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease—A brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Bolós, M.; Perea, J.R.; Avila, J. Alzheimer’s disease as an inflammatory disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef]
- Funk, K.E.; Kuret, J. Lysosomal fusion dysfunction as a unifying hypothesis for alzheimers disease pathology. Int. J. Alzheimer’s Dis. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Van Acker, Z.P.; Bretou, M.; Annaert, W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: Impact of genetic risk factors. Mol. Neurodegener. 2019, 14, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Voos, W. Mitochondria as potential targets in Alzheimer disease therapy: An update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; Marco, M.D.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial dysfunction in Alzheimer’s disease: A biomarker of the future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef]
- Morgen, K.; Frölich, L. The metabolism hypothesis of Alzheimer’s disease: From the concept of central insulin resistance and associated consequences to insulin therapy. J. Neural. Transm. 2015, 122, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Argentati, C.; Tortorella, I.; Bazzucchi, M.; Emiliani, C.; Morena, F.; Martino, S. The other side of Alzheimer’s disease: Influence of metabolic disorder features for novel diagnostic biomarkers. J. Pers. Med. 2020, 10, 115. [Google Scholar] [CrossRef]
- Paroni, G.; Bisceglia, P.; Seripa, D. Understanding the amyloid hypothesis in Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 68, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H. The beta amyloid dysfunction (BAD) hypothesis for Alzheimer’s disease. Front. Neurosci. 2019, 13, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanciu, G.D.; Ababei, D.C.; Bild, V.; Bild, W.; Paduraru, L.; Gutu, M.M.; Tamba, B.-I. Renal contributions in the pathophysiology and neuropathological substrates shared by chronic kidney disease and Alzheimer’s disease. Brain Sci. 2020, 10, 563. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer ’s disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A. Amyloid precursor protein & endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar]
- Lie, P.P.Y.; Nixon, R.A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 94–105. [Google Scholar] [CrossRef]
- Carosi, J.M.; Sargeant, T.J. Rapamycin and Alzheimer disease: A double-edged sword? Autophagy 2019, 15, 1460–1462. [Google Scholar] [CrossRef]
- Talboom, J.S.; Velazquez, R.; Oddo, S. The mammalian target of rapamycin at the crossroad between cognitive aging and Alzheimer’s disease. NPJ Aging Mech. Dis. 2015, 1, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Barbagallo, M. Type 2 diabetes mellitus and Alzheimer’s disease. World J. Diabetes 2014, 5, 889. [Google Scholar] [CrossRef] [Green Version]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1, 217–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivanesan, S.; Mundugaru, R.; Rajadas, J. Possible clues for brain energy translation via endolysosomal trafficking of APP-CTFs in Alzheimer’s disease. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Mueed, Z.; Tandon, P.; Maurya, S.K.; Deval, R.; Kamal, M.A.; Poddar, N.K. Tau and mTOR: The hotspots for multifarious diseases in Alzheimer’s development. Front. Neurosci. 2019, 13, 1017. [Google Scholar] [CrossRef]
- Oddo, S. The role of mTOR signaling in Alzheimer disease. Front. Biosci. 2012, 4, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.S.; Rahman, M.A.; Kabir, M.T.; Behl, T.; Mathew, B.; Perveen, A.; Barreto, G.E.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Multifarious roles of mTOR signaling in cognitive aging and cerebrovascular dysfunction of Alzheimer’s disease. IUBMB Life 2020, 72, 1843–1855. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Solcan, G. Acute idiopathic polyradiculoneuritis concurrent with acquired myasthenia gravis in a West Highland white terrier dog. BMC Vet. Res. 2016, 12, 111. [Google Scholar] [CrossRef] [Green Version]
- Praticò, D.; Clark, C.M.; Liun, F.; Lee, V.Y.M.; Trojanowski, J.Q. Increase of brain oxidative stress in mild cognitive impairment: A possible predictor of Alzheimer disease. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Zhong, M.B.; Toro, C.A.; Zhang, L.; Cai, D. Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer’s disease pathogenesis. Neurosci. Lett. 2019, 703, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Telpoukhovskaia, M.A.; Bahr, B.A.; Chen, X.; Gan, L. Endo-lysosomal dysfunction: A converging mechanism in neurodegenerative diseases. Curr. Opin. Neurobiol. 2018, 48, 52–58. [Google Scholar] [CrossRef]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O.M. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 2019, 12, 1–21. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Hamilton, D.J.; Nixon, R.A. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res. 1994, 640, 68–80. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Giau, V.V. Type 3 diabetes and its role implications in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, R.; Bolshette, N.; Gadhave, K.; Ahire, A.; Ahmed, S.; Cassano, T.; Gupta, V.B.; Lahkar, M. Insulin signaling: An opportunistic target to minify risk of Alzheimer’s disease. Psychoneuroendocrinology 2017, 83, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Esterline, R.; Oscarsson, J.; Burns, J. A Role for Sodium Glucose Cotransporter 2 Inhibitors (SGLT2is) in the Treatment of Alzheimer’s Disease? In International Review of Neurobiology; Academic Press Inc.: Cambridge, MA, USA, 2020; Volume 155, pp. 113–140. ISBN 9780128231210. [Google Scholar]
- Mao, Z.; Zhang, W. Role of mTOR in glucose and lipid metabolism. Int. J. Mol. Sci. 2018, 19, 2043. [Google Scholar] [CrossRef] [Green Version]
- Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378. [Google Scholar] [CrossRef]
- Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, Z.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578. [Google Scholar] [CrossRef]
- Hardigan, T.; Ward, R.; Ergul, A. Cerebrovascular complications of diabetes: Focus on cognitive dysfunction. Clin. Sci. 2016, 130, 1807–1822. [Google Scholar] [CrossRef] [Green Version]
- Calabrò, M.; Rinaldi, C.; Santoro, G.; Crisafulli, C. The biological pathways of Alzheimer disease: A review. AIMS Neurosci. 2021, 8, 86–132. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Livingston, N.R.; Raza, S.; Van der Doef, T.; Frangou, E.; Love, S.; Busza, G.; Calsolaro, V.; Carver, S.; Holmes, C.; et al. Does insulin resistance influence neurodegeneration in non-diabetic Alzheimer’s subjects? Alzheimer’s Res. Ther. 2021, 13, 47. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Rorbach-Dolata, A.; Piwowar, A. Neurometabolic evidence supporting the hypothesis of increased incidence of type 3 diabetes mellitus in the 21st century. Biomed. Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Esterline, R.L.; Vaag, A.; Oscarsson, J.; Vora, J. SGLT2 inhibitors: Clinical benefits by restoration of normal diurnal metabolism? Eur. J. Endocrinol. 2018, 178, R113–R125. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Domenico, F.D.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, C.; Hua, S.; Liao, H.; Wang, M.; Xiong, Y.; Cao, F. An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res. Clin. Pract. 2017, 124, 41–47. [Google Scholar] [CrossRef]
- Ninomiya, T. Diabetes mellitus and dementia. Curr. Diab. Rep. 2014, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martos, C.M.; Atkinson, R.A.K.; Chuah, M.I.; King, A.E.; Vickers, J.C. Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Imfeld, P.; Bodmer, M.; Jick, S.S.; Meier, C.R. Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: A population-based case-control study. J. Am. Geriatr. Soc. 2012, 60, 916–921. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain. Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, G.D.; Bild, V.; Ababei, D.C.; Rusu, R.N.; Beschea Chiriac, S.I.; Resus, E.; Luca, A. Relevance of surface neuronal protein autoantibodies as biomarkers in seizures-associated disorders 2019, 20, 4529. Int. J. Mol. Sci. 2019, 20, 4529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaribeygi, H.; Ashrafizadeh, M.; Henney, N.C.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. Neuromodulatory effects of anti-diabetes medications: A mechanistic review. Pharmacol. Res. 2020, 152, 104611. [Google Scholar] [CrossRef] [PubMed]
- Wium-Andersen, I.K.; Osler, M.; Jørgensen, M.B.; Rungby, J.; Wium-Andersen, M.K. Antidiabetic medication and risk of dementia in patients with type 2 diabetes: A nested case-control study. Eur. J. Endocrinol. 2019, 181, 499–507. [Google Scholar] [CrossRef]
- Oliveira, W.H.D.; Nunes, A.K.D.S.; França, M.E.R.D.; Santos, L.A.D.; Lós, D.B.; Rocha, S.W.S.; Barbosa, K.P.D.S.; Rodrigues, G.B.; Peixoto, C.A. Effects of metformin on inflammation and short-term memory in streptozotocin-induced diabetic mice. Brain Res. 2016, 1644, 149–160. [Google Scholar] [CrossRef]
- Mousavi, F.; Eidi, A.; Khalili, M.; Roghani, M. Metformin ameliorates learning and memory deficits in streptozotocin-induced diabetic rats. J. Basic Clin. Pathophysiol. 2018, 6, 17–22. [Google Scholar]
- Shingo, A.S.; Kanabayashi, T.; Kito, S.; Murase, T. Intracerebroventricular administration of an insulin analogue recovers STZ-induced cognitive decline in rats. Behav. Brain Res. 2013, 241, 105–111. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Frey, W.H.; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.S.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s Disease dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, T.M.; Ferreira de Sá, D.S.; Westerhausen, R.; Strelzyk, F.; Larra, M.F.; Hallschmid, M.; Savaskan, E.; Oitzl, M.S.; Busch, H.P.; Naumann, E.; et al. Intranasal insulin increases regional cerebral blood flow in the insular cortex in men independently of cortisol manipulation. Hum. Brain Mapp. 2014, 35, 1944–1956. [Google Scholar] [CrossRef]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Hallschmid, M.; Schmitz, K.; Schultes, B.; Ratter, F.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans: Superiority of insulin aspart. Neuropsychopharmacology 2007, 32, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Köhler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiTacchio, K.A.; Heinemann, S.F.; Dziewczapolski, G. Metformin treatment alters memory function in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1–42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with alzheimer disease. Arch. Neurol. 2011, 68, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Rosenblat, J.D.; Brietzke, E.; Park, C.; Lee, Y.; Musial, N.; Pan, Z.; Mansur, R.B.; McIntyre, R.S. Comparative efficacy and acceptability of antidiabetic agents for Alzheimer’s disease and mild cognitive impairment: A systematic review and network meta-analysis. Diabetes Obes. Metab. 2018, 20, 2467–2471. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.Y.; Yang, J.T.; Wang, Z.J.; Zhang, J.; Yang, W.; Wu, M.N.; Qi, J.S. Lixisenatide reduces amyloid plaques, neurofibrillary tangles and neuroinflammation in an APP/PS1/tau mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2018, 495, 1034–1040. [Google Scholar] [CrossRef]
- McClean, P.L.; Hölscher, C. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology 2014, 86, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Candeias, E.; Alves, I.N.; Mena, D.; Silva, D.F.; Machado, N.J.; Campos, E.J.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Liraglutide protects against brain amyloid-β1–42 accumulation in female mice with early Alzheimer’s disease-like pathology by partially rescuing oxidative/nitrosative stress and inflammation. Int. J. Mol. Sci. 2020, 21, 1746. [Google Scholar] [CrossRef] [Green Version]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: Randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Dong, Q.; Teng, S.W.; Wang, Y.; Qin, F.; Li, Y.; Ai, L.L.; Yu, H. Sitagliptin protects the cognition function of the Alzheimer’s disease mice through activating glucagon-like peptide-1 and BDNF-TrkB signalings. Neurosci. Lett. 2019, 696, 184–190. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; Filippo, C.D.; Marfella, R.; Abbatecola, A.M.; Ferraraccio, F.; Rossi, F.; Paolisso, G. Long-term inhibition of dipeptidyl peptidase-4 in Alzheimer’s prone mice. Exp. Gerontol. 2010, 45, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Kosaraju, J.; Holsinger, R.M.D.; Guo, L.; Tam, K.Y. Linagliptin, a dipeptidyl peptidase-4 inhibitor, mitigates cognitive deficits and pathology in the 3×Tg-AD mouse model of Alzheimer’s disease. Mol. Neurobiol. 2017, 54, 6074–6084. [Google Scholar] [CrossRef]
- Kosaraju, J.; Gali, C.C.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Holsinger, R.M.D.; Madhunapantula, V.S.R.; Nataraj, S.K.M.; Basavan, D. Saxagliptin: A dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 2013, 72, 291–300. [Google Scholar] [CrossRef]
- Kosaraju, J.; Murthy, V.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Nataraj, S.K.M.; Basavan, D. Vildagliptin: An anti-diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin-induced Alzheimer’s disease. J. Pharm. Pharmacol. 2013, 65, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Arafa, N.M.S.; Ali, E.H.A.; Hassan, M.K. Canagliflozin prevents scopolamine-induced memory impairment in rats: Comparison with galantamine hydrobromide action. Chem. Biol. Interact. 2017, 277, 195–203. [Google Scholar] [CrossRef]
- Felice, F.G.D.; Vieira, M.N.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury, N.B.E.; Gratuze, M.; Papon, M.A.; Bretteville, A.; Planel, E. Insulin dysfunction and Tau pathology. Front. Cell. Neurosci. 2014, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Bryan, M.R.; Bowman, A.B. Manganese and the Insulin-IGF Signaling Network in Huntington’s Disease and Other Neurodegenerative Disorders. In Advances in Neurobiology; Springer: New York, NY, USA, 2017; Volume 18, pp. 113–142. [Google Scholar]
- Hossain, M.F.; Wang, N.; Chen, R.; Li, S.; Roy, J.; Uddin, M.G.; Li, Z.; Lim, L.W.; Song, Y.Q. Exploring the multifunctional role of melatonin in regulating autophagy and sleep to mitigate Alzheimer’s disease neuropathology. Ageing Res. Rev. 2021, 67, 101304. [Google Scholar] [CrossRef]
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516. [Google Scholar] [CrossRef]
- Femminella, G.D.; Bencivenga, L.; Petraglia, L.; Visaggi, L.; Gioia, L.; Grieco, F.V.; Lucia, C.D.; Komici, K.; Corbi, G.; Edison, P.; et al. Antidiabetic drugs in Alzheimer’s disease: Mechanisms of action and future perspectives. J. Diabetes Res. 2017, 2017. [Google Scholar] [CrossRef]
- Sarkar, S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [Green Version]
- Burns, J. Dapagliflozin In Alzheimer’s Disease—Full Text View—ClinicalTrials. Available online: https://clinicaltrials.gov/ct2/show/NCT03801642 (accessed on 19 April 2021).
- Rizvi, S.; Shakil, S.; Biswas, D.; Shakil, S.; Shaikh, S.; Bagga, P.; Kamal, M. Invokana (canagliflozin) as a dual inhibitor of acetylcholinesterase and sodium glucose co-transporter 2: Advancement in Alzheimer’s disease—Diabetes type 2 linkage via an enzoinformatics study. CNS Neurol. Disord. Drug Targets 2014, 13, 447–451. [Google Scholar] [CrossRef]
- O’Neill, C. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 2013, 48, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.L.; Zheng, W.; Halloran, J.J.; Burbank, R.R.; Hussong, S.A.; Hart, M.J.; Javors, M.; Shih, Y.Y.I.; Muir, E.; Solano Fonseca, R.; et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1412–1421. [Google Scholar] [CrossRef]
- Gureev, A.P.; Popov, V.N.; Starkov, A.A. Crosstalk between the mTOR and Nrf2/ARE signaling pathways as a target in the improvement of long-term potentiation. Exp. Neurol. 2020, 328, 113285. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Stanfel, M.N.; Shamieh, L.S.; Kaeberlein, M.; Kennedy, B.K. The TOR pathway comes of age. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Garza-Lombó, C.; Schroder, A.; Reyes-Reyes, E.M.; Franco, R. mTOR/AMPK signaling in the brain: Cell metabolism, proteostasis and survival. Curr. Opin. Toxicol. 2018, 8, 102–110. [Google Scholar] [CrossRef]
- Guillén, C.; Benito, M. MTORC1 overactivation as a key aging factor in the progression to type 2 diabetes mellitus. Front. Endocrinol. 2018, 9, 621. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New insights into the role of mtor signaling in the cardiovascular system. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. MTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Zhao, B.; Li, K.; Zhang, L.; Li, C.; Quazi, S.H.; Tan, Y. Mammalian target of rapamycin: A valid therapeutic target through the autophagy pathway for alzheimer’s disease? J. Neurosci. Res. 2012, 90, 1105–1118. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11, eaar4289. [Google Scholar] [CrossRef] [PubMed]
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Aβ exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005, 94, 215–225. [Google Scholar] [CrossRef]
- Zhou, X.W.; Tanila, H.; Pei, J.J. Parallel increase in p70 kinase activation and tau phosphorylation (S262) with Aβ overproduction. FEBS Lett. 2008, 582, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Hoeffer, C.A.; Capetillo-Zarate, E.; Yu, F.; Wong, H.; Lin, M.T.; Tampellini, D.; Klann, E.; Blitzer, R.D.; Gouras, G.K. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e12845. [Google Scholar] [CrossRef]
- Caccamo, A.; Maldonado, M.A.; Majumder, S.; Medina, D.X.; Holbein, W.; Magrí, A.; Oddo, S. Naturally secreted amyloid-β increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J. Biol. Chem. 2011, 286, 8924–8932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; Magrì, A.; Medina, D.X.; Wisely, E.V.; López-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Onuki, R.; Bando, Y.; Suyama, E.; Katayama, T.; Kawasaki, H.; Baba, T.; Tohyama, M.; Taira, K. An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer’s disease. EMBO J. 2004, 23, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Tramutola, A.; Triplett, J.C.; Domenico, F.D.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.J.; Björkdahl, C.; Zhang, H.; Zhou, X.; Winblad, B. p70 S6 kinase and tau in Alzheimer’s disease. J. Alzheimer’s Dis. 2008, 14, 385–392. [Google Scholar] [CrossRef]
- An, W.L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.G.; Winblad, B.; Pei, J.J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, M.S.; Wang, H.F.; Cao, L.; Zhang, Q.Q.; Shi, J.Q.; Gao, L.; Qin, H.; et al. Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol. Res. 2014, 81, 54–63. [Google Scholar] [CrossRef]
- Domenico, F.D.; Tramutola, A.; Foppoli, C.; Head, E.; Perluigi, M.; Butterfield, D.A. mTOR in Down syndrome: Role in Aβ and tau neuropathology and transition to Alzheimer disease-like dementia. Free Radic. Biol. Med. 2018, 114, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Briaud, I.; Dickson, L.M.; Lingohr, M.K.; McCuaig, J.F.; Lawrence, J.C.; Rhodes, C.J. Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback down-regulates protein kinase B-mediated signaling pathway in β-cells. J. Biol. Chem. 2005, 280, 2282–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.J. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes. Metab. 2019, 21, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, F.; Scheen, A.J. Effects of SGLT2 inhibitors on systemic and tissue low-grade inflammation: The potential contribution to diabetes complications and cardiovascular disease. Diabetes Metab. 2018, 44, 457–464. [Google Scholar] [CrossRef]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli, E. Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes 2016, 65, 1190–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T. Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol. 2017, 27, 220–222. [Google Scholar] [CrossRef] [Green Version]
- White, C.S.; Lawrence, C.B.; Brough, D.; Rivers-Auty, J. Inflammasomes as therapeutic targets for Alzheimer’s disease. Brain Pathol. 2017, 27, 223–234. [Google Scholar] [CrossRef]
- Joosten, L.A.B.; Crişan, T.O.; Bjornstad, P.; Johnson, R.J. Asymptomatic hyperuricaemia: A silent activator of the innate immune system. Nat. Rev. Rheumatol. 2020, 16, 75–86. [Google Scholar] [CrossRef]
- Tana, C.; Ticinesi, A.; Prati, B.; Nouvenne, A.; Meschi, T. Uric acid and cognitive function in older individuals. Nutrients 2018, 10, 975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaaren, B.F.J.; Vernooij, M.W.; Dehghan, A.; Vrooman, H.A.; Boer, R.D.; Hofman, A.; Witteman, J.C.M.; Niessen, W.J.; Breteler, M.M.B.; Van Der Lugt, A.; et al. The relation of uric acid to brain atrophy and cognition: The rotterdam scan study. Neuroepidemiology 2013, 41, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Giannitsi, S.; Maria, B.; Bechlioulis, A.; Naka, K. Endothelial dysfunction and heart failure: A review of the existing bibliography with emphasis on flow mediated dilation. JRSM Cardiovasc. Dis. 2019, 8, 204800401984304. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, M.; Pedrinolla, A.; Galazzo, I.B.; Fonte, C.; Smania, N.; Tamburin, S.; Muti, E.; Crispoltoni, L.; Stabile, A.; Pistilli, A.; et al. Impact of nitric oxide bioavailability on the progressive cerebral and peripheral circulatory impairments during aging and Alzheimer’s disease. Front. Physiol. 2018, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Decker, B.; Pumiglia, K. mTORc1 activity is necessary and sufficient for phosphorylation of eNOSS1177. Physiol. Rep. 2018, 6, 13733. [Google Scholar] [CrossRef]
- Van Skike, C.E.; Jahrling, J.B.; Olson, A.B.; Sayre, N.L.; Hussong, S.A.; Ungvari, Z.; Lechleiter, J.D.; Galvan, V. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer’s disease and vascular cognitive impairment. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H693–H703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesniewski, L.A.; Seals, D.R.; Walker, A.E.; Henson, G.D.; Blimline, M.W.; Trott, D.W.; Bosshardt, G.C.; LaRocca, T.J.; Lawson, B.R.; Zigler, M.C.; et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 2017, 16, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Nwadike, C.; Williamson, L.E.; Gallagher, L.E.; Guan, J.-L.; Chan, E.Y.W. AMPK inhibits ULK1-dependent autophagosome formation and lysosomal acidification via distinct mechanisms. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.M.; Feng, Y.; Wang, J.; Shi, R.; Jiang, X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudaliar, S.; Henry, R.R.; Boden, G.; Smith, S.; Chalamandaris, A.G.; Duchesne, D.; Iqbal, N.; List, J. Changes in insulin sensitivity and insulin secretion with the sodium glucose cotransporter 2 inhibitor dapagliflozin. Diabetes Technol. Ther. 2014, 16, 137–144. [Google Scholar] [CrossRef]
- Kappel, B.A.; Lehrke, M.; Schütt, K.; Artati, A.; Adamski, J.; Lebherz, C.; Marx, N. Effect of empagliflozin on the metabolic signature of patients with type 2 diabetes mellitus and cardiovascular disease. Circulation 2017, 136, 969–972. [Google Scholar] [CrossRef]
- Majd, S.; Power, J.H.T. Oxidative stress and decreased mitochondrial superoxide dismutase 2 and peroxiredoxins 1 and 4 based mechanism of concurrent activation of AMPK and mTOR in Alzheimer’s disease. Curr. Alzheimer Res. 2018, 15, 764–776. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Antidiabetic Drugs | Experimental Model | Findings | References |
|---|---|---|---|
| Insulin | rat model of intracerebroventricular streptozotocin (STZ) injection-induced cognitive dysfunction, intraventricular delivery of 0.5 units = 12 nmol of detemir | rescued STZ-induced cognitive decline | [79] |
| patients with early AD or moderate cognitive impairment; intranasal delivery of 20 or 40 IU insulin | improved attention, verbal memory and functional status; modulation of Aβ peptide | [80,81,82,83] | |
| healthy volunteers, intranasal administration of 4 × 40 IU of insulin | improvement in memory and mood, increase regional cerebral blood flow in the putamen and the insular cortex | [84,85,86] | |
| Metformin | neuronal cell lines under prolonged hyperinsulinemic conditions, various concentrations of metformin (0.4–3.2 mM) | insulin signaling resensitization, prevention of the molecular and pathological changes observed in AD neurons | [87] |
| murine primary neurons (from tau transgenic mice and wild type), different concentration of metformin (2.5 mM or 10 nM) | reduction of tau phosphorylation | [88] | |
| transgenic mouse model of AD intraperitoneal delivery of 200 mg/kg metformin; or 350 mg/kg/day metformin delivered in drinking water for several months | amelioration of cognitive deficits, reduce Aβ plaque deposition attenuation of memory impairment | [73] [89] | |
| in older adults with an incident diagnosis of AD; 1–9, 10–29, 30–59, or ≥60 metformin prescriptions | more than 60 prescriptions were correlated with a slightly increased risk of developing AD | [72] | |
| Thiazolidinediones | transgenic AD mouse model 0.03 mg/kg/day of leptin intranasal delivery + intraperitoneal administration of 10 mg/kg/day pioglitazone for 2 weeks | reduce brain Aβ levels and spatial memory impairments | [71] |
| 7 days gavage therapy with 40 mg/kg/day of pioglitazone | decrease glial inflammation and soluble Aβ1–42 peptide levels by 27% | [90] | |
| control trial in patients with AD and diabetes, doses of 15–30 mg pioglitazone for 6 months | cognitive deficits amelioration and stabilization of the disease in diabetics with AD | [91] | |
| pilot trial with AD patients without diabetes; daily 45 mg of pioglitazone | no important efficacy data were detected | [92] | |
| clinical trials; 2 to 8 mg of rosiglitazone, as adjunct therapy in AD patients | pro-cognitive effects | [93] | |
| Glucagon-like peptide-1 receptor agonists | transgenic mouse model of AD intraperitoneal injection with 1 or 10 nmol/kg of lixisenatide for 10 weeks 10 nmol/kg lixisenatide for 60 days | prevented memory impairment, neuronal loss, and deterioration of synaptic plasticity reduction of amyloid plaques and neurofibrillary tangles | [94] [95] |
| intraperitoneal injection with 2.5 or 25 nmol/kg of liraglutide for 10 weeks | reduce Aβ deposition by 40–50%, and decrease inflammatory response | [96] | |
| a pilot clinical trial in AD patients; daily subcutaneously injections of 0.6 mg liraglutide in the first week; hereafter 1.2 mg daily for another week before finally increasing to 1.8 mg daily (week 26) | brain glucose metabolism decline prevention; no important cognitive changes compared with placebo group | [97] | |
| Dipeptidyl Peptidase-4 Inhibitors | transgenic mouse models of AD 20 mg/kg/day of sitagliptin for an 8-weeks period daily gavage of 5, 10 and 20 mg/kg sitagliptin for 12 weeks | pro-cognitive effects, reduction of Aβ deposits diminution of nitrosative stress and inflammation markers, reduction of Aβ deposition | [98] [99] |
| daily oral administration of 5, 10, and 20 mg/kg linagliptin for 8 weeks | amelioration of cognitive deficits, diminution of Aβ42 levels, reduction of tau phosphorylation and neuroinflammation | [100] | |
| STZ-induced rat model of AD; 0.25, 0.5 and 1 mg/kg of saxagliptin in gavage delivery for 60 days | reduction of Aβ formation, a marked decrease of Aβ42 level and tau phosphorylation | [101] | |
| STZ- induced rat model of AD; daily orally doses of 2.5, 5 and 10 mg/kg vildagliptin for 30 days | attenuation of tau phosphorylation, Aβ and inflammatory markers | [102] | |
| Sodium-glucose cotransporter 2 inhibitors | scopolamine-induced rat model of memory impairment; daily oral gavage of 10 mg/kg canagliflozin for 14 days | improvement of memory dysfunction | [103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanciu, G.D.; Rusu, R.N.; Bild, V.; Filipiuc, L.E.; Tamba, B.-I.; Ababei, D.C. Systemic Actions of SGLT2 Inhibition on Chronic mTOR Activation as a Shared Pathogenic Mechanism between Alzheimer’s Disease and Diabetes. Biomedicines 2021, 9, 576. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050576
Stanciu GD, Rusu RN, Bild V, Filipiuc LE, Tamba B-I, Ababei DC. Systemic Actions of SGLT2 Inhibition on Chronic mTOR Activation as a Shared Pathogenic Mechanism between Alzheimer’s Disease and Diabetes. Biomedicines. 2021; 9(5):576. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050576
Chicago/Turabian StyleStanciu, Gabriela Dumitrita, Razvan Nicolae Rusu, Veronica Bild, Leontina Elena Filipiuc, Bogdan-Ionel Tamba, and Daniela Carmen Ababei. 2021. "Systemic Actions of SGLT2 Inhibition on Chronic mTOR Activation as a Shared Pathogenic Mechanism between Alzheimer’s Disease and Diabetes" Biomedicines 9, no. 5: 576. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050576