
Macrophages and Foam Cells: Brief Overview of Their Role, Linkage, and Targeting Potential in Atherosclerosis

 ,
, {kind=link}
{kind=link}
Abstract
:1. Atherosclerosis
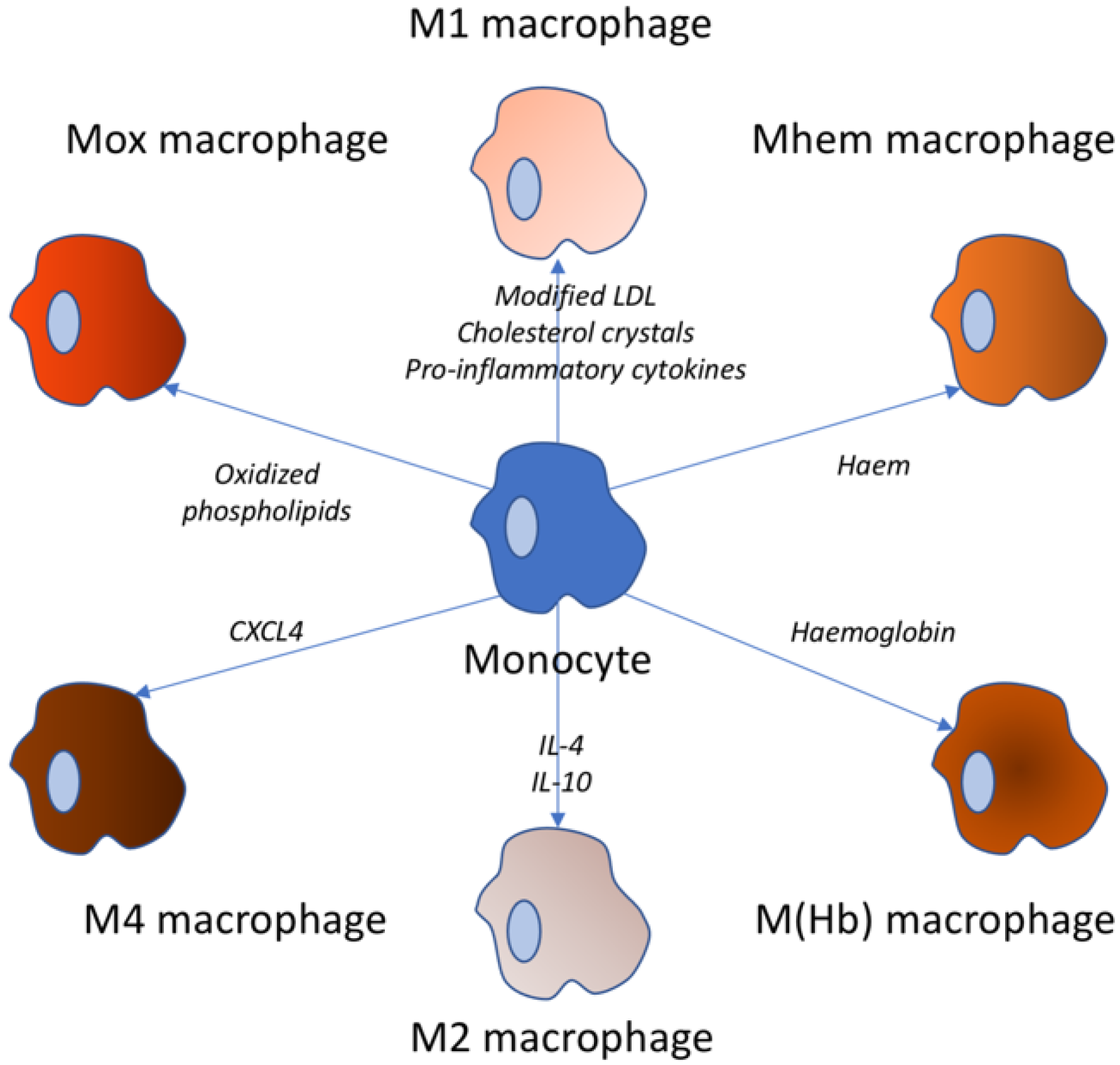
2. Inflammatory Role of Monocytes
3. Inflammatory Role of M1 and M2 Macrophages
- (1)
- M2a—M2a macrophages are induced by IL4 and IL 13 and generate increased levels of CD206 and IL-1 receptor antagonist [23];
- (2)
- M2b—M2b macrophages are extraordinary and trigger immune complexes, IL1β and PAMPs, and produce both the proinflammatory cytokines (IL-1, IL-6, and TNF-α) and the anti-inflammatory cytokine IL-10 [24];
- (3)
- M2c—the most prominent anti-inflammatory subtype triggered by IL10, TGFß, and glucocorticoids and produce IL10, TGFß and pentraxin 3 (PTX3) [25];
- (4)
- M2d—are triggered by diphtheria toxin receptor (DTR) signals and have angiogenic properties that play a role in both plaque growing and tumor development [26].
4. Inflammatory Role of Macrophages of Other Phenotypes
5. Macrophages in Atherosclerosis
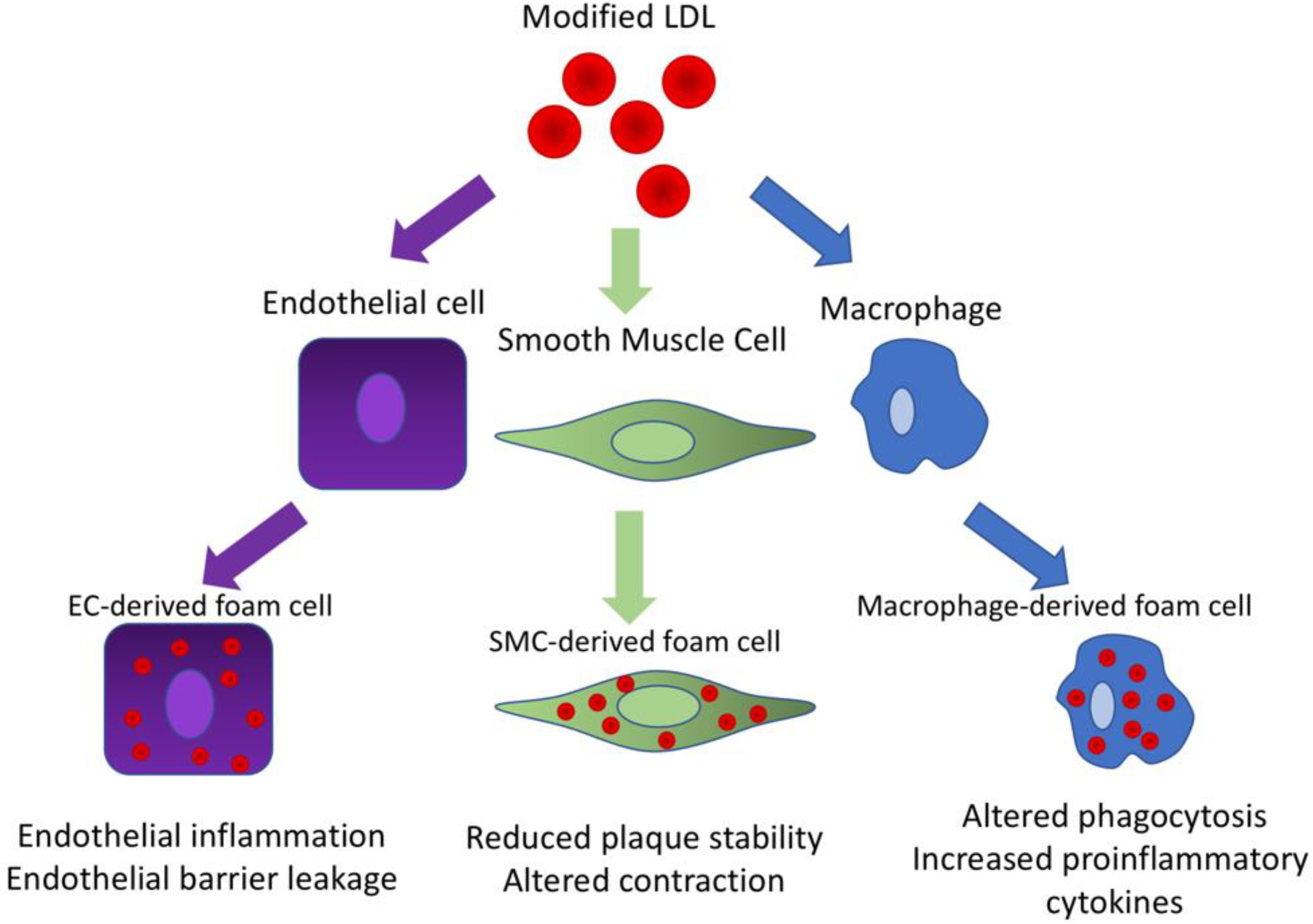
6. Foam Cells
7. Foam Cells Are Not Always Macrophage-Derived
8. Therapeutic Strategies Targeting Macrophages
9. Therapeutic Strategies Targeting Foam Cells
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, J.J.; Fang, C.H. Atheroscleritis is a more rational term for the pathological entity currently known as atherosclerosis. Med. Hypotheses 2004, 63, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Oberhuber, A.; Floros, N.; Busch, A.; Wagenhäuser, M.U.; Schelzig, H.; Duran, M. Acute Limb Ischemia-Much More Than Just a Lack of Oxygen. Int. J. Mol. Sci. 2018, 19, 374. [Google Scholar] [CrossRef] [Green Version]
- Linton, M.R.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000; Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK343489/ (accessed on 3 January 2019).
- Panuganti, K.K.; Tadi, P.; Lui, F. Transient Ischemic Attack. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021; Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK459143/ (accessed on 27 July 2021).
- Osawa, K.; Nakanishi, R.; McClelland, R.L.; Polak, J.F.; Bishop, W.; Sacco, R.L.; Ceponiene, I.; Nezarat, N.; Rahmani, S.; Qi, H.; et al. Ischemic stroke/transient ischemic attack events and carotid artery disease in the absence of or with minimal coronary artery calcification: Results from the Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2018, 275, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Mensah, G.A.; Wei, G.S.; Sorlie, P.D.; Fine, L.J.; Rosenberg, Y.; Kaufmann, P.G.; Mussolino, M.E.; Hsu, L.L.; Addou, E.; Engelgau, M.M.; et al. Decline in Cardiovascular Mortality: Possible Causes and Implications. Circ. Res. 2017, 120, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Zhang, F.; Zhang, C.; Zheng, L.R.; Yang, J. The Biomarkers for Acute Myocardial Infarction and Heart Failure. BioMed Res. Int. 2020, 17, 2018035. [Google Scholar] [CrossRef] [Green Version]
- Kapellos, T.S.; Bonaguro, L.; Gemünd, I.; Reusch, N.; Saglam, A.; Hinkley, E.R.; Schultze, J.L. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 2019, 30, 2035. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Gao, J.; Kaur, P.; Wang, Z. Neutrophils and Macrophages as Targets for Development of Nanotherapeutics in Inflammatory Diseases. Pharmaceutics 2020, 17, 1222. [Google Scholar] [CrossRef]
- Kuznetsova, T.; Prange, K.H.M.; Glass, C.K.; de Winther, M.P.J. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 216–228. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 3, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and Their Role in Atherosclerosis: Pathophysiology and Transcriptome Analysis. BioMed Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Rudra, D. Emerging Functions of Regulatory T Cells in Tissue Homeostasis. Front. Immunol. 2018, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yung, M.M.H.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression. Int. J. Mol. Sci. 2021, 22, 6560. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Corrigendum: Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2020, 11, 234, Erratum for: Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Weier, M.; Herderschee, J.; Perreau, M.; Calandra, T.; Roger, T.; Giannoni, E. IRF5 Is a Key Regulator of Macrophage Response to Lipopolysaccharide in Newborns. Front. Immunol. 2018, 9, 1597. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [Green Version]
- Raggi, F.; Pelassa, S.; Pierobon, D.; Penco, F.; Gattorno, M.; Novelli, F.; Eva, A.; Varesio, L.; Giovarelli, M.; Bosco, M.C. Regulation of Human Macrophage M1-M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front. Immunol. 2017, 8, 1097. [Google Scholar] [CrossRef]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Stoneman, V.; Braganza, D.; Figg, N.; Mercer, J.; Lang, R.; Goddard, M.; Bennett, M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ. Res. 2007, 100, 884–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, V.; Curina, A.; Genua, M.; Ghisletti, S.; Simonatto, M.; Sabò, A.; Amati, B.; Ostuni, R.; Natoli, G. Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross-talk. Nat. Immunol. 2017, 18, 530–540. [Google Scholar] [CrossRef] [Green Version]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef] [Green Version]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A.; et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.J. Heme and haemoglobin direct macrophage Mhem phenotype and counter foam cell formation in areas of intraplaque haemorrhage. Curr. Opin. Lipidol. 2012, 23, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Kondo, K.; Momiyama, Y. The Protective Role of Heme Oxygenase-1 in Atherosclerotic Diseases. Int. J. Mol. Sci. 2019, 20, 3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Jiang, J.; Chen, W.; Li, W.; Chen, Z. Vascular Macrophages in Atherosclerosis. J. Immunol. Res. 2019, 2019, 4354786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sousa, J.R.; Da Costa Vasconcelos, P.F.; Quaresma, J.A.S. Functional aspects, phenotypic heterogeneity, and tissue immune response of macrophages in infectious diseases. Infect. Drug Resist. 2019, 12, 2589–2611. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Wilson, D.P. Is Atherosclerosis a Pediatric Disease? In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000; Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK395576/ (accessed on 23 January 2020).
- Hong, Y.M. Atherosclerotic cardiovascular disease beginning in childhood. Korean Circ. J. 2010, 40, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Moss, J.W.; Ramji, D.P. Cytokines: Roles in atherosclerosis disease progression and potential therapeutic targets. Future Med. Chem. 2016, 8, 1317–1330. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Kavurma, M.M.; Rayner, K.J.; Karunakaran, D. The walking dead: Macrophage inflammation and death in atherosclerosis. Curr. Opin. Lipidol. 2017, 28, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Kim, S.Y.; Nair, M.G. Macrophages in wound healing: Activation and plasticity. Immunol. Cell Biol. 2019, 97, 258–267. [Google Scholar] [CrossRef]
- Orliaguet, L.; Ejlalmanesh, T.; Alzaid, F. Metabolic and Molecular Mechanisms of Macrophage Polarisation and Adipose Tissue Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 5731. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jiang, T.; Li, M.Q.; Zheng, X.L.; Zhao, G.J. Transcriptional Regulation of Macrophages Polarization by MicroRNAs. Front. Immunol. 2018, 9, 1175. [Google Scholar] [CrossRef]
- Bi, Y.; Chen, J.; Hu, F.; Liu, J.; Li, M.; Zhao, L. M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural. Plast. 2019, 2019, 6724903. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Palma, A.; Jarrah, A.S.; Tieri, P.; Cesareni, G.; Castiglione, F. Gene Regulatory Network Modeling of Macrophage Differentiation Corroborates the Continuum Hypothesis of Polarization States. Front. Physiol. 2018, 9, 1659. [Google Scholar] [CrossRef] [Green Version]
- He, P.; Gelissen, I.C.; Ammit, A.J. Regulation of ATP binding cassette transporter A1 (ABCA1) expression: Cholesterol-dependent and -independent signaling pathways with relevance to inflammatory lung disease. Respir. Res. 2020, 21, 250. [Google Scholar] [CrossRef]
- Joo Choi, R.; Cheng, M.S.; Shik Kim, Y. Desoxyrhapontigenin up-regulates Nrf2-mediated heme oxygenase-1 expression in macrophages and inflammatory lung injury. Redox Biol. 2014, 2, 504–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Ji, H.H.; Li, Y.J.; Guo, S.D. Macrophage Plasticity and Atherosclerosis Therapy. Front. Mol. Biosci. 2021, 8, 679797. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Changes in transcriptome of macrophages in atherosclerosis. J. Cell. Mol. Med. 2015, 19, 1163–1173. [Google Scholar] [CrossRef]
- Feig, J.E.; Fisher, E.A. Laser capture microdissection for analysis of macrophage gene expression from atherosclerotic lesions. Methods Mol. Biol. 2013, 1027, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yan, F.; Cui, J.; Wu, Y.; Luan, H.; Yin, M.; Zhao, Z.; Feng, J.; Zhang, J. Triggering Receptor Expressed on Myeloid Cells 2 Overexpression Inhibits Proinflammatory Cytokines in Lipopolysaccharide-Stimulated Microglia. Mediat. Inflamm. 2017, 2017, 9340610. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, L.; de Winther, M.P. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J. Pathol. 2020, 250, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Immunological Genome Consortium. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [Green Version]
- Zani, I.A.; Stephen, S.L.; Mughal, N.A.; Russell, D.; Homer-Vanniasinkam, S.; Wheatcroft, S.B.; Ponnambalam, S. Scavenger receptor structure and function in health and disease. Cells 2015, 4, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukhorukov, V.N.; Khotina, V.A.; Chegodaev, Y.S.; Ivanova, E.; Sobenin, I.A.; Orekhov, A.N. Lipid Metabolism in Macrophages: Focus on Atherosclerosis. Biomedicines 2020, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N. LDL and foam cell formation as the basis of atherogenesis. Curr. Opin. Lipidol. 2018, 29, 279–284. [Google Scholar] [CrossRef]
- Martin-Garrido, A.; Williams, H.C.; Lee, M.; Seidel-Rogol, B.; Ci, X.; Dong, J.T.; Lassègue, B.; Martín, A.S.; Griendling, K.K. Transforming growth factor β inhibits platelet derived growth factor-induced vascular smooth muscle cell proliferation via Akt-independent, Smad-mediated cyclin D1 downregulation. PLoS ONE 2013, 8, e79657. [Google Scholar] [CrossRef]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell. Signal. 2018, 52, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Baylis, R.A.; Durgin, B.G.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; St Hilaire, C.; Müller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef]
- Tang, R.Z.; Zhu, J.J.; Yang, F.F.; Zhang, Y.P.; Xie, S.A.; Liu, Y.F.; Yao, W.J.; Pang, W.; Han, L.L.; Kong, W.; et al. DNA methyltransferase 1 and Krüppel-like factor 4 axis regulates macrophage inflammation and atherosclerosis. J. Mol. Cell. Cardiol. 2019, 128, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Sukhovershin, R.A.; Toledano Furman, N.E.; Tasciotti, E.; Trachtenberg, B.H. Local Inhibition of Macrophage and Smooth Muscle Cell Proliferation to Suppress Plaque Progression. Methodist Debakey Cardiovasc. J. 2016, 12, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzerodt, A.; Maniecki, M.B.; Graversen, J.H.; Møller, H.J.; Torchilin, V.P.; Moestrup, S.K. Efficient intracellular drug-targeting of macrophages using stealth liposomes directed to the hemoglobin scavenger receptor CD163. J. Control. Release 2012, 160, 72–80. [Google Scholar] [CrossRef]
- Peterson, K.R.; Cottam, M.A.; Kennedy, A.J.; Hasty, A.H. Macrophage-Targeted Therapeutics for Metabolic Disease. Trends Pharmacol. Sci. 2018, 39, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Verheye, S.; Martinet, W.; Kockx, M.M.; Knaapen, M.W.; Salu, K.; Timmermans, J.P.; Ellis, J.T.; Kilpatrick, D.L.; De Meyer, G.R. Selective clearance of macrophages in atherosclerotic plaques by autophagy. J. Am. Coll. Cardiol. 2007, 49, 706–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.; César, F.A.; de Oliveira, E.M.; Turato, W.M.; Tripodi, G.L.; Castilho, G.; Machado-Lima, A.; de las Heras, B.; Boscá, L.; Rabello, M.M. New PPARγ partial agonist improves obesity-induced metabolic alterations and atherosclerosis in LDLr−/− mice. Pharmacol. Res. 2016, 104, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Javadifar, A.; Rastgoo, S.; Banach, M.; Jamialahmadi, T.; Johnston, T.P.; Sahebkar, A. Foam Cells as Therapeutic Targets in Atherosclerosis with a Focus on the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 2529. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Wong, M.M.; Potter, C.M.; Simpson, R.M.; Karamariti, E.; Zhang, Z.; Zeng, L.; Warren, D.; Hu, Y.; Wang, W.; et al. Vascular Stem/Progenitor Cell Migration Induced by Smooth Muscle Cell-Derived Chemokine (C-C Motif) Ligand 2 and Chemokine (C-X-C motif) Ligand 1 Contributes to Neointima Formation. Stem Cells 2016, 34, 2368–2380. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Nikiforov, N.G.; Starodubova, A.V.; Popkova, T.V.; Orekhov, A.N. Macrophages and Foam Cells: Brief Overview of Their Role, Linkage, and Targeting Potential in Atherosclerosis. Biomedicines 2021, 9, 1221. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9091221
Poznyak AV, Nikiforov NG, Starodubova AV, Popkova TV, Orekhov AN. Macrophages and Foam Cells: Brief Overview of Their Role, Linkage, and Targeting Potential in Atherosclerosis. Biomedicines. 2021; 9(9):1221. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9091221
Chicago/Turabian StylePoznyak, Anastasia V., Nikita G. Nikiforov, Antonina V. Starodubova, Tatyana V. Popkova, and Alexander N. Orekhov. 2021. "Macrophages and Foam Cells: Brief Overview of Their Role, Linkage, and Targeting Potential in Atherosclerosis" Biomedicines 9, no. 9: 1221. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9091221