Genetic Predisposition to Neuroblastoma


1
Department of Pediatrics, University of Chicago, Chicago, IL 60637, USA
2
Committee on Clinical Pharmacology and Pharmacogenomics, University of Chicago, Chicago, IL 60637, USA
*
Author to whom correspondence should be addressed.
Children 2018, 5(9), 119; https://0-doi-org.brum.beds.ac.uk/10.3390/children5090119
Submission received: 16 July 2018
/
Revised: 22 August 2018
/
Accepted: 28 August 2018
/
Published: 31 August 2018
(This article belongs to the Special Issue Recent Advances in Diagnosis and Treatment of Neuroblastoma)
Abstract
:Neuroblastoma is the most common solid tumor in children under the age of one. It displays remarkable phenotypic heterogeneity, resulting in differences in outcomes that correlate with clinical and biologic features at diagnosis. While neuroblastoma accounts for approximately 5% of all cancer diagnoses in pediatrics, it disproportionately results in about 9% of all childhood deaths. Research advances over the decades have led to an improved understanding of neuroblastoma biology. However, the initiating events that lead to the development of neuroblastoma remain to be fully elucidated. It has only been recently that advances in genetics and genomics have allowed researchers to unravel the predisposing factors enabling the development of neuroblastoma and fully appreciate the interplay between the genetics of tumor and host. In this review, we outline the current understanding of familial neuroblastoma and highlight germline variations that predispose children to sporadic disease. We also discuss promising future directions in neuroblastoma genomic research and potential clinical applications for these advances.
1. Introduction
Neuroblastoma is the most common tumor seen in the first year of life, and is the most frequent extracranial solid tumor of childhood [1]. It is derived from neural crest cells, and may arise in the adrenal medulla or anywhere along the paraspinal sympathetic ganglia [1]. Typically, neuroblastoma presents with bone pain, anemia, or, in babies, hepatomegaly, and has a median age at diagnosis of 18 months [1]. While neuroblastoma accounts for approximately 5% of all cancer diagnoses in pediatrics, it disproportionately results in about 9% of all childhood deaths from cancer [2].
Compared to other adult and pediatric malignancies, neuroblastoma has remarkable phenotypic heterogeneity, ranging from spontaneous regression with no treatment, to relentless progressive disease resistant, to intensive multimodal therapy [3,4,5]. Approximately 40% of children diagnosed with neuroblastoma are classified as high-risk, and only 50% of these patients will achieve long-term survival [4,6]. Conversely, patients with low- and intermediate-risk disease do quite well, achieving greater than 95% overall survival [4]. One consequence of this phenotypic heterogeneity has been the considerable effort placed on prognosticating outcome at the time of diagnosis to ensure that treatment is optimally tailored. The International Neuroblastoma Risk Group (INRG) classification system uses seven clinical and biologic factors associated with outcome at diagnosis to classify neuroblastoma tumors into four categories (very low-risk, low-risk, intermediate-risk, and high-risk) [6,7]. The expectation of this nonsurgical classification system is to allow for comparison of patient outcomes by risk group across international cooperative groups.
Despite the progress made in the treatment and risk stratification of neuroblastoma patients, it is only recently that the field has begun to appreciate the interplay between the genetics of tumor and host. Somatic genetic aberrations were identified more than three decades ago with the discovery of the prognostic significance of MYCN status and tumor ploidy [8]. The link between MYCN-amplification and aggressive disease remains one of the most significant components of risk stratification and treatment selection [6,9,10,11,12]. The importance of DNA ploidy was first described in 1984 [13] and while diploid and hypodiploid status is associated with MYCN-amplification, this genetic feature confers additional poor prognosis [1,8,14]. The late 1980s and 1990s saw the discovery of segmental chromosomal aberrations in 1p, 11q, and 17q as additional genetic markers of worse outcome [15,16,17]. Over the past 15 years, additional genomic features including chromosome copy number, transcriptomics, and epigenetics have all proven to have a role in neuroblastoma pathogenesis [18,19,20,21,22]. While these advances have allowed for an improved understanding of neuroblastoma biology, they are unable to answer a most basic question asked by patients and families: Why does my child have neuroblastoma? It has only been in the past ten years that advances in genetics have allowed researchers to begin to address this question in earnest. In this review, we will outline the current understanding of the genetics of familial neuroblastoma and will also highlight recent germline variations that predispose children to sporadic disease (Figure 1). In addition, we will discuss exciting future directions of genomic research in neuroblastoma and potential clinical applications for these advances.
2. Familial Neuroblastoma
Though the majority of neuroblastoma tumors arise sporadically, rarely this cancer is heritable. In 1967, Chatten and Voorhess were the first to recognize this heritability in their description of a family in which four out of five siblings developed early onset neuroblastoma [23]. Additional case reports of families with an unusually high incidence of the disease were described subsequently [24,25]. We now know that familial neuroblastoma occurs in approximately 1–2% of neuroblastoma cases [1]. Familial neuroblastoma tends to present at a younger age, with a family history of relatives with the same tumor, and an increased likelihood of multiple primary sites, particularly tumors involving both adrenal glands [26]. It is usually inherited in an autosomal dominant manner, with incomplete penetrance and, similar to retinoblastoma, conforms to the classic two-hit model [26,27]. Additionally, some family members harboring predisposing genetic mutations do not develop neuroblastoma [28,29]. Although familial neuroblastoma is rare, careful investigation of these cases has yielded important information on neuroblastoma tumorigenesis and identified avenues for novel targeted therapies.
2.1. PHOX2B
While the existence of familial neuroblastoma had been described for decades, it was not until 2004 that the first neuroblastoma predisposition gene, paired-like homeobox 2B gene (PHOX2B), was identified [30,31]. It was noted that patients with diseases of neural crest origin, such as congenital central hypoventilation syndrome (CCHS) and Hirschsprung disease, had a 5–10% chance of developing neuroblastic tumors, compared to 0.01% for the general population [32]. PHOX2B was first identified as a driver of these developmental disorders, and later identified in neuroblastoma patients with a family history of neuroblastic tumors or congenital malformations [30]. These insights culminated in the recognition that mutations in PHOX2B predispose children to familial neuroblastoma [30,31,33,34].
The PHOX2B gene regulates autonomic nervous system development [35] and, in children with CCHS, there is a link between specific mutations and neuroblastic tumor development [33,36]. Children with PHOX2B frameshift or missense mutations (nonpolyalanine repeat mutations) are more likely to develop more severe disease with central nervous system tumors, and Hirschsprung’s disease, than those with the more common polyalanine repeat expansion mutations (PARM) suggesting that these mutations may impart more severe disruption to gene function [33,36,37]. Both Trochet and Mosse took blood from families with familial neuroblastoma and sequenced the PHOX2B gene discovering mutations in PHOX2B that passed from parents to their children [30,34]. Of note, not all families with neuroblastoma had a PHOX2B mutation, suggesting that other predisposition genes were yet to be discovered [34], and we now know that about six to ten percent of familial neuroblastoma cases will have a mutation in PHOX2B [34,38]. Additionally, PHOX2B mutations also occur in about 2% of sporadic cases of neuroblastoma [39,40].
2.2. ALK
The anaplastic lymphoma kinase (ALK) gene was identified as a neuroblastoma predisposition gene in 2008 in two studies; one, a genetic linkage study across 20 families with neuroblastoma and the other, a genome-wide comparative genomic hybridization analysis of 592 neuroblastoma tumors [28,29]. Three germline missense mutations (R1192P, R1275Q, and G1128A), were identified in the tyrosine kinase domain of ALK that were present in the majority of familial neuroblastoma cases evaluated. Concurrently, mutations in ALK were also reported in 10–12% of sporadic cases of neuroblastoma [28,29,41,42]. In neuroblastoma, constitutive activation of the tyrosine kinase domain of ALK by multiple possible genetic mechanisms results in increased oncogenicity [28,43,44]. Linking both PHOX2B and ALK biology, some neuroblastoma cell lines, inducing overexpression of PHOX2B, led to increased ALK expression [45]. Germline ALK mutations have incomplete phenotypic penetrance, meaning that not all affected individuals will develop neuroblastoma [43]. As has been described [46], different mutations in ALK confer a specific susceptibility to targeted inhibitors such as crizotinib. The R1275Q mutation is found in both familial and sporadic tumors and has higher penetrance than the weaker activating mutation G1128A [28,47]. Cell lines with R1275Q mutations also have greater sensitivity to ALK inhibition when treated with the small molecule inhibitor of ALK, crizotinib, than those with the common somatic mutation found at F1174L [48]. The presence of ALK mutations, initially identified through the small number of familial neuroblastoma cases, has now become the basis for treatment stratification in the recently opened Children’s Oncology Group (COG) study ANBL1531 which integrates crizotinib into the upfront setting for individuals with ALK+ disease (NCT03126916).
2.3. KIF1Bβ
KIF1Bβ is a proposed tumor suppressor gene thought to be involved in the pathogenesis of neural crest tumors such as neuroblastoma and pheochromocytoma [49]. Germline mutations in this gene are proposed to provide a survival advantage to neuronal progenitor cells with malignant potential, ultimately allowing them to develop into an aggressive tumor [49]. Yeh et al. evaluated five related individuals who had both neural crest-derived tumors (neuroblastoma, ganglioneuroma, and pheochromocytomas) and non-neural crest-derived tumors (leiomyosarcoma and lung adenocarcinoma). KIF1Bβ germline mutations were found in individuals who developed neuronal tumors. Furthermore, these patients had germline findings consistent with known somatic neuroblastoma biology with haploinsufficiency or methylation of the wild-type allele of KIF1Bβ. Conversely, in a patient with lung adenocarcinoma, there was loss of the wild-type allele, consistent with classic two-hit inactivation. These results suggest a gene dosage effect for tumor development with different tissues requiring only mono-allelic inactivation, while others require bi-allelic inactivation of the KIF1Bβ gene [50].
2.4. RAS Pathway Mutations and Other Cancer Predisposition Syndromes
While the genes described above were identified in families with a predisposition specifically for neuroblastic tumors, those with other cancer predisposition syndromes can be at increased risk of developing neuroblastoma in addition to other malignancies. These syndromes primarily involve genes in the canonical RAS pathway including Costello syndrome, Noonan syndrome, and neurofibromatosis type 1 [32,51,52,53,54,55,56,57,58,59]. RAS proteins control intracellular growth, differentiation, and survival signaling, including the important downstream mitogen-activated protein (MAP) kinase pathway. Mutations that disrupt any of the key proteins in this important pathway may result in a RASopathy [60]. Neural crest cells, from which neuroblastoma originates, migrate and innervate multiple organs and rely on RAS signaling for terminal maturation. Failure of neural crest cells to develop correctly, due to RAS pathway mutations, may help explain the prevalence of neuroblastoma in these predisposition syndromes. Confirming the importance of the RAS-MAPK pathway in neuroblastoma is the observation that recurrent, sporadic tumors have high frequencies of mutations in this pathway, and these mutations are an indicator of more aggressive disease [61].
2.5. Other Predisposition Syndromes
Recently, neuroblastoma has been described in patients with Li–Fraumeni syndrome and hereditary pheochromocytoma/paragangliomas syndromes [51]. In Li–Fraumeni, the R337H mutation in TP53 appears to have a particularly strong association with the development of neuroblastoma [62,63]. ROHHAD syndrome (rapid-onset obesity, hypothalamic dysfunction, hypoventilation and autonomic dysfunction) is a rare disorder which is clinically similar to CCHS [64]. It is frequently associated with neural crest tumors, such as neuroblastoma, ganglioneuroma, or ganglioneuroblastoma; however, its genetic origins are yet to be elucidated [65,66,67]. Beckwith–Wiedemann syndrome is a syndrome of overgrowth, characterized by hemihypertrophy, macroglossia, midline abdominal wall defects, and macrosomia. It is caused by abnormal methylation of chromosome 11p15, or uniparental disomy of that region, which disrupts the expression of CKDN1C, an inhibitor of cell proliferation. Patients with this syndrome are predisposed to developing hepatoblastoma and Wilms tumor, but also have an increased incidence of neuroblastoma (2–5% risk) [68,69,70,71]. Weaver syndrome and familial paraganglioma/ pheochromocytoma have also been linked to neuroblastoma development, with mutations found in EZH2 and SDHB, respectively [72,73,74]. Fanconi anemia, a rare chromosomal instability disorder, has been associated with neuroblastoma in addition to many other cancers [75,76,77]. Although there are many genetic mutations associated with Fanconi anemia, the most common mutations associated with cancer predisposition are BRIP1, BRCA2, and PALB2 [75,78,79]. Though the exact excess risk of developing neuroblastoma has yet to be fully elucidated for these rare syndromes, a summary of known associations can be seen in Table 1. Additionally, the higher rates of neuroblastoma seen in some of these aforementioned genetic syndromes have been thought to warrant neuroblastoma surveillance, which has recently been outlined [51].
3. Sporadic Neuroblastoma
Though many cancers can be attributed to genetic changes induced by hereditary predisposition, as described in neuroblastoma for the PHOX2B and ALK genes, others may be the result of lifestyle or environmental factors. However, these modifiable factors are now thought to only account for about one-third of all cancers globally [80,81], and are not known to be contributory to the development of neuroblastoma. Additionally, perplexing in neuroblastoma tumorigenicity is the fact that only a small subset of tumors have an identifiable oncogenic driver mutation [82]. Together, these findings raise the fundamental question of why some children develop neuroblastoma, and what predisposing genetic factors may be present in these patients. Due to the inherent limitations of candidate gene and family association studies, this question could not be addressed until the development of genotyping arrays and high-performance computing allowed researchers to conduct Genome-wide association studies (GWAS) to identify germline genetic variation that may lead to development of neuroblastoma [83]. A summary of germline GWAS variants predisposing to neuroblastoma can be seen in Table 2.
3.1. Susceptibility to Sporadic Neuroblastoma
In 2008, the first successful GWAS in neuroblastoma compared 1032 neuroblastoma patients to 2043 healthy controls of European descent, using 464,934 single-nucleotide polymorphisms (SNPs) and a small replication cohort [84]. This study identified three variants in chromosome 6p22, which have been subsequently mapped to the genes CASC-15 and NBAT-1 (CASC-14), subsequently identified as long noncoding RNAs. The most statistically significant SNP was rs6939340, which was found to be more prevalent in patients who developed high-risk, MYCN-amplified, or stage 4 tumors. Subsequent studies established that this SNP is intronic to the long noncoding RNA CASC-15, and leads to a short isoform CASC15-S [85]. Additionally, expression of this isoform is decreased in high-risk tumors and patients with poor survival. Decreased expression of NBAT-1 has also been correlated with high-risk neuroblastoma [86]. Ten years after the initial GWAS, we now have a better mechanistic understanding of this genetic predisposition as CASC15 and NBAT-1 were shown to modulate the localization of the Ubiquitin-Specific Protease 36 (USP36) [87]. When these two long noncoding RNAs are lost, CHD7 is de-ubiquitinated by USP36 and then interacts with SOX9 to maintain a de-differentiated cellular state. Additional studies are ongoing to determine how this novel system can be therapeutically disrupted with the goal of differentiating neuroblastoma cells into a benign state.
As the locus in 6p22 was enriched primarily in patients who developed high-risk tumors, the next analysis of the same data set restricted the discovery cohort to those 397 high-risk patients from the original 1032 patient cohort and compared them to the same 2043 controls [88]. In addition to confirming the 6p22 locus, several new risk SNPs were identified in chromosome 2q35 that were intronic to the BARD1 gene. Moreover, these same risk SNPs have since been validated independently in an Italian cohort [89]. At the time of this discovery, it was recognized that BARD1 bound BRCA1 [90,91], though the role for BARD1 as a cancer predisposition gene was previously unknown. Functional studies of BARD1 demonstrated that a BARD1 isoform has high oncogenic activity and is sufficient for neoplastic transformation of mouse fibroblasts [92]. Furthermore, BARD1 has a mechanistic interaction with AURKA, which has an active role in stabilizing MYCN protein. In vitro analysis demonstrated that the AURKA is required for the growth of MYCN-amplified neuroblastoma cell lines [93] and the AURKA inhibitor alisertib (MLN8237) was able to inhibit the growth of neuroblastoma xenografts [94]. Unfortunately, while the drug was well tolerated, the objective response rate of 18.8% [95] was not sufficient for continued development of alisertib. This chain of research stemming from a GWAS result stands as clear proof of concept that GWAS can identify clinically actionable biology in neuroblastoma.
Over time, the cohort of genotyped neuroblastoma patients increased towards the ultimate goal of accruing 7500 affected children. Three years after the first analysis of this cohort, 1627 patients with any phenotype of neuroblastoma were compared to 3254 controls [96]. This study confirmed risk variants in CASC15 and BARD1, and identified a new locus on 11p15.4 mapping to the LMO1 gene. The LMO family of genes had been previously implicated in the development of leukemia and breast cancer [97], but this was the first time LMO1 had been linked to neuroblastoma. The variant allele, rs110419, found in 55% of neuroblastoma patients compared to 45% in healthy controls, results in a gain of function mutation that increases LMO1 expression [96]. In a zebrafish model of neuroblastoma, increased LMO1 synergizes with MYCN to promote tumorigenesis of aggressive phenotype neuroblastoma [98]. Further fine mapping and functional analysis then identified the protective variant SNP rs2168101 G > T to be more prevalent in healthy controls (31.3%) compared to neuroblastoma cases (24.2%, OR 0.65). This protective variant is located in a superenhancer element of LMO1 and the nucleotide conversion alters a GATA binding site, preventing the binding of multiple transcription factors which would otherwise lead to a malignant expression pattern [99].
Using a similar approach to what had been previously reported for the subgroup of high-risk tumors, the 574 low-risk patients were extracted from the larger cohort of 1627 neuroblastoma patients, and compared to 1722 controls matched 3:1 [100]. This GWAS led to the identification of one susceptibility SNP in DUSP12, and three SNPs in HSD17B12, statistically associated with the development of low-risk neuroblastoma [100]. Additional testing identified DDX4 and IL31RA as having interactions at the gene/variant level, suggesting that these variants also predispose children to low-risk neuroblastoma. While the mechanisms underlying these associations are not yet well understood, these findings again highlight the differing genetic underpinnings of high- and low-risk disease.
As the cohort of neuroblastoma genotyping grew, it became possible to identify variants with lower allele frequencies that were associated with the development of neuroblastoma. In 2012, Diskin et al. conducted a GWAS comparing a discovery cohort of 2101 patients, twice as many cases than four years prior, to 4202 European controls [101]. While confirming the previous findings already described, the investigators identified new risk variants in HACE1 and LIN28B that were associated with the development of neuroblastoma. Of note, this was the first GWAS in neuroblastoma to evaluate an African American cohort, confirming predisposition SNPs in HACE1 and identifying a trend towards significance for those in LIN28B. Additionally, even though it was in validation analysis, this was also the first neuroblastoma GWAS to impute SNPs based on phase 1 of the 1000 Genomes Project [102]. While HACE1 has been documented as a tumor suppressor in many cancers including neuroblastoma [103], the exact function of this gene and the associated variant have yet to be fully elucidated. In contrast, LIN28B has been shown to be overexpressed in high-risk neuroblastoma, leading to increased MYCN expression and stabilization by both inhibiting the miRNA let-7, and increasing RAN and AURKA expression [104,105].
Subsequently, this set of 2101 Caucasian patients was again compared to the same controls [106]. Due to advances in imputation and the availability of the 1000 Genomes phase 1 v3 release [107], it was possible to evaluate almost eight million SNPs, verifying previously identified loci and identifying new variants in the CPZ, MLF1, and RSRC1s genes. Combining these data with additional proteomic analysis also identified variants in NEFL that predispose children to developing high-risk neuroblastoma [108]. Capasso et al. also further analyzed this 2101 patient cohort and found a functional variant in CDKN1B that was associated with neuroblastoma risk [109]. Evaluation of copy number variants between cases and controls furthermore identified variation at 1q21.1 in the NBPF23 gene as another predisposing factor to neuroblastoma [110].
Recent efforts to better understand neuroblastoma tumor biology using next-generation sequencing of somatic tissue has also yielded new insights into predisposing germline variation from the peripheral blood. Several studies have reported a number of rare germline variants in neuroblastoma patients. The first such analysis by Pugh et al. found variants in CHEK2, PINK1, PALB2, and BARD1 [82]. Subsequent studies identified pathogenic germline variation in APC and BRCA2 [111] and SMARCA4 [112]. Despite these interesting findings, the extent to which these rare germline variants contribute to the development of neuroblastoma has yet to be fully ascertained.
3.2. Predisposition to Neuroblastoma Genotypes
The entirety of the studies reported to this point evaluated predisposition to any type of neuroblastoma or more narrowly focusing on the development of low- or high-risk disease. A recent report by Hungate et al. tested the hypothesis that in addition to phenotypic risk group, germline susceptibility loci are also associated with neuroblastoma genotype. Specifically, the hypothesis was that the development of MYCN-amplification in neuroblastoma cells and these loci would be different than those that predisposed patients to MYCN-nonamplified high-risk neuroblastoma [113]. Approximately 50% of high-risk patients have MYCN-amplified tumors, and while all tumors classified as high-risk are clinically aggressive, it is well known that the biology of MYCN-amplified and MYCN-nonamplified high-risk neuroblastoma are disparate [114,115]. This study implemented a unique case/control distribution, comparing patients with MYCN-amplified tumors to patients who developed non-high-risk neuroblastoma and then evaluating these results in conjunction with a second analysis of patients with MYCN-nonamplified high-risk disease, compared to the same controls. In this analysis of over ten million SNPs, rs80059929, which is intronic to KIF15, was found to be significantly associated with the development of MYCN-amplified, high-risk disease and not with MYCN-nonamplified high-risk disease. Additionally, while previously reported variants in BARD1 appeared to confer risk of developing high-risk disease in general, those in LMO1 were only associated with the development of MYCN-nonamplified, high-risk tumors. These results highlight potential to uncover a deeper understanding of cancer biology by utilizing alternate approaches to genomic analysis.
Shortly after these results were published, Chang et al. evaluated 113 patients with high-risk disease and loss of 11q, a genomic feature common in MYCN-nonamplified patients with poor outcomes [1], comparing them to 5109 Caucasian controls [116]. Three SNPs, the most significant being rs10895322, were identified in MMP20 that were not significant when comparing patients with MYCN-amplified tumors to the same controls. Loci at BARD1 and LMO1 were also associated with development of MYCN-nonamplified high-risk tumors, similar to previous analysis of this subgroup.
4. Future Directions
4.1. Germline Genetics Predisposing to Adverse Events
In addition to genetics predisposing children to developing neuroblastoma, additional efforts have uncovered whether germline genetics can also predispose patients to specific adverse events, such as treatment failure and the development of second cancers. Using Epstein–Barr Virus, immortalized lymphoblastoid cell lines treated with the cyclophosphamide derivative phosphoramide mustard, researchers identified two linked SNPs, rs9908694 and rs1453560, that were associated with resistance to therapy. Using a validation in a cohort of 2709 neuroblastoma patients, these SNPs were also significantly associated with decreased event-free survival (p = 0.01). rs9908694 is intronic to IKZF3, although the functional role of this variant remains to be elucidated. Additional studies are ongoing to determine if these or other SNPs are associated with survival in larger COG trials.
Epidemiologic studies have determined that neuroblastoma patients develop second malignant neoplasms (SMNs) at much higher rates than population controls [118]. Additionally, low-risk patients who received little to no chemotherapy or radiation also develop SMNs at increased rates, suggesting there may be additional genetic risk factors for this outcome beyond those described for conventional cytotoxic treatments [119]. Indeed, this hypothesis has been tested and validated in other tumor types [120]. To evaluate this question in neuroblastoma, data from the INRG Database were mined for patients who developed a second malignancy and had genotyping information to compare to similar patients who did not develop this outcome [121]. Using a candidate SNP approach, variants in two DNA repair genes XRCC3 (rs861539) and MSH2 (rs17036651) were identified as having the potential to predispose patients to developing second malignant neoplasms. However, these findings still require validation in larger cohorts.
4.2. Beyond Caucasians in Sporadic Neuroblastoma
To date, most GWAS in neuroblastoma has been restricted to patients of European decent. We do know, however, that African American and Native American patients are more likely to have high-risk neuroblastoma and worse event-free survival compared to Caucasians [122]. This suggests that different ethnic and racial groups are likely to have unique predisposing genetic factors [123,124]. Furthermore, deficiencies of understanding the differences in genetic predispositions between ethnicities can lead to unsuccessful applications of genomics in the clinic [125,126]. While some studies in neuroblastoma have used an African American cohort to validate findings from Caucasians [99,101,127], primary genome-wide analyses of race and ethnicities other than Caucasians have been sparse. This is in part because of the difficulties assembling a large non-Caucasian neuroblastoma cohort in the United States and, until recently, limited availability of haplotype blocks for non-Caucasians. Despite these challenges, Gamazon et al. performed a candidate SNP evaluation comparing 310 African American patients to 2709 neuroblastoma patients of European decent [117]. This analysis identified a novel variant in SPAG16 that is significantly more common in African American children who developed high-risk neuroblastoma and rarely present in Caucasians. Furthermore, this variant was also associated with worse event-free survival (p = 0.007) and incorporation of this genotype into a Cox model of outcome negates the difference in survival previously observed between racial/ethnic groups. Additionally, a cohort of southern Chinese patients have been evaluated to validate specific neuroblastoma predisposition variants [128]. However, despite almost 1000 patients in the cohort, a true GWAS of this population has not been reported and risk variants specific to this population have yet to be identified. Overall predisposing factors to developing neuroblastoma in patients of non-European decent should be a priority for the field as these studies may find additional biologic features of this disease.
4.3. Collaboration with Therapeutic and Technological Advances
Insights into both familial and sporadic cases of neuroblastoma have led to an improved understanding of the fundamental biology that promotes neuroblastoma tumorigenesis. These discoveries continue to drive hypotheses for clinical translation and have generated avenues for targeted therapeutic development. Targeted therapies have been developed for ALK+ neuroblastoma and others are in development for many of the genetic aberrations described. In addition to the phase 3 trial evaluating crizotinib in newly diagnosed patients (NCT03126916), there is the recently closed phase 2 study combining crizotinib with conventional chemotherapy in pediatric solid tumors and anaplastic large-cell lymphoma (NCT01606878). Newer generations of ALK inhibitors such as ceritinib and lorlatinib, which can overcome resistance to crizotinib, are also under investigation (NCT03107988 and NCT01742286). Although these trials focus on children already diagnosed with neuroblastoma, it is also conceivable that children diagnosed with a germline ALK mutation early in life could benefit from regular screening for neuroblastoma and early initiation of an ALK inhibitor may be warranted.
Recognizing the need to direct more funds towards understanding genetic predispositions to pediatric diseases, the Gabriella Miller Kids First Research Act was signed in 2014. The law eliminates taxpayer dollars from financing presidential campaigns and party conventions, instead directing these funds into an approximately 126 million-dollar, 10-year pediatric research initiative through the National Institutes of Health (NIH). Since it launched in 2015, more than 18,000 samples from patients and their families have been collected and sequencing data is publicly available for more than 2000 patients in the database of Genotypes and Phenotypes (dbGaP). This fund is currently providing support to sequence 563 neuroblastoma patients and their parents. The impact of the Gabriella Miller Kids First Program is likely to be vast and represents another exciting collaborative environment to further our understanding of the genetic basis of pediatric cancer as a whole.
Massively paralleled genomic technologies, such as whole exome and whole genome sequencing, have improved and become more affordable, leading to massive amounts of data that are often siloed within institutions and difficult to compare between data sets. This is true not only for analysis of tumor tissue, but also for germline variance as demonstrated by the Gabriella Miller project. Issues of interoperability and data harmonization between studies have necessitated the recent development of data commons to house, merge, and serve these genomic data to researchers. To date, the largest such effort is the Genomic Data Commons (GDC), sponsored by the NIH and the National Cancer Institute (NCI). This data commons contains raw and processed somatic and germline genomic data linked to clinical and histological information from NCI-funded projects [129]. Neuroblastoma is fundamentally a rare disease, and pooling information in this manner allows researchers to have the sample sizes required to detect important biology from germline sequencing data [129].
While the GDC currently hosts data for neuroblastoma and other pediatric and adult tumors, neuroblastoma has long led the field in these types of efforts. The INRG database has been collating and harmonizing clinical data for almost a decade [6]. As the database has grown to over 20,000 neuroblastoma patients treated worldwide, efforts have been undertaken to link these rich clinical data to available genomic datasets. Germline genetics are available on a large subset of these patients allowing researchers to more readily link germline variation with clinical phenotype and genotype. Recognizing the importance of such efforts, additional pediatric oncology disease groups such as soft tissue sarcoma and germ cell tumors have begun to form their own data commons to better harness the power of combining genomic datasets. These efforts underscore the importance of data sharing within the scientific community to advance the care for children with neuroblastoma and identify those who may be predisposed to the disease.
5. Conclusions
Despite considerable advances in classification and treatment of neuroblastoma, our understanding of the genetic causes of neuroblastoma is still modest, and we struggle to clinically identify which children are at highest risk of the disease. While the full genetic underpinnings of neuroblastoma have yet to be fully elucidated, meaningful progress has been made. The development of new technologies and laboratory methods, combined with improved data sharing, is sure to deliver many more discoveries, allowing for an ever-greater understanding of the genetic complexity of the disease. Ultimately, this knowledge must be applied towards the goal of curing children with neuroblastoma with minimal therapeutic toxicity.
Author Contributions
Conceptualization, E.K.B. and M.A.A.; Writing—Original Draft Preparation, E.K.B. and M.A.A.; Writing—Review & Editing E.K.B. and M.A.A.; Visualization, E.K.B. and M.A.A.
Funding
This research was supported in part by the Conquer Cancer Foundation, Cancer Research Foundation, and NIH Grant Numbers K12CA139160 and K08CA226237 (MAA). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.L.; Schmidt, M.L.; Cohn, S.L.; Maris, J.M.; London, W.B.; Buxton, A.; Stram, D.; Castleberry, R.P.; Shimada, H.; Sandler, A.; et al. Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N. Engl. J. Med. 2010, 363, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in risk classification and treatment strategies for neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Nuchtern, J.G.; London, W.B.; Barnewolt, C.E.; Naranjo, A.; McGrady, P.W.; Geiger, J.D.; Diller, L.; Schmidt, M.L.; Maris, J.M.; Cohn, S.L.; et al. A prospective study of expectant observation as primary therapy for neuroblastoma in young infants a children’s oncology group study. Ann. Surg. 2012, 256, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The international neuroblastoma risk group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The international neuroblastoma risk group (INRG) staging system: An INRG task force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Look, A.T.; Hayes, F.A.; Shuster, J.J.; Douglass, E.C.; Castleberry, R.P.; Bowman, L.C.; Smith, E.I.; Brodeur, G.M. Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma: A pediatric oncology group study. J. Clin. Oncol. 1991, 9, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Varmus, H.E.; Bishop, J.M. Human N-myc gene contributes to neoplastic transformation of mammalian cells in culture. Nature 1985, 316, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Alitalo, K.; Klempnauer, K.-H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Look, A.T.; Hayes, F.A.; Nitschke, R.; McWilliams, N.B.; Green, A.A. Cellular DNA content as a predictor of response to chemotherapy in infants with unresectable neuroblastoma. N. Engl. J. Med. 1984, 311, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Oppedal, B.R.; Storm-Mathisen, I.; Lie, S.O.; Brandtzaeg, P. Prognostic factors in neuroblastoma. Clinical, histopathologic, and immunohistochemical features and DNA ploidy in relation to prognosis. Cancer 1988, 62, 772–780. [Google Scholar] [CrossRef]
- Fong, C.T.; Dracopoli, N.C.; White, P.S.; Merrill, P.T.; Griffith, R.C.; Housman, D.E.; Brodeur, G.M. Loss of heterozygosity for the short arm of chromosome 1 in human neuroblastomas: Correlation with N-myc amplification. Proc. Natl. Acad. Sci. USA 1989, 86, 3753–3757. [Google Scholar] [CrossRef] [PubMed]
- Bown, N.; Cotterill, S.; Lastowska, M.; O’Neill, S.; Pearson, A.D.; Plantaz, D.; Meddeb, M.; Danglot, G.; Brinkschmidt, C.; Christiansen, H.; et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 1999, 340, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; White, P.S.; Weiss, M.J.; Hogarty, M.D.; Thompson, P.M.; Stram, D.O.; Gerbing, R.; Matthay, K.K.; Seeger, R.C.; Brodeur, G.M.; et al. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene 1999, 18, 4948–4957. [Google Scholar] [CrossRef] [PubMed]
- Kaat, D.; Frank, S. Epigenetic regulation of neuroblastoma development. Cell Tissue Res. 2018, 372, 309–324. [Google Scholar] [Green Version]
- Olsson, M.; Beck, S.; Kogner, P.; Martinsson, T.; Carén, H. Genome-wide methylation profiling identifies novel methylated genes in neuroblastoma tumors. Epigenetics 2016, 11, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleiermacher, G.; Mosseri, V.; London, W.B.; Maris, J.M.; Brodeur, G.M.; Attiyeh, E.; Haber, M.; Khan, J.; Nakagawara, A.; Speleman, F.; et al. Segmental chromosomal alterations have prognostic impact in neuroblastoma: A report from the INRG project. Br. J. Cancer 2012, 107, 1418–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decock, A.; Ongenaert, M.; Cannoodt, R.; Verniers, K.; De Wilde, B.; Laureys, G.; Van Roy, N.; Berbegall, A.P.; Bienertova-Vasku, J.; Bown, N. Methyl-CpG-binding domain sequencing reveals a prognostic methylation signature in neuroblastoma. Oncotarget 2016, 7, 1960–1972. [Google Scholar] [CrossRef] [PubMed]
- Westermann, F.; Schwab, M. Genetic parameters of neuroblastomas. Cancer Lett. 2002, 184, 127–147. [Google Scholar] [CrossRef]
- Chatten, J.; Voorhess, M.L. Familial neuroblastoma. N. Engl. J. Med. 1967, 277, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Hanenson, I.B.; Lampkin, B.C. Familial neuroblastoma. Am. J. Dis. Child. 1971, 121, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Hardy, P.C.; Nesbit, M.E., Jr. Familial neuroblastoma: Report of a kindred with a high incidence of infantile tumors. J. Pediatr. 1972, 80, 74–77. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr.; Strong, L.C. Mutation and cancer: Neuroblastoma and pheochromocytoma. Am. J. Hum. Genet. 1972, 24, 514–532. [Google Scholar] [PubMed]
- Maris, J.M.; Weiss, M.J.; Mosse, Y.; Hii, G.; Guo, C.; White, P.S.; Hogarty, M.D.; Mirensky, T.; Brodeur, G.M.; Rebbeck, T.R.; et al. Evidence for a hereditary neuroblastoma predisposition locus at chromosome 16p12-13. Cancer Res. 2002, 62, 6651–6658. [Google Scholar] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; Bourdeaut, F.; Janoueix-Lerosey, I.; Deville, A.; de Pontual, L.; Schleiermacher, G.; Coze, C.; Philip, N.; Frebourg, T.; Munnich, A.; et al. Germline mutations of the paired-like homeobox 2b (PHOX2B) gene in neuroblastoma. Am. J. Hum. Genet. 2004, 74, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Bourdeaut, F.; Trochet, D.; Janoueix-Lerosey, I.; Ribeiro, A.; Deville, A.; Coz, C.; Michiels, J.F.; Lyonnet, S.; Amiel, J.; Delattre, O. Germline mutations of the paired-like homeobox 2b (PHOX2B) gene in neuroblastoma. Cancer Lett. 2005, 228, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, T.; Trachsel, D.; Engelcke, G.; Hammer, J. Congenital central hypoventilation syndrome associated with hirschsprung’s disease and neuroblastoma: Case of multiple neurocristopathies. Pediatr. Pulmonol. 2002, 33, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; Hong, S.J.; Lim, J.K.; Brunet, J.F.; Munnich, A.; Kim, K.S.; Lyonnet, S.; Goridis, C.; Amiel, J. Molecular consequences of PHOX2B missense, frameshift and alanine expansion mutations leading to autonomic dysfunction. Hum. Mol. Genet. 2005, 14, 3697–3708. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Khazi, D.; Carlisle, A.J.; Winter, C.L.; Rappaport, E.; Maris, J.M. Germline PHOX2B mutation in hereditary neuroblastoma. Am. J. Hum. Genet. 2004, 75, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Pattyn, A.; Morin, X.; Cremer, H.; Goridis, C.; Brunet, J.F. The homeobox gene PHOX2B is essential for the development of autonomic neural crest derivatives. Nature 1999, 399, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; O’Brien, L.M.; Gozal, D.; Trang, H.; Nordenskjold, A.; Laudier, B.; Svensson, P.J.; Uhrig, S.; Cole, T.; Niemann, S.; et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am. J. Hum. Genet. 2005, 76, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.M.; Zhou, L.; Rand, C.M.; Weese-Mayer, D.E. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am. J. Respir. Crit. Care Med. 2006, 174, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Raabe, E.H.; Laudenslager, M.; Winter, C.; Wasserman, N.; Cole, K.; LaQuaglia, M.; Maris, D.J.; Mosse, Y.P.; Maris, J.M. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 2008, 27, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Van Limpt, V.; Schramm, A.; van Lakeman, A.; Sluis, P.; Chan, A.; van Noesel, M.; Baas, F.; Caron, H.; Eggert, A.; Versteeg, R. The PHOX2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 2004, 23, 9280–9288. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Haberle, B.; Konig, I.R.; Kappler, R.; Suttorp, M.; Schackert, H.K.; Roesner, D.; Fitze, G. Rare occurrence of PHOX2B mutations in sporadic neuroblastomas. J. Pediatr. Hematol. Oncol. 2008, 30, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azarova, A.M.; Gautam, G.; George, R.E. Emerging importance of ALK in neuroblastoma. Semin. Cancer Boil. 2011, 21, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransson, S.; Hansson, M.; Ruuth, K.; Djos, A.; Berbegall, A.; Javanmardi, N.; Abrahamsson, J.; Palmer, R.H.; Noguera, R.; Hallberg, B.; et al. Intragenic anaplastic lymphoma kinase (ALK) rearrangements: Translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosom. Cancer 2015, 54, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Bachetti, T.; Di Paolo, D.; Di Lascio, S.; Mirisola, V.; Brignole, C.; Bellotti, M.; Caffa, I.; Ferraris, C.; Fiore, M.; Fornasari, D.; et al. Phox2b-mediated regulation of ALK expression: In vitro identification of a functional relationship between two genes involved in neuroblastoma. PLoS ONE 2010, 5, e13108. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, M.A.; Desai, A.V.; Bender, J.L.G.; Cohn, S.L. Emerging and investigational therapies for neuroblastoma. Expert Opin. Orphan Drugs 2017, 5, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, V.P.; Coggins, G.E.; Maris, J.M. Genetic susceptibility to neuroblastoma. Curr. Opin. Genet. Dev. 2017, 42, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, A.C.; Laudenslager, M.; Haglund, E.A.; Attiyeh, E.F.; Pawel, B.; Courtright, J.; Plegaria, J.; Christensen, J.G.; Maris, J.M.; Mosse, Y.P. Inhibition of ALK mutated neuroblastomas by the selective inhibitor PF-02341066. J. Clin. Oncol. 2009, 27, 10008b. [Google Scholar] [CrossRef]
- Schlisio, S.; Kenchappa, R.S.; Vredeveld, L.C.; George, R.E.; Stewart, R.; Greulich, H.; Shahriari, K.; Nguyen, N.V.; Pigny, P.; Dahia, P.L.; et al. The kinesin KIF1Bβ acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008, 22, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.T.; Lenci, R.E.; Qin, Y.; Buddavarapu, K.; Ligon, A.H.; Leteurtre, E.; Cao, C.D.; Cardot-Bauters, C.; Pigny, P.; Dahia, P.L.M. A germline mutation of the KIF1Bβ gene on 1p36 in a family with neural and nonneural tumors. Hum. Genet. 2008, 124, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Kamihara, J.; Bourdeaut, F.; Foulkes, W.D.; Molenaar, J.J.; Mosse, Y.P.; Nakagawara, A.; Parareda, A.; Scollon, S.R.; Schneider, K.W.; Skalet, A.H.; et al. Retinoblastoma and neuroblastoma predisposition and surveillance. Clin. Cancer Res. 2017, 23, e98–e106. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Miranda, B.; Westra, S.J.; Boechat, M.I.; Yazdani, S. Noonan syndrome associated with neuroblastoma: A case report. Pediatr. Radiol. 1997, 27, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Cotton, J.L.; Williams, R.G. Noonan syndrome and neuroblastoma. Arch. Pediatr. Adolesc. Med. 1995, 149, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.A.; Allanson, J.E.; Dahlgren, J.; Gelb, B.D.; Hall, B.; Pierpont, M.E.; Roberts, A.E.; Robinson, W.; Takemoto, C.M.; Noonan, J.A. Noonan syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics 2010, 126, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Origone, P.; Defferrari, R.; Mazzocco, K.; Lo Cunsolo, C.; De Bernardi, B.; Tonini, G.P. Homozygous inactivation of nf1 gene in a patient with familial NF1 and disseminated neuroblastoma. Am. J. Med. Genet. A 2003, 118A, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Martinsson, T.; Sjöberg, R.-M.; Hedborg, F.; Kogner, P. Homozygous deletion of the neurofibromatosis-1 gene in the tumor of a patient with neuroblastoma. Cancer Genet. Cytogenet. 1997, 95, 183–189. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef] [PubMed]
- Moroni, I.; Bedeschi, F.; Luksch, R.; Casanova, M.; D’Incerti, L.; Uziel, G.; Selicorni, A. Costello syndrome: A cancer predisposing syndrome? Clin. Dysmorphol. 2000, 9, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W.; Lin, A.E. Costello syndrome: A Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet. Med. 2012, 14, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Tidyman, W.E.; Rauen, K.A. The rasopathies: Developmental syndromes of RAS/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009, 19, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Seidinger, A.L.; Fortes, F.P.; Mastellaro, M.J.; Cardinalli, I.A.; Zambaldi, L.G.; Aguiar, S.S.; Yunes, J.A. Occurrence of neuroblastoma among TP53 p.R337H carriers. PLoS ONE 2015, 10, e0140356. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Capasso, M.; Diamond, M.; Oldridge, D.A.; Conkrite, K.; Bosse, K.R.; Russell, M.R.; Iolascon, A.; Hakonarson, H.; Devoto, M.; et al. Rare variants in TP53 and susceptibility to neuroblastoma. J. Natl. Cancer Inst. 2014, 106, dju047. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.; Priante, E.; Pettenazzo, A.; Marson, P.; Suppiej, A.; Benini, F.; Perilongo, G.; Toldo, I. Intrathecal synthesis of oligoclonal bands in rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation syndrome: New evidence supporting immunological pathogenesis. J. Child. Neurol. 2013, 29, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, N.; Berard, E.; Chastagner, P.; Feillet, F.; Wagner, K.; Sommelet, D. Hypothalamic dysfunction associated with neuroblastoma: Evidence for a new paraneoplastic syndrome? Med. Pediatr. Oncol. 2003, 40, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Bougneres, P.; Pantalone, L.; Linglart, A.; Rothenbuhler, A.; Le Stunff, C. Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J. Clin. Endocrinol. Metab. 2008, 93, 3971–3980. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.S.; McGrath, S.; Marcus, C.L. Late-onset central hypoventilation with hypothalamic dysfunction: A distinct clinical syndrome. Pediatr. Pulmonol. 2000, 29, 62–68. [Google Scholar] [CrossRef]
- Emery, L.G.; Shields, M.; Shah, N.R.; Garbes, A. Neuroblastoma associated with Beckwith-Wiedemann syndrome. Cancer 1983, 52, 176–179. [Google Scholar] [CrossRef]
- Chitayat, D.; Friedman, J.M.; Dimmick, J.E. Neuroblastoma in a child with Wiedemann-Beckwith syndrome. Am. J. Med. Genet. 1990, 35, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.M.; Vansenne, F.; Kadouch, D.J.; Ibrahim, A.; Bliek, J.; Hopman, S.; Mannens, M.M.; Merks, J.H.; Maher, E.R.; Hennekam, R.C. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am. J. Med. Genet. A 2016, 170, 2248–2260. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Molinatto, C.; Baldassarre, G.; Riberi, E.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G.B. Cancer risk in Beckwith-Wiedemann syndrome: A systematic review and meta-analysis outlining a novel (epi)genotype specific histotype targeted screening protocol. J. Pediatr. 2016, 176, 142–149.e141. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Murray, A.; Hanks, S.; Douglas, J.; Armstrong, R.; Banka, S.; Bird, L.M.; Clericuzio, C.L.; Cormier-Daire, V.; Cushing, T.; et al. Weaver syndrome and EZH2 mutations: Clarifying the clinical phenotype. Am. J. Med. Genet. Part A 2013, 161, 2972–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimke, R.N.; Collins, D.L.; Stolle, C.A. Paraganglioma, neuroblastoma, and a SDHB mutation: Resolution of a 30-year-old mystery. Am. J. Med. Genet. Part A 2010, 152A, 1531–1535. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Gimenez-Roqueplo, A.P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Fanconi’s anemia and malignancies. Am. J. Hematol. 1996, 53, 99–110. [Google Scholar] [CrossRef]
- Malric, A.; Defachelles, A.-S.; Leblanc, T.; Lescoeur, B.; Lacour, B.; Peuchmaur, M.; Maurage, C.-A.; Pierron, G.; Guillemot, D.; d’Enghien, C.D.; et al. Fanconi anemia and solid malignancies in childhood: A national retrospective study. Pediatr. Blood Cancer 2015, 62, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2006, 39, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Mathew, C.G. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006, 25, 5875–5884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritenour, L.E.; Randall, M.P.; Bosse, K.R.; Diskin, S.J. Genetic susceptibility to neuroblastoma: Current knowledge and future directions. Cell Tissue Res. 2018, 372, 287–307. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Mosse, Y.P.; Bradfield, J.P.; Hou, C.; Monni, S.; Scott, R.H.; Asgharzadeh, S.; Attiyeh, E.F.; Diskin, S.J.; Laudenslager, M.; et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N. Engl. J. Med. 2008, 358, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Penikis, A.; Oldridge, D.A.; Alvarez-Dominguez, J.R.; McDaniel, L.; Diamond, M.; Padovan, O.; Raman, P.; Li, Y.; Wei, J.S.; et al. CASC15-S is a tumor suppressor lncRNA at the 6p22 neuroblastoma susceptibility locus. Cancer Res. 2015, 75, 3155–3166. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.K.; Mitra, S.; Subhash, S.; Hertwig, F.; Kanduri, M.; Mishra, K.; Fransson, S.; Ganeshram, A.; Mondal, T.; Bandaru, S.; et al. The risk-associated long noncoding RNA NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell 2014, 26, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Juvvuna, P.K.; Kirkeby, A.; Mitra, S.; Kosalai, S.T.; Traxler, L.; Hertwig, F.; Wernig-Zorc, S.; Miranda, C.; Deland, L.; et al. Sense-antisense lncRNA pair encoded by locus 6p22.3 determines neuroblastoma susceptibility via the USP36-CHD7-SOX9 regulatory axis. Cancer Cell 2018, 33, 417–434.e417. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Devoto, M.; Hou, C.; Asgharzadeh, S.; Glessner, J.T.; Attiyeh, E.F.; Mosse, Y.P.; Kim, C.; Diskin, S.J.; Cole, K.A.; et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat. Genet. 2009, 41, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Diskin, S.J.; Totaro, F.; Longo, L.; De Mariano, M.; Russo, R.; Cimmino, F.; Hakonarson, H.; Tonini, G.P.; Devoto, M.; et al. Replication of GWAS-identified neuroblastoma risk loci strengthens the role of BARD1 and affirms the cumulative effect of genetic variations on disease susceptibility. Carcinogenesis 2013, 34, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Xu, X.L.; Yang, M.C.; Wei, F.; Ayi, T.C.; Bowcock, A.M.; Baer, R. Cell cycle-dependent colocalization of BARD1 and BRCA1 proteins in discrete nuclear domains. Proc. Natl. Acad. Sci. USA 1997, 94, 12075–12080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irminger-Finger, I.; Jefford, C.E. Is there more to BARD1 than BRCA1? Nat. Rev. Cancer 2006, 6, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Diskin, S.J.; Cole, K.A.; Wood, A.C.; Schnepp, R.W.; Norris, G.; Nguyen le, B.; Jagannathan, J.; Laquaglia, M.; Winter, C.; et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res. 2012, 72, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schuttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc is a critical function of aurora a in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Wu, J.; et al. Initial testing of the aurora kinase a inhibitor MLN8237 by the pediatric preclinical testing program (PPTP). Pediatr. Blood Cancer 2010, 55, 26–34. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Marachelian, A.; Fox, E.; Kudgus, R.A.; Reid, J.M.; Groshen, S.; Malvar, J.; Bagatell, R.; Wagner, L.; Maris, J.M.; et al. Phase I study of the aurora a kinase inhibitor alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma: A NANT (new approaches to neuroblastoma therapy) trial. J. Clin. Oncol. 2016, 34, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Diskin, S.J.; Zhang, H.; Attiyeh, E.F.; Winter, C.; Hou, C.; Schnepp, R.W.; Diamond, M.; Bosse, K.; Mayes, P.A.; et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 2011, 469, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.M.; Lester, K.; Joseph, S.; Curtis, D.J. LIM-domain-only proteins in cancer. Nat. Rev. Cancer 2013, 13, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, X.; Weichert-Leahey, N.; Dong, Z.; Zhang, C.; Lopez, G.; Tao, T.; He, S.; Wood, A.C.; Oldridge, D.; et al. LMO1 synergizes with MYCN to promote neuroblastoma initiation and metastasis. Cancer Cell 2017, 32, 310–323.e315. [Google Scholar] [CrossRef] [PubMed]
- Oldridge, D.A.; Wood, A.C.; Weichert-Leahey, N.; Crimmins, I.; Sussman, R.; Winter, C.; McDaniel, L.D.; Diamond, M.; Hart, L.S.; Zhu, S.; et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015, 528, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Nguyen le, B.; Diskin, S.J.; Capasso, M.; Wang, K.; Diamond, M.A.; Glessner, J.; Kim, C.; Attiyeh, E.F.; Mosse, Y.P.; Cole, K.; et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility loci. PLoS Genet. 2011, 7, e1002026. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Capasso, M.; Schnepp, R.W.; Cole, K.A.; Attiyeh, E.F.; Hou, C.; Diamond, M.; Carpenter, E.L.; Winter, C.; Lee, H.; et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat. Genet. 2012, 44, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [Green Version]
- Zhang, L.; Anglesio, M.S.; O’Sullivan, M.; Zhang, F.; Yang, G.; Sarao, R.; Mai, P.N.; Cronin, S.; Hara, H.; Melnyk, N.; et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat. Med. 2007, 13, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Domingo-Fernandez, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; van Sluis, P.; Valentijn, L.J.; van Nes, J.; et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012, 44, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Schnepp, R.W.; Khurana, P.; Attiyeh, E.F.; Raman, P.; Chodosh, S.E.; Oldridge, D.A.; Gagliardi, M.E.; Conkrite, K.L.; Asgharzadeh, S.; Seeger, R.C.; et al. A LIN28B-RAN-AURKA signaling network promotes neuroblastoma tumorigenesis. Cancer Cell 2015, 28, 599–609. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.D.; Conkrite, K.L.; Chang, X.; Capasso, M.; Vaksman, Z.; Oldridge, D.A.; Zachariou, A.; Horn, M.; Diamond, M.; Hou, C.; et al. Common variants upstream of MLF1 at 3q25 and within CPZ at 4p16 associated with neuroblastoma. PLoS Genet. 2017, 13, e1006787. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [Green Version]
- Capasso, M.; Diskin, S.; Cimmino, F.; Acierno, G.; Totaro, F.; Petrosino, G.; Pezone, L.; Diamond, M.; McDaniel, L.; Hakonarson, H.; et al. Common genetic variants in NEFL influence gene expression and neuroblastoma risk. Cancer Res. 2014, 74, 6913–6924. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; McDaniel, L.D.; Cimmino, F.; Cirino, A.; Formicola, D.; Russell, M.R.; Raman, P.; Cole, K.A.; Diskin, S.J. The functional variant rs34330 of CDKN1B is associated with risk of neuroblastoma. J. Cell. Mol. Med. 2017, 21, 3224–3230. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Hou, C.; Glessner, J.T.; Attiyeh, E.F.; Laudenslager, M.; Bosse, K.; Cole, K.; Mosse, Y.P.; Wood, A.; Lynch, J.E.; et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature 2009, 459, 987–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hungate, E.A.; Applebaum, M.A.; Skol, A.D.; Vaksman, Z.; Diamond, M.; McDaniel, L.; Volchenboum, S.L.; Stranger, B.E.; Maris, J.M.; Diskin, S.J.; et al. Evaluation of genetic predisposition for MYCN-amplified neuroblastoma. J. Natl. Cancer Inst. 2017, 109, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Shohet, J.M.; Ghosh, R.; Coarfa, C.; Ludwig, A.; Benham, A.L.; Chen, Z.; Patterson, D.M.; Barbieri, E.; Mestdagh, P.; Sikorski, D.N.; et al. A genome-wide search for promoters that respond to increased MYCN reveals both new oncogenic and tumor suppressor microRNAs associated with aggressive neuroblastoma. Cancer Res. 2011, 71, 3841–3851. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, L.J.; Koster, J.; Haneveld, F.; Aissa, R.A.; van Sluis, P.; Broekmans, M.E.; Molenaar, J.J.; van Nes, J.; Versteeg, R. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc. Natl. Acad. Sci. USA 2012, 109, 19190–19195. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Zhao, Y.; Hou, C.; Glessner, J.; McDaniel, L.; Diamond, M.A.; Thomas, K.; Li, J.; Wei, Z.; Liu, Y.; et al. Common variants in MMP20 at 11q22.2 predispose to 11q deletion and neuroblastoma risk. Nat. Commun. 2017, 8, 569. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Pinto, N.; Konkashbaev, A.; Im, H.K.; Diskin, S.J.; London, W.B.; Maris, J.M.; Dolan, M.E.; Cox, N.J.; Cohn, S.L. Trans-population analysis of genetic mechanisms of ethnic disparities in neuroblastoma survival. J. Natl. Cancer Inst. 2013, 105, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, M.A.; Henderson, T.O.; Lee, S.M.; Pinto, N.; Volchenboum, S.L.; Cohn, S.L. Second malignancies in patients with neuroblastoma: The effects of risk-based therapy. Pediatr. Blood Cancer 2015, 62, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, L.M.; Neglia, J.P.; Reulen, R.C.; Ronckers, C.M.; van Leeuwen, F.E.; Morton, L.M.; Hodgson, D.C.; Yasui, Y.; Oeffinger, K.C.; Henderson, T.O. Risk, risk factors, and surveillance of subsequent malignant neoplasms in survivors of childhood cancer: A review. J. Clin. Oncol. 2018, 36, 2145–2152. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S. Genetic variation as a modifier of association between therapeutic exposure and subsequent malignant neoplasms in cancer survivors. Cancer 2015, 121, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, M.A.; Vaksman, Z.; Lee, S.M.; Hungate, E.A.; Henderson, T.O.; London, W.B.; Pinto, N.; Volchenboum, S.L.; Park, J.R.; Naranjo, A.; et al. Neuroblastoma survivors are at increased risk for second malignancies: A report from the international neuroblastoma risk group project. Eur. J. Cancer 2017, 72, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.O.; Bhatia, S.; Pinto, N.; London, W.B.; McGrady, P.; Crotty, C.; Sun, C.L.; Cohn, S.L. Racial and ethnic disparities in risk and survival in children with neuroblastoma: A children’s oncology group study. J. Clin. Oncol. 2011, 29, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.D.; Burchard, E.G.; De la Vega, F.M. Genomics for the world. Nature 2011, 475, 163–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, S.E.; French, B.; Kasner, S.E.; Johnson, J.A.; Anderson, J.L.; Gage, B.F.; Rosenberg, Y.D.; Eby, C.S.; Madigan, R.A.; McBane, R.B.; et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N. Engl. J. Med. 2013, 369, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Mapes, B.; El Charif, O.; Al-Sawwaf, S.; Dolan, M.E. Genome-wide association studies of chemotherapeutic toxicities: Genomics of inequality. Clin. Cancer Res. 2017, 23, 4010–4019. [Google Scholar] [CrossRef] [PubMed]
- Latorre, V.; Diskin, S.J.; Diamond, M.A.; Zhang, H.; Hakonarson, H.; Maris, J.M.; Devoto, M. Replication of neuroblastoma SNP association at the BARD1 locus in African-Americans. Cancer Epidemiol. Biomark. Prev. 2012, 21, 658–663. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zou, Y.; Wang, T.; Zhang, R.; Yang, T.; Zhu, J.; Wang, F.; Xia, H. Genetic variations of GWAS-identified genes and neuroblastoma susceptibility: A replication study in southern Chinese children. Transl. Oncol. 2017, 10, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
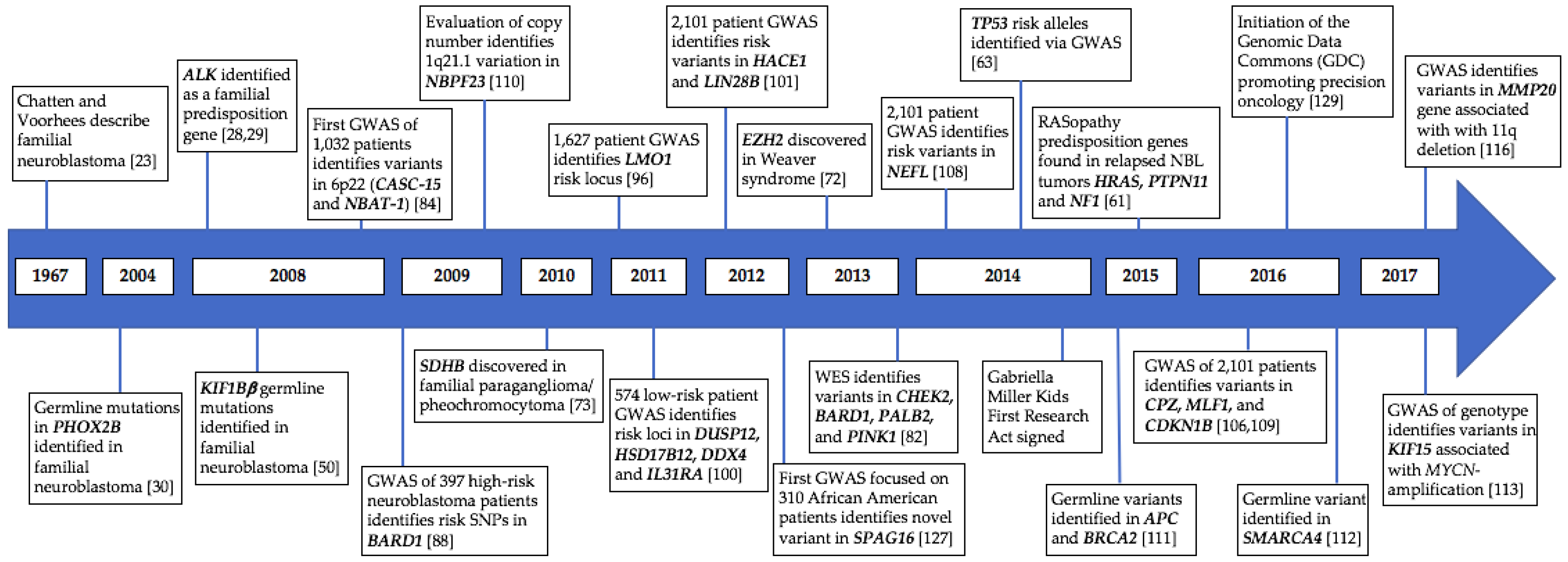
Figure 1.
Timeline of identified genetic variation predisposing to neuroblastoma. GWAS, genome-wide association studies.
Figure 1.
Timeline of identified genetic variation predisposing to neuroblastoma. GWAS, genome-wide association studies.


{kind=link}
Table 1.
Heritable conditions that predispose patients to developing neuroblastoma.
| Syndrome/Disease | Gene | Typical Genetic Alterations | Clinical Findings | Pre-Disposed Tumors |
|---|---|---|---|---|
| Congenital Central Hypoventilation Syndrome (CCHS) [32,33,36] | PHOX2B | Polyalanine and nonpolyalanine repeat expansion (frameshift or missense) | Respiratory dysfunction, autonomic dysfunction, Hirschsprung disease, neural crest tumors | Neuroblastoma, ganglioneuroma, ganglioneuroblastoma |
| ROHHAD [64,65,66,67] | Unknown | Unknown | Autonomic dysfunction, endocrinopathies, alveolar hypoventilation | Neuroblastoma, ganglioneuroma, ganglioneuroblastoma |
| Costello [58,59,60] | HRAS | Activating missense | Intellectual disability, coarse facial features, loose folds of skin, heart abnormalities, joint flexibility | Papilloma, rhabdomyosarcoma, neuroblastoma, transitional cell carcinoma |
| Noonan [52,53,54,60] | PTPN11, SOS1, RAF1, KRAS | Activating | Short stature, heat abnormalities, skeletal abnormalities, bleeding | Leukemia, neuroblastoma |
| Neurofibromatosis type 1 [55,56,57,60] | NF1 | Activating | Abnormal skin pigmentation, neurofibromas, scoliosis | Neurofibroma, MPNST, brain tumors, leukemia, optic glioma, neuroblastoma |
| Beckwith–Wiedemann [68,69,70,71] | CDKN1C *, H19, IGF2, KNBQOT1 | Abnormal methylation of chromosome 11 or uniparental disomy | Macrosomia, hemihypertrophy, abdominal wall defects, visceromegaly | Wilms tumor, hepatoblastoma, neuroblastoma |
| Li–Fraumeni [51,62,63] | TP53 | Missense | Increased cancer risk | Breast cancer, osteosarcoma, brain tumors, leukemia, neuroblastoma, adrenocortical carcinoma, soft tissue sarcoma |
| Weaver Syndrome [72] | EZH2 | Missense and truncating | Tall stature, intellectual disability, joint deformities, hypertelorism, micrognathia | Neuroblastoma |
| Familial Paraganglioma/Pheochromocytoma [73,74] | SDHB *, SDHAF2, SDHC, SDHD | Splice site, frameshift, nonsense | Growth of benign tumors in paraganglia | Paraganglioma, pheochromocytoma, neuroblastoma |
| Fanconi Anemia [75,76,77,78,79] | FANCA, FANCC, FANCG, BRCA1, BRCA2 *, PALB2 *, BRIP1 *, and many others | Truncating, frameshift, missense | Bone marrow failure, organ defects, skeletal abnormalities | Leukemia, Wilms tumor, medulloblastoma, neuroblastoma, embryonal tumors, sarcomas, nephroblastoma |
* mutations in this gene confer the greatest susceptibility to neuroblastoma in this syndrome. Abbreviations: MPNST, malignant peripheral nerve sheath tumor; CCHS, congenital hypoventilation syndrome; ROHHAD, rapid-onset obesity, hypothalamic dysfunction, hypoventilation and autonomic dysfunction.
Table 2.
Germline variants that increase risk of developing neuroblastoma.
| Candidate Gene(s) | Variant * | Genomic Location |
|---|---|---|
| TP53 [63] | rs35850753 | 17p13.1 |
| CASC-15 and NBAT-1 [84] | rs6939340 | 6p22 |
| BARD1 [88] | rs6435862 | 2q35 |
| LMO1 [96] | rs2168101 | 11p15.4 |
| DUSP12 [100] | rs1027702 | 1q23.3 |
| DDX4 [100] | rs2619046 | 5q11.2 |
| IL31RA [100] | rs10055201 | 5q11.2 |
| HSD17B12 [100] | rs11037575 | 11p11.2 |
| HACE1 [101] | rs4336470 | 6q16 |
| LIN28B [101] | rs17065417 | 6q16 |
| CPZ [106] | rs3796727 | 4p16 |
| MLF1 [106] | rs6441201 | 3q25 |
| NEFL [108] | rs1059111 | 8q21 |
| CDKN1B [109] | rs34330 | 12p13 |
| KIF15 [113] | rs80059929 | 3p21.31 |
| MMP20 [116] | rs10895322 | 11q22.2 |
| SPAG16 [117] | rs1033069 | 2q34 |
* Most significant single-nucleotide polymorphisms (SNPs).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Barr, E.K.; Applebaum, M.A. Genetic Predisposition to Neuroblastoma. Children 2018, 5, 119. https://0-doi-org.brum.beds.ac.uk/10.3390/children5090119
AMA Style
Barr EK, Applebaum MA. Genetic Predisposition to Neuroblastoma. Children. 2018; 5(9):119. https://0-doi-org.brum.beds.ac.uk/10.3390/children5090119
Chicago/Turabian StyleBarr, Erin K., and Mark A. Applebaum. 2018. "Genetic Predisposition to Neuroblastoma" Children 5, no. 9: 119. https://0-doi-org.brum.beds.ac.uk/10.3390/children5090119
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.