
Proatherogenic Sialidases and Desialylated Lipoproteins: 35 Years of Research and Current State from Bench to Bedside

 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Atherogenicity
2. Desialylation of Lipoproteins as a Risk Factor in Atherosclerosis
3. Chemical Modification of Lipoproteins and Its Impact
4. Immunogenicity of Desialylated Lipoproteins
5. Enzymes Implicated in Desialylation
6. Contribution of Viral Sialidases
7. Abzymes Exhibiting Sialidase Activity
8. Multiple Modification of LDL in the Blood
9. Changes in Glycosylation of HDL
9.1. Coronary Artery Disease
9.2. Type II Diabetes
9.3. Familial Apolipoprotein A-I (ApoAI) Deficiency
10. Animal Models of Desialylation
11. Clinical Impact of Desialylation
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chazov, E.I.; Tertov, V.V.; Orekhov, A.N.; Lyakishev, A.A.; Perova, N.V.; Kurdanov, K.A.; Khashimov, K.A.; Novikov, I.D.; Smirnov, V.N. Atherogenicity of blood serum from patients with coronary heart disease. Lancet 1986, 2, 595–598. [Google Scholar] [CrossRef]
- Packard, R.R.; Libby, P. Inflammation in atherosclerosis: From vascular biology to biomarker discovery and risk prediction. Clin. Chem. 2008, 54, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N.; Mikhailenko, I.A. Modification of low density lipoprotein by desialylation causes lipid accumulation in cultured cells: Discovery of desialylated lipoprotein with altered cellular metabolism in the blood of atherosclerotic patients. Biochem. Biophys. Res. Commun. 1989, 162, 206–211. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Sobenin, I.A.; Smirnov, V.N.; Via, D.P.; Guevara, J., Jr.; Gotto, A.M., Jr.; Morrisett, J.D. Sialic acid content of human low density lipoproteins affects their interaction with cell receptors and intracellular lipid accumulation. J. Lipid Res. 1992, 33, 805–817. [Google Scholar] [CrossRef]
- Kotelianskiĭ, V.E.; Orekhov, A.N.; Tertov, V.V.; Khashimov Kh, A.; Glukhova, M.A. Effect of components of the extracellular matrix on the accumulation of lipids in human cells. Biulleten Eksperimental Biologii Meditsiny 1987, 104, 562–564. [Google Scholar] [CrossRef]
- Lehmann, F.; Tiralongo, E.; Tiralongo, J. Sialic acid-specific lectins: Occurrence, specificity and function. Cell. Mol. Life Sci. Cmls 2006, 63, 1331–1354. [Google Scholar] [CrossRef] [PubMed]
- Weigel, P.H.; Yik, J.H. Glycans as endocytosis signals: The cases of the asialoglycoprotein and hyaluronan/chondroitin sulfate receptors. Biochim. Biophys. Acta 2002, 1572, 341–363. [Google Scholar] [CrossRef]
- Millar, J.S. The sialylation of plasma lipoproteins. Atherosclerosis 2001, 154, 1–13. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N. Desialylated low density lipoprotein--naturally occurring modified lipoprotein with atherogenic potency. Atherosclerosis 1991, 86, 153–161. [Google Scholar] [CrossRef]
- Mel’nichenko, A.A.; Tertov, V.V.; Ivanova, O.A.; Aksenov, D.V.; Sobenin, I.A.; Popov, E.V.; Kaplun, V.V.; Suprun, I.V.; Panasenko, O.M.; Orekhov, A.N. Desialylation decreases the resistance of apo B-containing lipoproteins to aggregation and increases their atherogenic potential. Bull. Exp. Biol. Med. 2005, 140, 51–54. [Google Scholar] [CrossRef]
- Aksenov, D.V.; Medvedeva, L.A.; Skalbe, T.A.; Sobenin, I.A.; Tertov, V.V.; Gabbasov, Z.A.; Popov, E.V.; Orekhov, A.N. Deglycosylation of apo B-containing lipoproteins increase their ability to aggregate and to promote intracellular cholesterol accumulation in vitro. Arch. Physiol. Biochem. 2008, 114, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Tertov, V.V.; Kaplun, V.V.; Sobenin, I.A.; Orekhov, A.N. Low-density lipoprotein modification occurring in human plasma possible mechanism of in vivo lipoprotein desialylation as a primary step of atherogenic modification. Atherosclerosis 1998, 138, 183–195. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Tertov, V.V.; Orekhov, A.N.; Smirnov, V.N. Synergetic effect of desialylated and glycated low density lipoproteins on cholesterol accumulation in cultured smooth muscle intimal cells. Atherosclerosis 1991, 89, 151–154. [Google Scholar] [CrossRef]
- Hunt, J.V.; Bottoms, M.A.; Clare, K.; Skamarauskas, J.T.; Mitchinson, M.J. Glucose oxidation and low-density lipoprotein-induced macrophage ceroid accumulation: Possible implications for diabetic atherosclerosis. Biochem. J. 1994, 300 Pt 1, 243–249. [Google Scholar] [CrossRef]
- Ravandi, A.; Kuksis, A.; Shaikh, N.A. Glucosylated glycerophosphoethanolamines are the major LDL glycation products and increase LDL susceptibility to oxidation: Evidence of their presence in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 467–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chisolm, G.M.; Steinberg, D. The oxidative modification hypothesis of atherogenesis: An overview. Free Radic. Biol. Med. 2000, 28, 1815–1826. [Google Scholar] [CrossRef]
- Sukhorukov, V.; Gudelj, I.; Pučić-Baković, M.; Zakiev, E.; Orekhov, A.; Kontush, A.; Lauc, G. Glycosylation of human plasma lipoproteins reveals a high level of diversity, which directly impacts their functional properties. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2019, 1864, 643–653. [Google Scholar] [CrossRef]
- Cuniberti, L.A.; Martinez, V.; Schachter, J.; Magariños, G.; Meckert, P.C.; Laguens, R.P.; Levenson, J.; Werba, J.P. Sialic acid as a protective barrier against neointima development. Atherosclerosis 2005, 181, 225–231. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Kudryashov, S.A.; Smirnov, V.N. Triggerlike stimulation of cholesterol accumulation and DNA and extracellular matrix synthesis induced by atherogenic serum or low density lipoprotein in cultured cells. Circ. Res. 1990, 66, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, A.N.; Tertov, V.V.; Kabakov, A.E.; Adamova, I.; Pokrovsky, S.N.; Smirnov, V.N. Autoantibodies against modified low density lipoprotein. Nonlipid factor of blood plasma that stimulates foam cell formation. Arterioscler. Thromb. A J. Vasc. Biol. 1991, 11, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Palinski, W.; Rosenfeld, M.E.; Ylä-Herttuala, S.; Gurtner, G.C.; Socher, S.S.; Butler, S.W.; Parthasarathy, S.; Carew, T.E.; Steinberg, D.; Witztum, J.L. Low density lipoprotein undergoes oxidative modification in vivo. Proc. Natl. Acad. Sci. USA 1989, 86, 1372–1376. [Google Scholar] [CrossRef] [Green Version]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef] [Green Version]
- Sobenin, I.A.; Salonen, J.T.; Zhelankin, A.V.; Melnichenko, A.A.; Kaikkonen, J.; Bobryshev, Y.V.; Orekhov, A.N. Low density lipoprotein-containing circulating immune complexes: Role in atherosclerosis and diagnostic value. Biomed Res. Int. 2014, 2014, 205697. [Google Scholar] [CrossRef] [PubMed]
- Hollander, W.; Colombo, M.A.; Kirkpatrick, B.; Paddock, J. Soluble proteins in the human atherosclerotic plaque. With spectral reference to immunoglobulins, C3-complement component, alpha 1-antitrypsin and alpha 2-macroglobulin. Atherosclerosis 1979, 34, 391–405. [Google Scholar] [CrossRef]
- Hansson, G.K.; Bondjers, G.; Bylock, A.; Hjalmarsson, L. Ultrastructural studies on the localization of IgG in the aortic endothelium and subendothelial intima of atherosclerotic and nonatherosclerotic rabbits. Exp. Mol. Pathol. 1980, 33, 302–315. [Google Scholar] [CrossRef]
- Parums, D.; Mitchinson, M.J. Demonstration of immunoglobulin in the neighbourhood of advanced atherosclerotic plaques. Atherosclerosis 1981, 38, 211–216. [Google Scholar] [CrossRef]
- Tertov, V.V.; Orekhov, A.N.; Kacharava, A.G.; Sobenin, I.A.; Perova, N.V.; Smirnov, V.N. Low density lipoprotein-containing circulating immune complexes and coronary atherosclerosis. Exp. Mol. Pathol. 1990, 52, 300–308. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N.; Jaakkola, O.; Solakivi, T.; Nikkari, T. Characteristics of low density lipoprotein isolated from circulating immune complexes. Atherosclerosis 1996, 122, 191–199. [Google Scholar] [CrossRef]
- Burut, D.F.; Karim, Y.; Ferns, G.A. The role of immune complexes in atherogenesis. Angiology 2010, 61, 679–689. [Google Scholar] [CrossRef]
- Salonen, J.T. Markers of oxidative damage and antioxidant protection: Assessment of LDL oxidation. Free Radic. Res. 2000, 33, S41–S46. [Google Scholar]
- Witztum, J.L.; Steinbrecher, U.P.; Kesaniemi, Y.A.; Fisher, M. Autoantibodies to glucosylated proteins in the plasma of patients with diabetes mellitus. Proc. Natl. Acad. Sci. USA 1984, 81, 3204–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maverakis, E.; Kim, K.; Shimoda, M.; Gershwin, M.E.; Patel, F.; Wilken, R.; Raychaudhuri, S.; Ruhaak, L.R.; Lebrilla, C.B. Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: A critical review. J. Autoimmun. 2015, 57, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Vasiljevic, S.; Mitchell, D.A.; Crispin, M.; Scanlan, C.N. Dissecting the molecular mechanism of IVIg therapy: The interaction between serum IgG and DC-SIGN is independent of antibody glycoform or Fc domain. J. Mol. Biol. 2013, 425, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, Y.; Ise, W.; Harazono, A.; Takakura, D.; Fukuyama, H.; Baba, Y.; Narazaki, M.; Shoda, H.; Takahashi, N.; Ohkawa, Y.; et al. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat. Commun. 2016, 7, 11205. [Google Scholar] [CrossRef] [Green Version]
- Menni, C.; Gudelj, I.; Macdonald-Dunlop, E.; Mangino, M.; Zierer, J.; Bešić, E.; Joshi, P.K.; Trbojević-Akmačić, I.; Chowienczyk, P.J.; Spector, T.D.; et al. Glycosylation Profile of Immunoglobulin G Is Cross-Sectionally Associated With Cardiovascular Disease Risk Score and Subclinical Atherosclerosis in Two Independent Cohorts. Circ. Res. 2018, 122, 1555–1564. [Google Scholar] [CrossRef]
- Washburn, N.; Schwab, I.; Ortiz, D.; Bhatnagar, N.; Lansing, J.C.; Medeiros, A.; Tyler, S.; Mekala, D.; Cochran, E.; Sarvaiya, H.; et al. Controlled tetra-Fc sialylation of IVIg results in a drug candidate with consistent enhanced anti-inflammatory activity. Proc. Natl. Acad. Sci. USA 2015, 112, E1297–E1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raju, T.S. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr. Opin. Immunol. 2008, 20, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.L.; Chung, A.W.; Rosebrock, T.R.; Ghebremichael, M.; Yu, W.H.; Grace, P.S.; Schoen, M.K.; Tafesse, F.; Martin, C.; Leung, V.; et al. A Functional Role for Antibodies in Tuberculosis. Cell 2016, 167, 433–443 e414. [Google Scholar] [CrossRef] [Green Version]
- Vestrheim, A.C.; Moen, A.; Egge-Jacobsen, W.; Reubsaet, L.; Halvorsen, T.G.; Bratlie, D.B.; Paulsen, B.S.; Michaelsen, T.E. A pilot study showing differences in glycosylation patterns of IgG subclasses induced by pneumococcal, meningococcal, and two types of influenza vaccines. Immun. Inflamm. Dis. 2014, 2, 76–91. [Google Scholar] [CrossRef]
- Markina, Y.V.; Gerasimova, E.V.; Markin, A.M.; Glanz, V.Y.; Wu, W.K.; Sobenin, I.A.; Orekhov, A.N. Sialylated Immunoglobulins for the Treatment of Immuno-Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 5472. [Google Scholar] [CrossRef]
- Goulabchand, R.; Vincent, T.; Batteux, F.; Eliaou, J.F.; Guilpain, P. Impact of autoantibody glycosylation in autoimmune diseases. Autoimmun. Rev. 2014, 13, 742–750. [Google Scholar] [CrossRef]
- Schwab, I.; Nimmerjahn, F. Intravenous immunoglobulin therapy: How does IgG modulate the immune system? Nat. Reviews. Immunol. 2013, 13, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Ito, K.; Furukawa, J.; Nakata, J.; Alvarez, M.; Verbeek, J.S.; Shinohara, Y.; Izui, S. Galactosylation of IgG1 modulates FcγRIIB-mediated inhibition of murine autoimmune hemolytic anemia. J. Autoimmun. 2013, 47, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Gornik, O.; Lauc, G. Glycosylation of serum proteins in inflammatory diseases. Dis. Markers 2008, 25, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Tertov, V.V.; Kaplun, V.V.; Sobenin, I.A.; Boytsova, E.Y.; Bovin, N.V.; Orekhov, A.N. Human plasma trans-sialidase causes atherogenic modification of low density lipoprotein. Atherosclerosis 2001, 159, 103–115. [Google Scholar] [CrossRef]
- Zhang, Z.; Wuhrer, M.; Holst, S. Serum sialylation changes in cancer. Glycoconj. J. 2018, 35, 139–160. [Google Scholar] [CrossRef] [Green Version]
- Pickup, J.C.; Mattock, M.B.; Crook, M.A.; Chusney, G.D.; Burt, D.; Fitzgerald, A.P. Serum sialic acid concentration and coronary heart disease in NIDDM. Diabetes Care 1995, 18, 1100–1103. [Google Scholar] [CrossRef]
- Afzali, B.; Bakri, R.S.; Bharma-Ariza, P.; Lumb, P.J.; Dalton, N.; Turner, N.C.; Wierzbicki, A.S.; Crook, M.A.; Goldsmith, D.J. Raised plasma total sialic acid levels are markers of cardiovascular disease in renal dialysis patients. J. Nephrol. 2003, 16, 540–545. [Google Scholar] [PubMed]
- Glanz, V.Y.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Trans-sialidase Associated with Atherosclerosis: Defining the Identity of a Key Enzyme Involved in the Pathology. Curr. Drug Targets 2019, 20, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Mehr, K.; Withers, S.G. Mechanisms of the sialidase and trans-sialidase activities of bacterial sialyltransferases from glycosyltransferase family 80. Glycobiology 2016, 26, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Kuro-o, M. Klotho and aging. Biochim. Biophys. Acta 2009, 1790, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, M.V.; Michaud, L.; Mezentsev, A.V.; Lukong, K.E.; El-Alfy, M.; Morales, C.R.; Potier, M.; Pshezhetsky, A.V. Molecular mechanism of lysosomal sialidase deficiency in galactosialidosis involves its rapid degradation. Biochem. J. 1998, 330 Pt 2, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Lukong, K.E.; Seyrantepe, V.; Landry, K.; Trudel, S.; Ahmad, A.; Gahl, W.A.; Lefrancois, S.; Morales, C.R.; Pshezhetsky, A.V. Intracellular distribution of lysosomal sialidase is controlled by the internalization signal in its cytoplasmic tail. J. Biol. Chem. 2001, 276, 46172–46181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanchetti, G.; Colombi, P.; Manzoni, M.; Anastasia, L.; Caimi, L.; Borsani, G.; Venerando, B.; Tettamanti, G.; Preti, A.; Monti, E.; et al. Sialidase NEU3 is a peripheral membrane protein localized on the cell surface and in endosomal structures. Biochem. J. 2007, 408, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Hinek, A.; Pshezhetsky, A.V.; von Itzstein, M.; Starcher, B. Lysosomal sialidase (neuraminidase-1) is targeted to the cell surface in a multiprotein complex that facilitates elastic fiber assembly. J. Biol. Chem. 2006, 281, 3698–3710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolini, L.; Orizio, F.; Busatto, S.; Radeghieri, A.; Bresciani, R.; Bergese, P.; Monti, E. Exosomes Secreted by HeLa Cells Shuttle on Their Surface the Plasma Membrane-Associated Sialidase NEU3. Biochemistry 2017, 56, 6401–6408. [Google Scholar] [CrossRef]
- Demina, E.P.; Smutova, V.; Pan, X.; Fougerat, A.; Guo, T.; Zou, C.; Chakraberty, R.; Snarr, B.D.; Shiao, T.C.; Roy, R.; et al. Neuraminidases 1 and 3 Trigger Atherosclerosis by Desialylating Low-Density Lipoproteins and Increasing Their Uptake by Macrophages. J. Am. Heart Assoc. 2021, 10, e018756. [Google Scholar] [CrossRef]
- Yang, J.; Liu, S.; Du, L.; Jiang, S. A new role of neuraminidase (NA) in the influenza virus life cycle: Implication for developing NA inhibitors with novel mechanism of action. Rev. Med. Virol. 2016, 26, 242–250. [Google Scholar] [CrossRef]
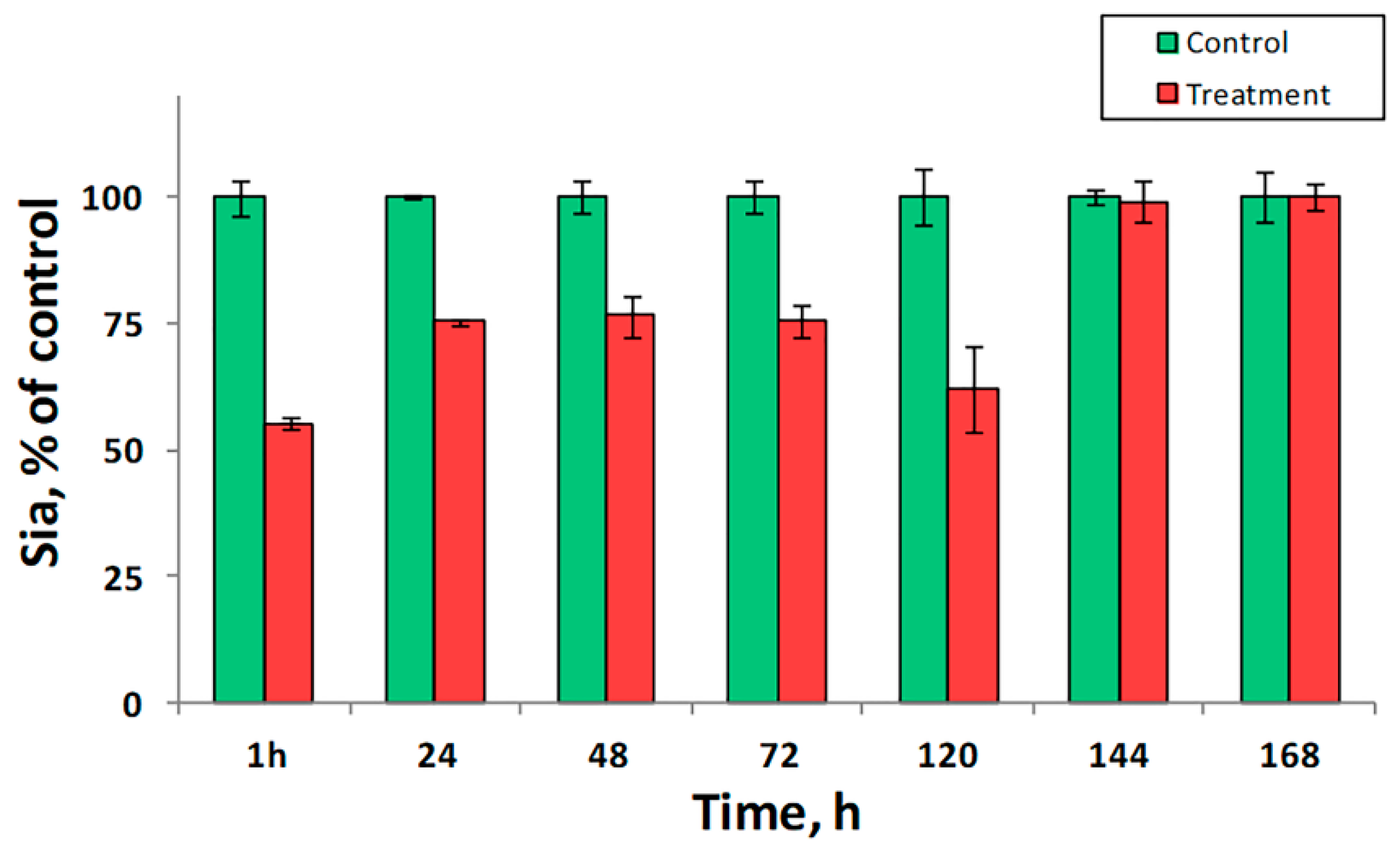
- Glanz, V.Y.; Kashirskikh, D.A.; Grechko, A.V.; Yet, S.F.; Sobenin, I.A.; Orekhov, A.N. Sialidase Activity in Human Blood Serum Has a Distinct Seasonal Pattern: A Pilot Study. Biology 2020, 9, 184. [Google Scholar] [CrossRef]
- Suo, J.; Zhao, L.; Wang, J.; Zhu, Z.; Zhang, H.; Gao, R. Influenza virus aggravates the ox-LDL-induced apoptosis of human endothelial cells via promoting p53 signaling. J. Med. Virol. 2015, 87, 1113–1123. [Google Scholar] [CrossRef]
- Birck, M.M.; Saraste, A.; Hyttel, P.; Odermarsky, M.; Liuba, P.; Saukko, P.; Hansen, A.K.; Pesonen, E. Endothelial cell death and intimal foam cell accumulation in the coronary artery of infected hypercholesterolemic minipigs. J. Cardiovasc. Transl. Res. 2013, 6, 579–587. [Google Scholar] [CrossRef]
- Peretz, A.; Azrad, M.; Blum, A. Influenza virus and atherosclerosis. QJM Mon. J. Assoc. Physicians 2019, 112, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Madjid, M.; Awan, I.; Ali, M.; Frazier, L.; Casscells, W. Influenza and atherosclerosis: Vaccination for cardiovascular disease prevention. Expert Opin. Biol. Ther. 2005, 5, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Aidoud, A.; Marlet, J.; Angoulvant, D.; Debacq, C.; Gavazzi, G.; Fougère, B. Influenza vaccination as a novel means of preventing coronary heart disease: Effectiveness in older adults. Vaccine 2020, 38, 4944–4955. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.; Barlas, Z.; Siadaty, S.; Naguib, S.; Madjid, M.; Casscells, W. Association of influenza vaccination and reduced risk of recurrent myocardial infarction. Circulation 2000, 102, 3039–3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, F.; Chen, T.L.; Shih, C.C.; Lin, C.S.; Yeh, C.C.; Lee, Y.J.; Hu, C.J.; Chiou, H.Y.; Liao, C.C. Protective effect of influenza vaccination on outcomes in geriatric stroke patients: A nationwide matched cohort study. Atherosclerosis 2019, 282, 85–90. [Google Scholar] [CrossRef]
- Phrommintikul, A.; Kuanprasert, S.; Wongcharoen, W.; Kanjanavanit, R.; Chaiwarith, R.; Sukonthasarn, A. Influenza vaccination reduces cardiovascular events in patients with acute coronary syndrome. Eur. Heart J. 2011, 32, 1730–1735. [Google Scholar] [CrossRef] [Green Version]
- Gopal, R.; Marinelli, M.A.; Alcorn, J.F. Immune Mechanisms in Cardiovascular Diseases Associated With Viral Infection. Front. Immunol. 2020, 11, 570681. [Google Scholar] [CrossRef]
- Sessa, R.; Pietro, M.D.; Filardo, S.; Turriziani, O. Infectious burden and atherosclerosis: A clinical issue. World J. Clin. Cases 2014, 2, 240–249. [Google Scholar] [CrossRef]
- Ciszewski, A. Cardioprotective effect of influenza and pneumococcal vaccination in patients with cardiovascular diseases. Vaccine 2018, 36, 202–206. [Google Scholar] [CrossRef]
- Dwarakanath, A.D.; Tsai, H.H.; Sunderland, D.; Hart, C.A.; Figura, N.; Crabtree, J.E.; Rhodes, J.M. The production of neuraminidase and fucosidase by Helicobacter pylori: Their possible relationship to pathogenicity. FEMS Immunol. Med. Microbiol. 1995, 12, 213–216. [Google Scholar] [CrossRef]
- Charakida, M.; Tousoulis, D. Infections and atheromatous plaque: Current therapeutic implications. Curr. Pharm. Des. 2013, 19, 1638–1650. [Google Scholar] [PubMed]
- Ford, P.J.; Gemmell, E.; Hamlet, S.M.; Hasan, A.; Walker, P.J.; West, M.J.; Cullinan, M.P.; Seymour, G.J. Cross-reactivity of GroEL antibodies with human heat shock protein 60 and quantification of pathogens in atherosclerosis. Oral Microbiol. Immunol. 2005, 20, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Grau, A.J.; Marquardt, L.; Lichy, C. The effect of infections and vaccinations on stroke risk. Expert Rev. Neurother. 2006, 6, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Tramontano, A.; Janda, K.D.; Lerner, R.A. Catalytic antibodies. Science 1986, 234, 1566–1570. [Google Scholar] [CrossRef] [PubMed]
- Gabibov, A.G.; Gololobov, G.V.; Makarevich, O.I.; Schourov, D.V.; Chernova, E.A.; Yadav, R.P. DNA-hydrolyzing autoantibodies. Appl. Biochem. Biotechnol. 1994, 47, 293–302; discussion 303. [Google Scholar] [CrossRef]
- Ponomarenko, N.A.; Durova, O.M.; Vorobiev, I.I.; Belogurov, A.A., Jr.; Kurkova, I.N.; Petrenko, A.G.; Telegin, G.B.; Suchkov, S.V.; Kiselev, S.L.; Lagarkova, M.A.; et al. Autoantibodies to myelin basic protein catalyze site-specific degradation of their antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Berisha, H.I.; Bratut, M.; Bangale, Y.; Colasurdo, G.; Paul, S.; Said, S.I. New evidence for transmitter role of VIP in the airways: Impaired relaxation by a catalytic antibody. Pulm. Pharmacol. Ther. 2002, 15, 121–127. [Google Scholar] [CrossRef]
- Bilyy, R.; Tomin, A.; Mahorivska, I.; Shalay, O.; Lohinskyy, V.; Stoika, R.; Kit, Y. Antibody-mediated sialidase activity in blood serum of patients with multiple myeloma. J. Mol. Recognit. 2011, 24, 576–584. [Google Scholar] [CrossRef]
- Tomin, A.; Dumych, T.; Tolstyak, Y.; Kril, I.; Mahorivska, I.; Bila, E.; Stoika, R.; Herrmann, M.; Kit, Y.; Bilyy, R. Desialylation of dying cells with catalytically active antibodies possessing sialidase activity facilitate their clearance by human macrophages. Clin. Exp. Immunol. 2015, 179, 17–23. [Google Scholar] [CrossRef]
- Alipov, V.I.; Sukhorukov, V.N.; Karagodin, V.P.; Grechko, A.V.; Orekhov, A.N. Chemical composition of circulating native and desialylated low density lipoprotein: What is the difference? Vessel Plus 2017, 1, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Rosales, C.; Gillard, B.K.; Xu, B.; Gotto, A.M., Jr.; Pownall, H.J. Revisiting Reverse Cholesterol Transport in the Context of High-Density Lipoprotein Free Cholesterol Bioavailability. Methodist Debakey Cardiovasc. J. 2019, 15, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Barenghi, L.; Bradamante, S.; Pagani, F. Modulators of oxidized LDL-induced hyperadhesiveness in human endothelial cells. Biochem. Biophys. Res. Commun. 1994, 204, 673–677. [Google Scholar] [CrossRef]
- Dimayuga, P.; Zhu, J.; Oguchi, S.; Chyu, K.Y.; Xu, X.O.; Yano, J.; Shah, P.K.; Nilsson, J.; Cercek, B. Reconstituted HDL containing human apolipoprotein A-1 reduces VCAM-1 expression and neointima formation following periadventitial cuff-induced carotid injury in apoE null mice. Biochem. Biophys. Res. Commun. 1999, 264, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Gomez Rosso, L.; Lhomme, M.; Meroño, T.; Dellepiane, A.; Sorroche, P.; Hedjazi, L.; Zakiev, E.; Sukhorukov, V.; Orekhov, A.; Gasparri, J.; et al. Poor glycemic control in type 2 diabetes enhances functional and compositional alterations of small, dense HDL3c. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Munford, R.S.; Andersen, J.M.; Dietschy, J.M. Sites of tissue binding and uptake in vivo of bacterial lipopolysaccharide-high density lipoprotein complexes: Studies in the rat and squirrel monkey. J. Clin. Investig. 1981, 68, 1503–1513. [Google Scholar] [CrossRef]
- Zeiher, A.M.; Schächlinger, V.; Hohnloser, S.H.; Saurbier, B.; Just, H. Coronary atherosclerotic wall thickening and vascular reactivity in humans. Elevated high-density lipoprotein levels ameliorate abnormal vasoconstriction in early atherosclerosis. Circulation 1994, 89, 2525–2532. [Google Scholar] [CrossRef] [Green Version]
- Kontush, A.; Chapman, M.J. Antiatherogenic small, dense HDL--guardian angel of the arterial wall? Nat. Clin. Practice. Cardiovasc. Med. 2006, 3, 144–153. [Google Scholar] [CrossRef]
- Kontush, A. HDL particle number and size as predictors of cardiovascular disease. Front. Pharmacol. 2015, 6, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin. 2017, 8, 66–77. [Google Scholar] [CrossRef]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, A.N.; Pushkarsky, T.; Oishi, Y.; Nikiforov, N.G.; Zhelankin, A.V.; Dubrovsky, L.; Makeev, V.J.; Foxx, K.; Jin, X.; Kruth, H.S.; et al. HDL activates expression of genes stimulating cholesterol efflux in human monocyte-derived macrophages. Exp. Mol. Pathol. 2018, 105, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Harada, L.M.; Carvalho, M.D.; Passarelli, M.; Quintão, E.C. Lipoprotein desialylation simultaneously enhances the cell cholesterol uptake and impairs the reverse cholesterol transport system: In vitro evidences utilizing neuraminidase-treated lipoproteins and mouse peritoneal macrophages. Atherosclerosis 1998, 139, 65–75. [Google Scholar] [CrossRef]
- Marmillot, P.; Rao, M.N.; Liu, Q.H.; Lakshman, M.R. Desialylation of human apolipoprotein E decreases its binding to human high-density lipoprotein and its ability to deliver esterified cholesterol to the liver. Metab. Clin. Exp. 1999, 48, 1184–1192. [Google Scholar] [CrossRef]
- Dobiásová, M. Lecithin: Cholesterol acyltransferase and the regulation of endogenous cholesterol transport. Adv. Lipid Res. 1983, 20, 107–194. [Google Scholar]
- Alwaili, K.; Bailey, D.; Awan, Z.; Bailey, S.D.; Ruel, I.; Hafiane, A.; Krimbou, L.; Laboissiere, S.; Genest, J. The HDL proteome in acute coronary syndromes shifts to an inflammatory profile. Biochim. Biophys. Acta 2012, 1821, 405–415. [Google Scholar] [CrossRef]
- Zakiev, E.; Rached, F.; Lhomme, M.; Darabi-Amin, M.; Ponnaiah, M.; Becker, P.H.; Therond, P.; Serrano, C.V., Jr.; Santos, R.D.; Chapman, M.J.; et al. Distinct phospholipid and sphingolipid species are linked to altered HDL function in apolipoprotein A-I deficiency. J. Clin. Lipidol. 2019, 13, 468–480 e468. [Google Scholar] [CrossRef] [PubMed]
- Artl, A.; Marsche, G.; Lestavel, S.; Sattler, W.; Malle, E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Sung, K.C.; Ryu, S.; Wild, S.H.; Byrne, C.D. An increased high-density lipoprotein cholesterol/apolipoprotein A-I ratio is associated with increased cardiovascular and all-cause mortality. Heart 2015, 101, 553–558. [Google Scholar] [CrossRef]
- Iatan, I.; Palmyre, A.; Alrasheed, S.; Ruel, I.; Genest, J. Genetics of cholesterol efflux. Curr. Atheroscler. Rep. 2012, 14, 235–246. [Google Scholar] [CrossRef]
- Sorci-Thomas, M.G.; Thomas, M.J. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc. Med. 2002, 12, 121–128. [Google Scholar] [CrossRef]
- von Eckardstein, A. Differential diagnosis of familial high density lipoprotein deficiency syndromes. Atherosclerosis 2006, 186, 231–239. [Google Scholar] [CrossRef]
- Camont, L.; Lhomme, M.; Rached, F.; Le Goff, W.; Nègre-Salvayre, A.; Salvayre, R.; Calzada, C.; Lagarde, M.; Chapman, M.J.; Kontush, A. Small, dense high-density lipoprotein-3 particles are enriched in negatively charged phospholipids: Relevance to cellular cholesterol efflux, antioxidative, antithrombotic, anti-inflammatory, and antiapoptotic functionalities. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2715–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef] [PubMed]
- Hansel, B.; Kontush, A.; Bonnefont-Rousselot, D.; Bruckert, E.; Chapman, M.J. Alterations in lipoprotein defense against oxidative stress in metabolic syndrome. Curr. Atheroscler. Rep. 2006, 8, 501–509. [Google Scholar] [CrossRef]
- Nobécourt, E.; Jacqueminet, S.; Hansel, B.; Chantepie, S.; Grimaldi, A.; Chapman, M.J.; Kontush, A. Defective antioxidative activity of small dense HDL3 particles in type 2 diabetes: Relationship to elevated oxidative stress and hyperglycaemia. Diabetologia 2005, 48, 529–538. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Why is HDL functionally deficient in type 2 diabetes? Curr. Diabetes Rep. 2008, 8, 51–59. [Google Scholar] [CrossRef]
- Curtiss, L.K.; Bonnet, D.J.; Rye, K.A. The conformation of apolipoprotein A-I in high-density lipoproteins is influenced by core lipid composition and particle size: A surface plasmon resonance study. Biochemistry 2000, 39, 5712–5721. [Google Scholar] [CrossRef] [PubMed]
- Hansel, B.; Giral, P.; Nobecourt, E.; Chantepie, S.; Bruckert, E.; Chapman, M.J.; Kontush, A. Metabolic syndrome is associated with elevated oxidative stress and dysfunctional dense high-density lipoprotein particles displaying impaired antioxidative activity. J. Clin. Endocrinol. Metab. 2004, 89, 4963–4971. [Google Scholar] [CrossRef] [PubMed]
- Perségol, L.; Vergès, B.; Foissac, M.; Gambert, P.; Duvillard, L. Inability of HDL from type 2 diabetic patients to counteract the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia 2006, 49, 1380–1386. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, E.J.; Anthanont, P.; Diffenderfer, M.R.; Polisecki, E.; Asztalos, B.F. Diagnosis and treatment of high density lipoprotein deficiency. Prog. Cardiovasc. Dis. 2016, 59, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rached, F.; Santos, R.D.; Camont, L.; Miname, M.H.; Lhomme, M.; Dauteuille, C.; Lecocq, S.; Serrano, C.V., Jr.; Chapman, M.J.; Kontush, A. Defective functionality of HDL particles in familial apoA-I deficiency: Relevance of alterations in HDL lipidome and proteome. J. Lipid Res. 2014, 55, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.D.; Schaefer, E.J.; Asztalos, B.F.; Polisecki, E.; Wang, J.; Hegele, R.A.; Martinez, L.R.; Miname, M.H.; Rochitte, C.E.; Da Luz, P.L.; et al. Characterization of high density lipoprotein particles in familial apolipoprotein A-I deficiency. J. Lipid Res. 2008, 49, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Poznyak, A.V.; Grechko, A.V.; Wetzker, R.; Orekhov, A.N. In Search for Genes Related to Atherosclerosis and Dyslipidemia Using Animal Models. Int. J. Mol. Sci. 2020, 21, 2097. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, S.; Brown, M.S.; Goldstein, J.L.; Gerard, R.D.; Hammer, R.E.; Herz, J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Investig. 1993, 92, 883–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nioi, P.; Sigurdsson, A.; Thorleifsson, G.; Helgason, H.; Agustsdottir, A.B.; Norddahl, G.L.; Helgadottir, A.; Magnusdottir, A.; Jonasdottir, A.; Gretarsdottir, S.; et al. Variant ASGR1 Associated with a Reduced Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 2131–2141. [Google Scholar] [CrossRef] [Green Version]
- Gayral, S.; Garnotel, R.; Castaing-Berthou, A.; Blaise, S.; Fougerat, A.; Berge, E.; Montheil, A.; Malet, N.; Wymann, M.P.; Maurice, P.; et al. Elastin-derived peptides potentiate atherosclerosis through the immune Neu1-PI3Kγ pathway. Cardiovasc. Res. 2014, 102, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.J.; Gyulay, G.; Lhoták, Š.; Szewczyk, M.M.; Chong, T.; Fuller, M.T.; Dadoo, O.; Fox-Robichaud, A.E.; Austin, R.C.; Trigatti, B.L.; et al. Sialidase down-regulation reduces non-HDL cholesterol, inhibits leukocyte transmigration, and attenuates atherosclerosis in ApoE knockout mice. J. Biol. Chem. 2018, 293, 14689–14706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocquet, O.; Wahart, A.; Sarazin, T.; Vincent, E.; Schneider, C.; Fougerat, A.; Gayral, S.; Henry, A.; Blaise, S.; Romier-Crouzet, B.; et al. Adverse effects of oseltamivir phosphate therapy on the liver of LDLR-/- mice without any benefit on atherosclerosis and thrombosis. J. Cardiovasc. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Ivanova, E.A. Cellular models of atherosclerosis and their implication for testing natural substances with anti-atherosclerotic potential. Phytomedicine Int. J. Phytother. Phytopharm. 2016, 23, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Sobenin, I.A.; Revin, V.V.; Bobryshev, Y.V. Development of Antiatherosclerotic Drugs on the basis of Natural Products Using Cell Model Approach. Oxidative Med. Cell. Longev. 2015, 2015, 463797. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, V.A.; Ivashinnikova, G.A.; Sobenin, I.A.; Ivanova, E.A.; Orekhov, A.N. Blood Serum Atherogenicity: Cellular Test for the Development of Anti- Atherosclerotic Therapy. Curr. Pharm. Des. 2017, 23, 1195–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukhorukov, V.N.; Karagodin, V.P.; Zakiev, E.R.; Grechko, A.V.; Orekhov, A.N. Sialidases: Therapeutic and Antiatherogenic Potential. Curr. Pharm. Des. 2017, 23, 4696–4701. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Bobryshev, Y.V.; Sobenin, I.A.; Melnichenko, A.A.; Chistiakov, D.A. Modified low density lipoprotein and lipoprotein-containing circulating immune complexes as diagnostic and prognostic biomarkers of atherosclerosis and type 1 diabetes macrovascular disease. Int. J. Mol. Sci. 2014, 15, 12807–12841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraley, A.E.; Schwartz, G.G.; Olsson, A.G.; Kinlay, S.; Szarek, M.; Rifai, N.; Libby, P.; Ganz, P.; Witztum, J.L.; Tsimikas, S. Relationship of oxidized phospholipids and biomarkers of oxidized low-density lipoprotein with cardiovascular risk factors, inflammatory biomarkers, and effect of statin therapy in patients with acute coronary syndromes: Results from the MIRACL (Myocardial Ischemia Reduction With Aggressive Cholesterol Lowering) trial. J. Am. Coll. Cardiol. 2009, 53, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Fraley, A.E.; Tsimikas, S. Clinical applications of circulating oxidized low-density lipoprotein biomarkers in cardiovascular disease. Curr. Opin. Lipidol. 2006, 17, 502–509. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Karagodin, V.P.; Melnichenko, A.C.; Bobryshev, Y.V.; Orekhov, A.N. Diagnostic and prognostic value of low density lipoprotein-containing circulating immune complexes in atherosclerosis. J. Clin. Immunol. 2013, 33, 489–495. [Google Scholar] [CrossRef]
- Wang, J.; Qiang, H.; Zhang, C.; Liu, X.; Chen, D.; Wang, S. Detection of IgG-bound lipoprotein(a) immune complexes in patients with coronary heart disease. Clin. Chim. Acta Int. J. Clin. Chem. 2003, 327, 115–122. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Sobenin, I.A.; Korneev, N.V.; Kirichenko, T.V.; Myasoedova, V.A.; Melnichenko, A.A.; Balcells, M.; Edelman, E.R.; Bobryshev, Y.V. Anti-atherosclerotic therapy based on botanicals. Recent Pat. Cardiovasc. Drug Discov. 2013, 8, 56–66. [Google Scholar] [CrossRef]
- Doo, Y.C.; Han, S.J.; Lee, J.H.; Cho, G.Y.; Hong, K.S.; Han, K.R.; Lee, N.H.; Oh, D.J.; Ryu, K.H.; Rhim, C.Y.; et al. Associations among oxidized low-density lipoprotein antibody, C-reactive protein, interleukin-6, and circulating cell adhesion molecules in patients with unstable angina pectoris. Am. J. Cardiol. 2004, 93, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Murayama, N.; Asano, Y.; Kato, K.; Sakamoto, Y.; Hosoda, S.; Yamada, N.; Kodama, T.; Murase, T.; Akanuma, Y. Effects of plasma infusion on plasma lipids, apoproteins and plasma enzyme activities in familial lecithin: Cholesterol acyltransferase deficiency. Eur. J. Clin. Investig. 1984, 14, 122–129. [Google Scholar] [CrossRef]
- Shamburek, R.D.; Bakker-Arkema, R.; Auerbach, B.J.; Krause, B.R.; Homan, R.; Amar, M.J.; Freeman, L.A.; Remaley, A.T. Familial lecithin:cholesterol acyltransferase deficiency: First-in-human treatment with enzyme replacement. J. Clin. Lipidol. 2016, 10, 356–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kootte, R.S.; Smits, L.P.; van der Valk, F.M.; Dasseux, J.L.; Keyserling, C.H.; Barbaras, R.; Paolini, J.F.; Santos, R.D.; van Dijk, T.H.; Dallinga-van Thie, G.M.; et al. Effect of open-label infusion of an apoA-I-containing particle (CER-001) on RCT and artery wall thickness in patients with FHA. J. Lipid Res. 2015, 56, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Bambauer, R.; Olbricht, C.J.; Schoeppe, E. Low-density lipoprotein apheresis for prevention and regression of atherosclerosis: Clinical results. Ther. Apher. Off. J. Int. Soc. Apher. Jpn. Soc. Apher. 1997, 1, 242–248. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Melnichenko, A.A.; Sobenin, I.A. Approach to reduction of blood atherogenicity. Oxidative Med. Cell. Longev. 2014, 2014, 738679. [Google Scholar] [CrossRef]
- Zhang, Y.; Albohy, A.; Zou, Y.; Smutova, V.; Pshezhetsky, A.V.; Cairo, C.W. Identification of selective inhibitors for human neuraminidase isoenzymes using C4,C7-modified 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA) analogues. J. Med. Chem. 2013, 56, 2948–2958. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezentsev, A.; Bezsonov, E.; Kashirskikh, D.; Baig, M.S.; Eid, A.H.; Orekhov, A. Proatherogenic Sialidases and Desialylated Lipoproteins: 35 Years of Research and Current State from Bench to Bedside. Biomedicines 2021, 9, 600. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9060600
Mezentsev A, Bezsonov E, Kashirskikh D, Baig MS, Eid AH, Orekhov A. Proatherogenic Sialidases and Desialylated Lipoproteins: 35 Years of Research and Current State from Bench to Bedside. Biomedicines. 2021; 9(6):600. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9060600
Chicago/Turabian StyleMezentsev, Alexandre, Evgeny Bezsonov, Dmitry Kashirskikh, Mirza S. Baig, Ali H. Eid, and Alexander Orekhov. 2021. "Proatherogenic Sialidases and Desialylated Lipoproteins: 35 Years of Research and Current State from Bench to Bedside" Biomedicines 9, no. 6: 600. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9060600