
Environmental Enrichment Rescues Endocannabinoid-Dependent Synaptic Plasticity Lost in Young Adult Male Mice after Ethanol Exposure during Adolescence

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Ethics Statement
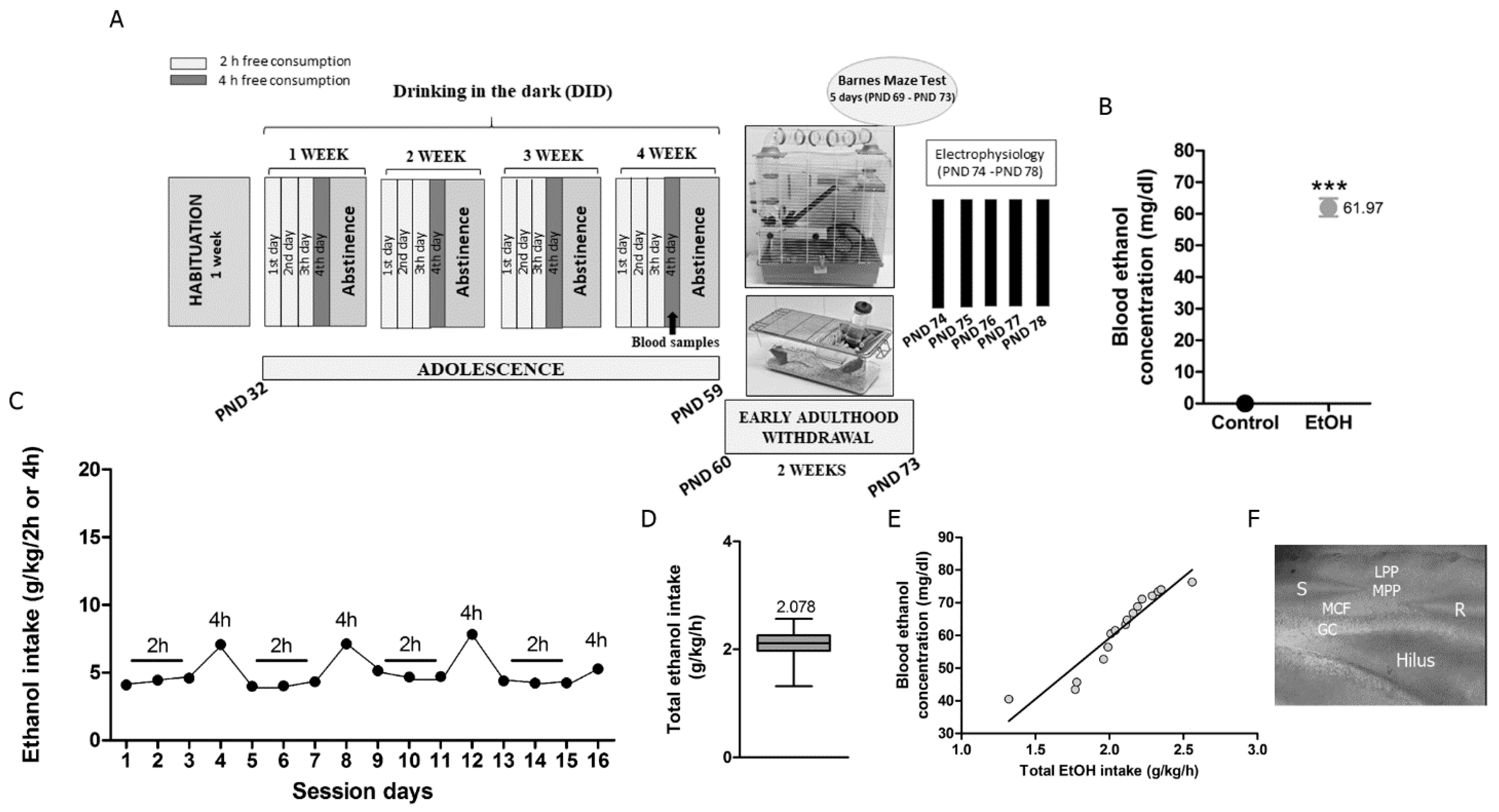
2.2. DID Procedure and Timeline
2.3. Enriched Environment
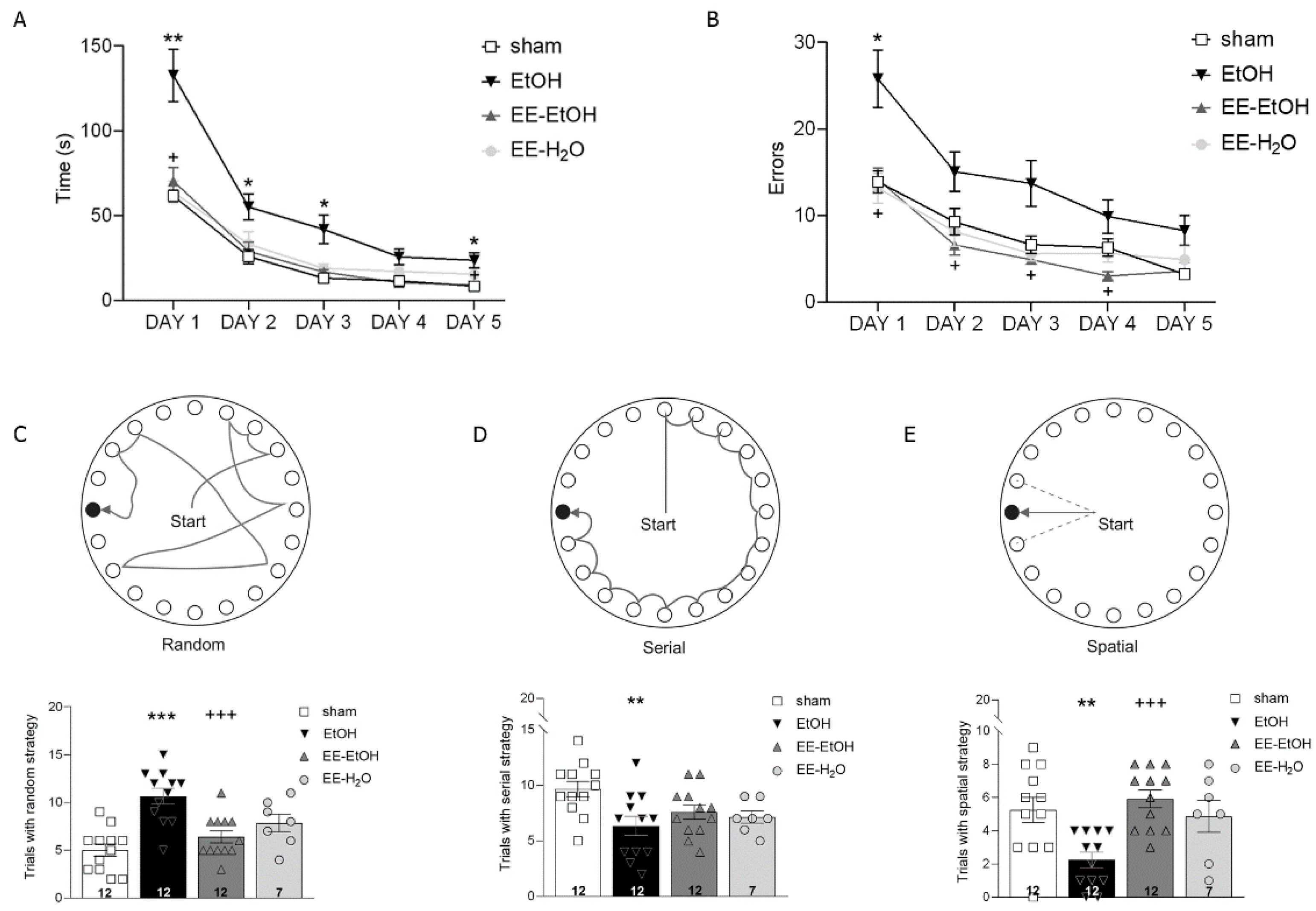
2.4. Barnes Maze Test
2.5. Slice Preparation and Extracellular Field Recordings
2.6. Statistical Analysis
3. Results
3.1. EE Recovers EtOH-Induced Memory Impairment
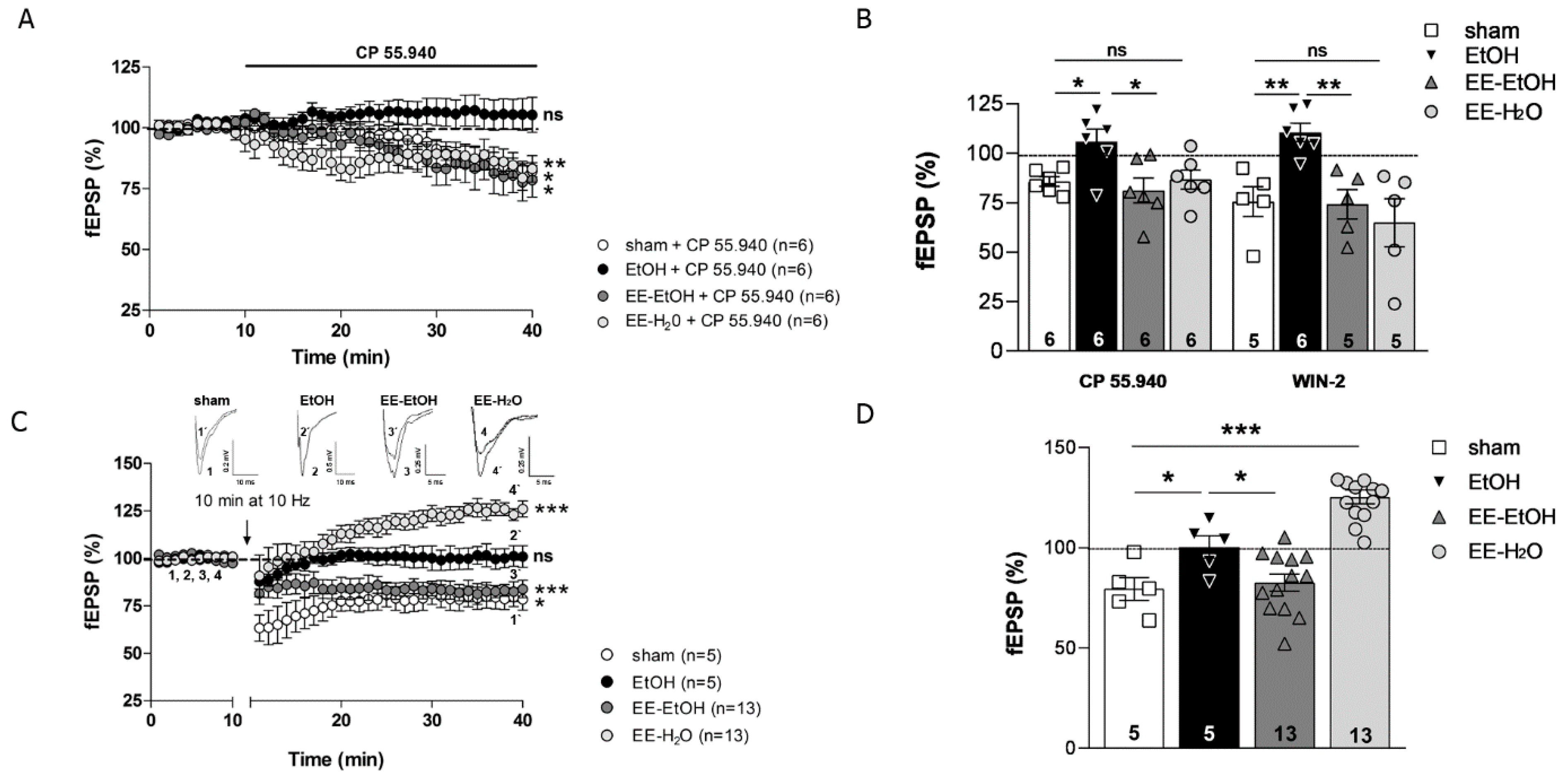
3.2. EE Recovers the CB1 Receptor-Mediated Excitatory Synaptic Transmission and MPP-LTD Lost after EtOH
3.3. EE Promotes MPP-LTP in Young Adult Mice
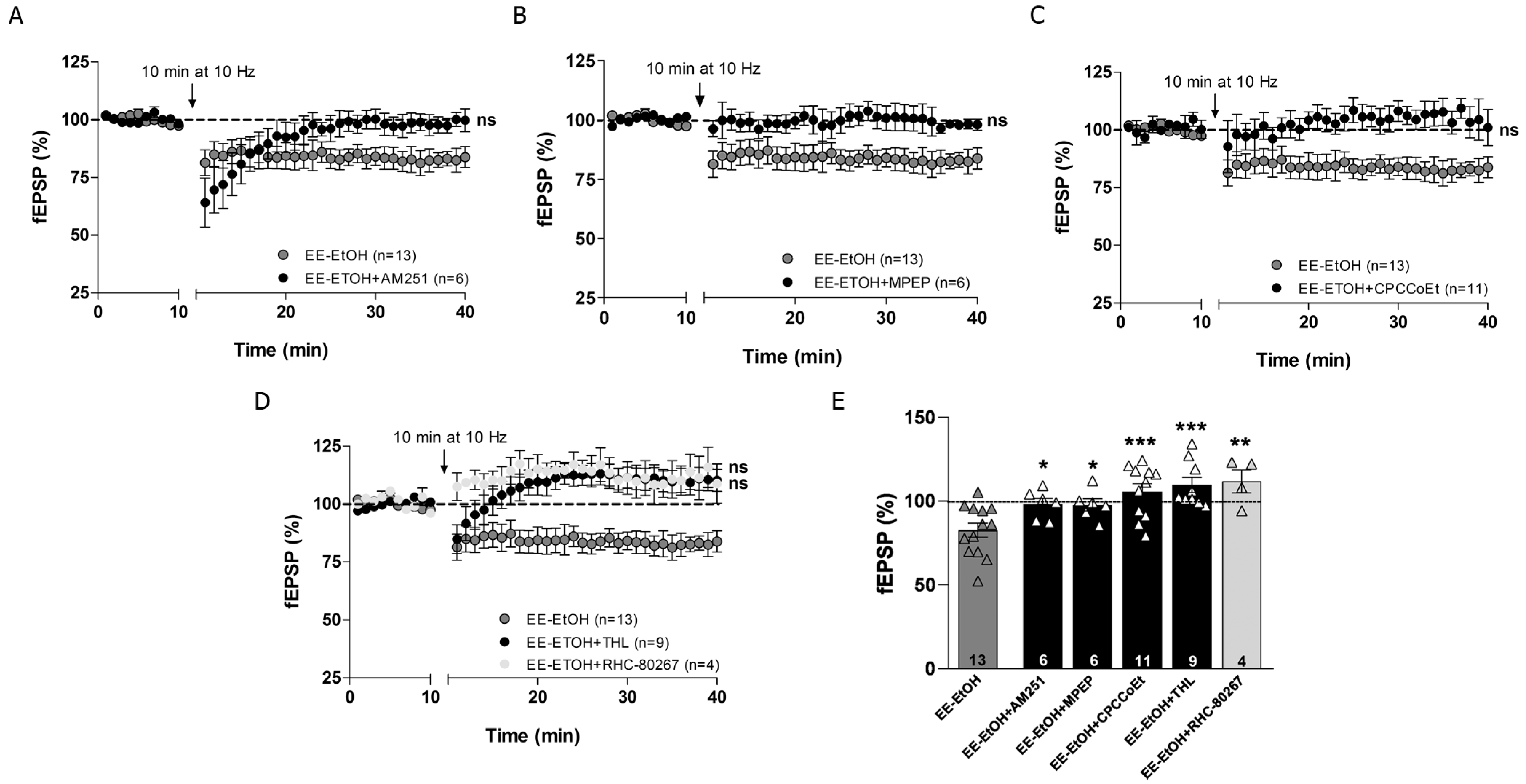
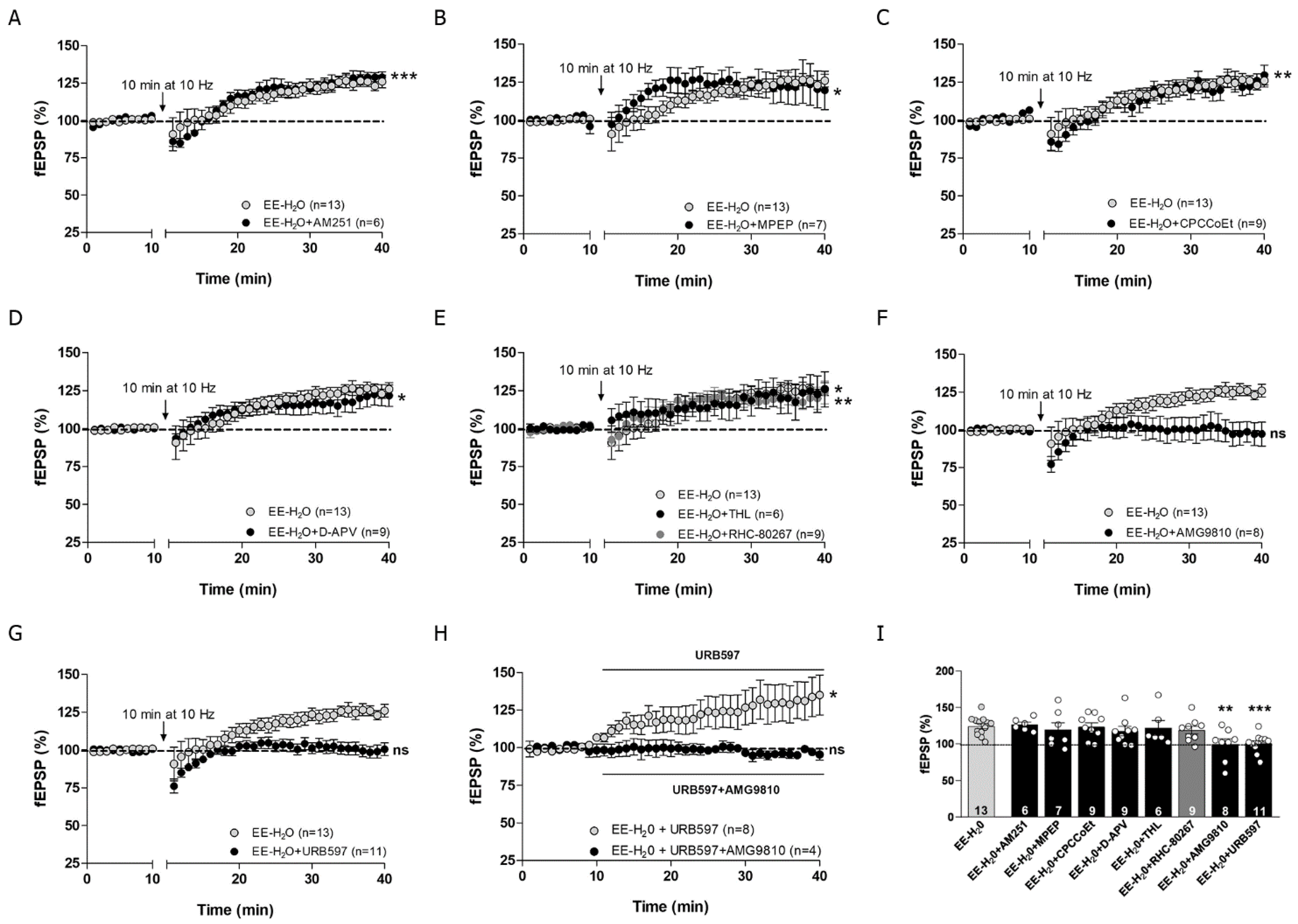
3.4. EE Involves CB1R, Group I mGluRs and 2-AG in MPP-LTD Recovery after Adolescent EtOH
3.5. MPP-LTP Elicited by EE Requires TRPV1 and AEA
4. Discussion
4.1. Mechanisms of MPP-LTD Rescue by EE in Adult Mice after Adolescent EtOH Intake
4.2. EE Prompts MPP-LTP in Water-Exposed Mice
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miguel-Hidalgo, J.J. Brain structural and functional changes in adolescents with psychiatric disorders. Int. J. Adolesc. Med. Health 2013, 25, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiller-Sturmhöfel, S.; Spear, L.P. Binge drinking’s effects on the developing brain-animal models. Alcohol Res. 2018, 39, 77–86. [Google Scholar] [PubMed]
- Crews, F.T.; Vetreno, R.P.; Broadwater, M.A.; Robinson, D.L. Adolescent alcohol exposure persistently impacts adult neurobiology and behavior. Pharmacol. Rev. 2016, 68, 1074–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briones, T.L.; Woods, J. Chronic binge-like alcohol consumption in adolescence causes depression-like symptoms possibly mediated by the effects of BDNF on neurogenesis. Neuroscience 2013, 254, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Rico-Barrio, I.; Peñasco, S.; Puente, N.; Ramos, A.; Fontaine, C.J.; Reguero, L.; Elvira, M.E.; Buceta, I.; Terradillos, I.; Lekuberri, L.; et al. Cognitive neurobehavioral benefits of an enriched environment on young adult mice after chronic ethanol consumption during adolescence. Addict. Biol. 2019, 24, 969–980. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Pereira, M.C.; Santana, L.N.D.S.; Fernandes, R.M.; Teixeira, F.B.; Oliveira, G.B.; Fernandes, L.M.; Fontes-Júnior, E.A.; Prediger, R.D.; Crespo-López, M.E.; et al. Chronic ethanol exposure during adolescence through early adulthood in female rats induces emotional and memory deficits associated with morphological and molecular alterations in hippocampus. J. Psychopharmacol. 2015, 29, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.J.M.; Phelan, K.D.; Douglas, J.C.; Wagoner, G.; Johnson, J.W.; Xu, J.; Phelan, P.S.; Drew, P.D. Effects of ethanol on immune response in the brain: Region-specific changes in adolescent versus adult mice. Alcohol Clin. Exp. Res. 2014, 38, 384–391. [Google Scholar] [CrossRef]
- Montesinos, J.; Pascual, M.; Rodríguez-Arias, M.; Miñarro, J.; Guerri, C. Involvement of TLR4 in the long-term epigenetic changes, rewarding and anxiety effects induced by intermittent ethanol treatment in adolescence. Brain Behav. Immun. 2016, 53, 159–171. [Google Scholar] [CrossRef]
- Drissi, I.; Deschamps, C.; Fouquet, G.; Alary, R.; Peineau, S.; Gosset, P.; Sueur, H.; Marcq, I.; Debuysscher, V.; Naassila, M.; et al. Memory and plasticity impairment after binge drinking in adolescent rat hippocampus: GluN2A/GluN2B NMDA receptor subunits imbalance through HDAC2. Addict. Biol. 2020, 25, e12760. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Iannotti, F.A.; Di Marzo, V.; Petrosino, S. Endocannabinoids and endocannabinoid-related mediators: Targets, metabolism and role in neurological disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Cooper, T.B.; Hungund, B.L. Chronic ethanol administration down-regulates cannabinoid receptors in mouse brain synaptic plasma membrane. Brain Res. 1998, 793, 212–218. [Google Scholar] [CrossRef]
- Ortiz, S.; Oliva, J.M.; Pérez-Rial, S.; Palomo, T.; Manzanares, J. Chronic ethanol consumption regulates cannabinoid CB1 receptor gene expression in selected regions of rat brain. Alcohol Alcohol. 2004, 39, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Vinod, K.Y.; Yalamanchili, R.; Xie, S.; Cooper, T.B.; Hungund, B.L. Effect of chronic ethanol exposure and its withdrawal on the endocannabinoid system. Expert Opin. Ther. Targets 2006, 10, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Mitrirattanakul, S.; López-Valdés, H.E.; Liang, J.; Matsuka, Y.; Mackie, K.; Faull, K.F.; Spigelman, I. Bidirectional alterations of hippocampal cannabinoid 1 receptors and their endogenous ligands in a rat model of alcohol withdrawal and dependence. Alcohol Clin. Exp. Res. 2007, 31, 855–867. [Google Scholar] [CrossRef]
- Pava, M.J.; Woodward, J.J. Chronic ethanol alters network activity and endocannabinoid signaling in the prefrontal cortex. Front. Integr. Neurosci. 2014, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peñasco, S.; Rico-Barrio, I.; Puente, N.; Fontaine, C.J.; Ramos, A.; Reguero, L.; Gerrikagoitia, I.; Rodríguez de Fonseca, F.; Suarez, J.; Barrondo, S.; et al. Intermittent ethanol exposure during adolescence impairs cannabinoid type 1 receptor-dependent long-term depression and recognition memory in adult mice. Neuropsychopharmacology 2020, 45, 309–318. [Google Scholar] [CrossRef]
- Peñasco, S.; Rico-Barrio, I.; Puente, N.; Gómez-Urquijo, S.M.; Fontaine, C.J.; Egaña-Huguet, J.; Achicallende, S.; Ramos, A.; Reguero, L.; Elezgarai, I.; et al. Endocannabinoid long-term depression revealed at medial perforant path excitatory synapses in the dentate gyrus. Neuropharmacology 2019, 153, 32–40. [Google Scholar] [CrossRef]
- Marco, E.M.; Peñasco, S.; Hernández, M.D.; Gil, A.; Borcel, E.; Moya, M.; Giné, E.; López-Moreno, J.A.; Guerri, C.; López-Gallardo, M.; et al. Long-term effects of intermittent adolescent alcohol exposure in male and female rats. Front. Behav. Neurosci. 2017, 11, 233. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, M.R.; Bennett, E.L.; Hebert, M.; Morimoto, H. Social grouping cannot account for cerebral effects of enriched environments. Brain Res. 1978, 153, 563–576. [Google Scholar] [CrossRef]
- Ohline, S.M.; Abraham, W.C. Environmental enrichment effects on synaptic and cellular physiology of hippocampal neurons. Neuropharmacology 2019, 145, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mahati, K.; Bhagya, V.; Christofer, T.; Sneha, A.; Shankaranarayana Rao, B.S. Enriched environment ameliorates depression-induced cognitive deficits and restores abnormal hippocampal synaptic plasticity. Neurobiol. Learn. Mem. 2016, 134, 379–391. [Google Scholar] [CrossRef]
- Meng, F.T.; Zhao, J.; Ni, R.J.; Fang, H.; Zhang, L.F.; Zhang, Z.; Yiu, Y.J. Beneficial effects of enriched environment on behaviors were correlated with decreased estrogen and increased BDNF in the hippocampus of male mice. Neuroendocrinol. Lett. 2015, 36, 490–497. [Google Scholar]
- Eisinger, B.E.; Zhao, X. Identifying molecular mediators of environmentally-enhanced neurogenesis. Cell Tissue Res. 2018, 371, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Garthe, A.; Roeder, I.; Kempermann, G. Mice in an enriched environment learn more flexibly because of adult hippocampal neurogenesis. Hippocampus 2016, 26, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindu, B.; Alladi, P.A.; Mansooralikhan, B.M.; Strikumar, B.N.; Raju, T.R.; Kutty, B.M. Short-term exposure to an enriched environment enhances dendritic branching but not brain-derived neurotrophic factor expression in the hippocampus of rats with ventral subicular lesions. Neuroscience 2007, 144, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Beauquis, J.; Roig, P.; De Nicola, A.F.; Saravia, F. Short-term environmental enrichment enhances adult neurogenesis, vascular network and dendritic complexity in the hippocampus of type 1 diabetic mice. PLoS ONE 2019, 5, e13993. [Google Scholar] [CrossRef]
- Bayat, M.; Sharifi, M.D.; Haghani, M.; Shabani, M. Enriched environment improves synaptic plasticity and cognitive deficiency in chronic cerebral hypoperfused rats. Brain Res. Bull. 2015, 119, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Hosseiny, S.; Pietri, M.; Petit-Paitel, A.; Zarif, H.; Heurteaux, C.; Chabry, J.; Guyon, A. Differential neuronal plasticity in mouse hippocampus associated with various periods of enriched environment during postnatal development. Brain Struct. Funct. 2015, 220, 3435–3448. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, J.; Sun, H.; Zhang, L.; Liu, H.; Zeng, X.; Yang, Y.; Yao, Z. An enriched environment reverses the synaptic plasticity deficit induced by chronic cerebral hypoperfusion. Neurosci. Lett. 2011, 502, 71–75. [Google Scholar] [CrossRef]
- Solinas, M.; Thiriet, N.; Chauvet, C.; Jaber, M. Prevention and treatment of drug addiction by environmental enrichment. Prog. Neurobiol. 2010, 92, 572–592. [Google Scholar] [CrossRef] [PubMed]
- Bonilla-Del Río, I.; Puente, N.; Peñasco, S.; Rico, I.; Gutiérrez-Rodrίguez, A.; Elezgarai, I.; Ramos, A.; Reguero, L.; Gerrikagoitia, I.; Christie, B.R.; et al. Adolescent ethanol intake alters cannabinoid type-1 receptor localization in astrocytes of the adult mouse hippocampus. Addict. Biol. 2019, 24, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.A. Memory deficits associated with senescence: A neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 1979, 93, 1–74. [Google Scholar] [CrossRef] [PubMed]
- Novkovic, T.; Mittmann, T.; Manahan-Vaughan, D. BDNF contributes to the facilitation of hippocampal synaptic plasticity and learning enabled by environmental enrichment. Hippocampus 2015, 25, 1–15. [Google Scholar] [CrossRef]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef] [Green Version]
- Merrill, J.E.; Carey, K.B. Drinking over the Lifespan: Focus on college ages. Alcohol Res. 2016, 38, 103–114. [Google Scholar]
- Pava, M.J.; Woodward, J.J. A review of the interactions between alcohol and the endocannabinoid system: Implications for alcohol dependence and future directions for research. Alcohol 2012, 46, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Nobre, M.J. Environmental enrichment may protect against neural and behavioural damage caused by withdrawal from chronic alcohol intake. Int. J. Dev. Neurosci. 2016, 55, 15–27. [Google Scholar] [CrossRef]
- Nithianantharajah, J.; Hannan, A.J. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat. Rev. Neurosci. 2006, 7, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Ickes, B.R.; Pham, T.M.; Sanders, L.A.; Albeck, D.S.; Mohammed, A.H.; Granholm, A.C. Long-term environmental enrichment leads to regional increases in neurotrophin levels in rat brain. Exp. Neurol. 2000, 164, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; De Bartolo, P.; Gelfo, F.; Foti, F.; Cutuli, D.; Bossù, P.; Caltagirone, C.; Petrosini, L. Increased concentrations of nerve growth factor and brain-derived neurotrophic factor in the rat cerebellum after exposure to environmental enrichment. Cerebellum 2009, 8, 499–506. [Google Scholar] [CrossRef]
- Alessandro, S.; Nicoletta, B.; Lamberto, M. Environment and brain plasticity: Towards an endogenous pharmacotherapy. Physiol. Rev. 2014, 94, 189–234. [Google Scholar] [CrossRef]
- Clemenson, D.C.; Wei, G.; Fred, H. Environmental enrichment and neurogenesis: From mice to humans. Curr. Opin. Behav. Sci. 2015, 4, 56–62. [Google Scholar] [CrossRef]
- Birch, A.M.; McGarry, N.B.; Kelly, A.M. Short-term environmental enrichment, in the absence of exercise, improves memory, and increases NGF concentration, early neuronal survival, and synaptogenesis in the dentate gyrus in a time-dependent manner. Hippocampus 2013, 23, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Buschler, A.; Manahan-Vaughan, D. Metabotropic glutamate receptor, mGlu5, mediates enhancements of hippocampal long-term potentiation after environmental enrichment in young and old mice. Neuropharmacology 2017, 115, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Melendez, R.I.; Gregory, M.L.; Bardo, M.T.; Kalivas, P.W. Impoverished rearing environment alters metabotropic glutamate receptor expression and function in the prefrontal cortex. Neuropsychopharmacology 2004, 29, 1980–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segovia, G.; Yagüe, A.G.; García-Verdugo, J.M.; Mora, F. Environmental enrichment promotes neurogenesis and changes the extracellular concentrations of glutamate and GABA in the hippocampus of aged rats. Brain Res. Bull. 2006, 70, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Andin, J.; Hallbeck, M.; Mohammed, A.H.; Marcusson, J. Influence of environmental enrichment on steady-state mRNA levels for EAAC1, AMPA1 and NMDA2A receptor subunits in rat hippocampus. Brain Res. 2007, 1174, 18–27. [Google Scholar] [CrossRef]
- Neyman, S.; Manahan-Vaughan, D. Metabotropic glutamate receptor 1 (mGluR1) and 5 (mGluR5) regulate late phases of LTP and LTD in the hippocampal CA1 region in vitro. Eur. J. Neurosci. 2008, 27, 1345–1352. [Google Scholar] [CrossRef]
- Popkirov, S.G.; Manahan-Vaughan, D. Involvement of the Metabotropic Glutamate Receptor mGluR5 in NMDA Receptor-Dependent, Learning-Facilitated Long-Term Depression in CA1 Synapses. Cereb. Cortex 2011, 21, 501–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basavarajappa, B.S.; Saito, M.; Cooper, T.B.; Hungund, B.L. Stimulation of cannabinoid receptor agonist 2-arachidonylglycerol by chronic ethanol and its modulation by specific neuromodulators in cerebellar granule neurons. Biochim. Biophys. Acta 2000, 1535, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Subbanna, S.; Psychoyos, D.; Xie, S.; Basavarajappa, B.S. Postnatal ethanol exposure alters levels of 2-arachidonylglycerol-metabolizing enzymes and pharmacological inhibition of monoacylglycerol lipase does not cause neurodegeneration in neonatal mice. J. Neurochem. 2015, 134, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Nomura, D.K.; Cravatt, B.F. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 2009, 16, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steindel, F.; Lerner, R.; Häring, M.; Ruehle, S.; Marsicano, G.; Lutz, B.; Monory, K. Neuron-type specific cannabinoid-mediated G protein signalling in mouse hippocampus. J. Neurochem. 2013, 124, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Green, E.J.; Greenough, W.T. Altered synaptic transmission in dentate gyrus of rats reared in complex environments: Evidence from hippocampal slices maintained in vitro. J. Neurophysiol. 1986, 55, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H.; Christie, B.R.; Sejnowski, T.J. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 13427–13431. [Google Scholar] [CrossRef] [Green Version]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Bogoslavski, D.L.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 8, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Stein, L.R.; O’Dell, K.A.; Funatsu, M. Short-term environmental enrichment enhances synaptic plasticity in hippocampal slices from aged rats. Neuroscience 2016, 329, 294–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Paillé, V.; Xu, H.; Genet, S.; Delord, B.; Fino, E.; Berry, H.; Venance, L. Endocannabinoids mediate bidirectional striatal spike-timing-dependent plasticity. J. Physiol. 2015, 593, 2833–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavanaugh, D.J.; Chesler, A.T.; Bráz, J.M.; Shah, N.M.; Julius, D.; Basbaum, A.I. Restriction of transient receptor potential Vanilloid-1 to the peptidergic subset of primary afferent neurons follows its developmental downregulation in nonpeptidergic neurons. J. Neurosci. 2011, 31, 10119–10127. [Google Scholar] [CrossRef]
- Gibson, H.E.; Edwards, J.G.; Page, R.S.; Van Hook, M.J.; Kauer, J.A. TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron 2008, 57, 746–759. [Google Scholar] [CrossRef] [Green Version]
- Chávez, A.E.; Chiu, C.Q.; Castillo, P.E. TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat. Neurosci. 2010, 13, 1511–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egaña-Huguet, J.; Bonilla-Del Río, I.; Gómez-Urquijo, S.M.; Mimenza, A.; Saumell-Esnaola, M.; Borrega-Roman, L.; García Del Caño, G.; Sallés, J.; Puente, N.; Gerrikagoitia, I.; et al. The absence of the transient receptor potential vanilloid 1 directly impacts on the expression and localization of the endocannabinoid system in the mouse hippocampus. Front. Neuroanat. 2021, 22, 645940. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gonzalo, M.; Navarrete, M.; Perea, G.; Covelo, A.; Martín-Fernández, M.; Shigemoto, R.; Luján, R.; Araque, A. Endocannabinoids induce lateral long-term potentiation of transmitter release by stimulation of gliotransmission. Cereb. Cortex 2015, 25, 3699–3712. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Prokin, I.; Xu, H.M.; Delord, B.; Genet, S.; Venance, L.; Berry, H. Endocannabinoid dynamics gate spike-timing dependent depression and potentiation. eLife 2016, 5, e13185. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Zygmunt, P.M.; Ermund, A.; Movahed, P.; Andersson, D.A.; Simonsen, C.; Jönsson, B.A.G.; Blomgren, A.; Birnir, B.; Bevan, S.; Eschalier, A.; et al. Monoacylglycerols activate TRPV1—A link between phospholipase C and TRPV1. PLoS ONE 2013, 8, e81618. [Google Scholar] [CrossRef] [Green Version]
- Caires, R.; Bell, B.; Lee, J.; Romero, L.O.; Vásquez, V.; Cordero-Morales, J.F. Deficiency of inositol monophosphatase activity decreases phosphoinositide lipids and enhances TRPV1 function in vivo. J. Neurosci. 2021, 41, 408–423. [Google Scholar] [CrossRef]
- Puente, N.; Reguero, L.; Elezgarai, I.; Canduela, M.J.; Mendizabal-Zubiaga, J.; Ramos-Uriarte, A.; Fernández-Espejo, E.; Grandes, P. The transient receptor potential vanilloid-1 is localized at excitatory synapses in the mouse dentate gyrus. Brain Struct. Funct. 2015, 220, 1187–1194. [Google Scholar] [CrossRef]
- Egertová, M.; Simon, G.M.; Cravatt, B.F.; Elphick, M.R. Localization of N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) expression in mouse brain: A new perspective on N-acylethanolamines as neural signaling molecules. J. Comp. Neurol. 2008, 506, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Nyilas, R.; Dudok, B.; Urbán, G.M.; Mackie, K.; Watanabe, M.; Cravatt, B.F.; Freund, T.F.; Katona, I. Enzymatic machinery for endocannabinoid biosynthesis associated with calcium stores in glutamatergic axon terminals. J. Neurosci. 2008, 28, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Puente, N.; Cui, Y.; Lassalle, O.; Lafourcade, M.; Georges, F.; Venance, L.; Grandes, P.; Manzoni, O.J. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat. Neurosci. 2011, 14, 1542–1547. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rico-Barrio, I.; Peñasco, S.; Lekunberri, L.; Serrano, M.; Egaña-Huguet, J.; Mimenza, A.; Soria-Gomez, E.; Ramos, A.; Buceta, I.; Gerrikagoitia, I.; et al. Environmental Enrichment Rescues Endocannabinoid-Dependent Synaptic Plasticity Lost in Young Adult Male Mice after Ethanol Exposure during Adolescence. Biomedicines 2021, 9, 825. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9070825
Rico-Barrio I, Peñasco S, Lekunberri L, Serrano M, Egaña-Huguet J, Mimenza A, Soria-Gomez E, Ramos A, Buceta I, Gerrikagoitia I, et al. Environmental Enrichment Rescues Endocannabinoid-Dependent Synaptic Plasticity Lost in Young Adult Male Mice after Ethanol Exposure during Adolescence. Biomedicines. 2021; 9(7):825. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9070825
Chicago/Turabian StyleRico-Barrio, Irantzu, Sara Peñasco, Leire Lekunberri, Maitane Serrano, Jon Egaña-Huguet, Amaia Mimenza, Edgar Soria-Gomez, Almudena Ramos, Ianire Buceta, Inmaculada Gerrikagoitia, and et al. 2021. "Environmental Enrichment Rescues Endocannabinoid-Dependent Synaptic Plasticity Lost in Young Adult Male Mice after Ethanol Exposure during Adolescence" Biomedicines 9, no. 7: 825. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9070825