Identifying Downregulation of Autophagy Markers in Kawasaki Disease



1
Department of Pediatrics, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 833, Taiwan
2
Kawasaki Disease Center, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung 833, Taiwan
3
Genomics and Proteomics Core Laboratory, Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 833, Taiwan
*
Author to whom correspondence should be addressed.
Children 2020, 7(10), 166; https://0-doi-org.brum.beds.ac.uk/10.3390/children7100166
Submission received: 5 September 2020
/
Revised: 27 September 2020
/
Accepted: 29 September 2020
/
Published: 4 October 2020
Abstract
:Kawasaki disease (KD) is the most common cause of heart disease acquired in childhood. Even if treated with high-dose intravenous immunoglobulin G (IVIG) at the early stage; children are still at risk of developing coronary artery lesions. Accumulating evidence suggests that autophagy is enhanced in various heart diseases. Evaluating the pathogenic role of autophagy in KD and coronary artery lesions (CAL) may aid in identifying a potential therapeutic target for the treatment or prevention of the disease. Blood samples were obtained from 20 children with KD at the onset of disease and 21 days after IVIG therapy. Twenty children with other causes of febrile disease and 20 healthy children were included as controls. Total RNA was extracted from white blood cells; and autophagy-related gene mRNA expression levels were measured using real-time polymerase chain reaction. The patients with KD had downregulated levels of LC3B mRNA (0.50 ± 0.06 vs. 1.67 ± 0.15; p < 0.001), BECN 1 mRNA (0.70 ± 0.08 vs. 1.43 ± 0.23; p < 0.05), and ATG16L1 mRNA (0.28 ± 0.04 vs. 0.96 ± 0.16; p < 0.01) compared to the febrile control group. The values of these parameters all increased significantly 21 days after the IVIG therapy as follows: LC3B mRNA (1.77 ± 0.29 vs. 0.50 ± 0.06; p < 0.001), BECN 1 mRNA (1.67 ± 0.36 vs. 0.70 ± 0.08; p < 0.05), and ATG16L1 mRNA (2.96 ± 0.43 vs. 0.28 ± 0.04; p < 0.001), while the level of ATG16L1 mRNA persists low in KD patients with CAL. Our results showed the autophagy-related genes expressions in KD and their change after IVIG administration. This suggests that autophagy may have a protective effect on KD.
1. Introduction
Kawasaki disease (KD) is the most common cause of heart disease in childhood in the developed nations [1]. It is also known as mucocutaneous lymph node syndrome [2] and typically presents with fever for more than 5 days in children. Without treatment, a subset of patients (24%) tend to develop coronary artery lesions (CAL) with aneurysms in 8% [3]; these patients are inclined to complications of the heart and sudden death [4] and the mortality rate for this patient group is approximately 1–2% [5]. Administration of high-dose intravenous immunoglobulin (IVIG) plus aspirin early in the course of KD reduces the prevalence of coronary artery abnormalities from 18 to 4% [6]. Despite this success, children with persistent or recurrent fever are at an increased risk of developing CAL [7]. Additional therapies administered to these patients include retreatment with IVIG, use of immunomodulatory agents, such as cyclosporine, cyclophosphamide, and infliximab, plasma exchange, and pulsed steroid therapy with methylprednisolone [4]. Identification of the pathogenesis of KD and its complications, as well as evaluation of more effective therapy, are mandatory to prevent complications of the coronary artery in the patients with KD.
Autophagy is the major intracellular degradation system that removes unnecessary or dysfunctional components and serves as a dynamic recycling system for cellular renovation and homeostasis. Autophagy at low basal levels maintains the cardiac function and morphology of the normal heart. Autophagy is activated in response to the cellular stress that develops in almost all forms of heart disease [8]. However, studies on the functional role of autophagy in the diseased heart have conflicting results. In some cases, enhanced autophagic responses provide protective effects on cardiovascular diseases, while in others, it promotes deterioration in disease. There is increasing evidence that autophagy is enhanced in various cardiovascular diseases, including cardiac hypertrophy [9], cardiomyopathy [10], valvular and hypertensive heart disease [11], heart failure [12], and during myocardial ischemia and reperfusion [13]. Taken together, these studies support the current role of autophagy in cardiovascular diseases though the results are conflicting [8]. Thus, evaluating the pathogenic role of autophagy in KD and CAL may aid in identifying a potential therapeutic target for the treatment or prevention of the disease.
Therefore, this study aimed to investigate the expression of autophagy markers, including Beclin 1, LC3, and autophagy related 16 like 1 (ATG16L1), in the peripheral leukocytes in children with KD.
2. Patients and Methods
2.1. Patients
This study was approved by the Institutional Review Board of the Chang Gung Memorial Hospital (no. 201601024B0), and informed consent was obtained from either the parents or guardians of the children. All children diagnosed with KD were treated with IVIG (2 gm/kg) infusion slowly. Blood samples were obtained at the onset (before IVIG treatment; KD1) and 21 days after IVIG treatment (KD3). Patients who did not fulfill the diagnostic criteria of KD were excluded. We measured the internal lumen diameters of the coronary artery using two-dimensional echocardiography. CAL were defined as when the internal diameters of the coronary artery were greater than 3 mm (or 4 mm if the subject was aged more than 5 years) or when the internal diameter of a segment was at least 1.5 times that of the adjacent segment, and by a clearly irregular luminal contour pursuant to the guidelines of the Japanese Ministry of Health [14]. Age-matched patients who had fever caused by other etiologies, including herpangina, acute bronchiolitis, acute pharyngotonsillitis and acute sinusitis, and did not have a history of KD constituted the febrile control group. From the outpatient clinics, we enrolled another 20 healthy children (without any history of KD) who tested negative for an allergen test and volunteered to participate in our study as the healthy control group. The demographic and clinical characteristics of these children are shown in the Tables.
2.2. Real-Time PCR
We collected 3–5 mL whole blood samples from all of the participants and submitted them to white blood cell (WBC) enrichment. Total RNA was extracted from the leukocytes of patients with KD and from the leukocytes of the febrile and healthy control groups as previously reported [15]. Reverse transcription step was using TaKaRa PrimeScript™ RT reagent Kit (TaKaRaCat #RR037A; Takara Bio Inc., Shiga, Japan) in 20-μL reactions and a final concentration of 1 μg total RNA, 5x PrimeScript buffer, 50 μM Oligo dT primer, 100 μM Random 6 mers, PrimeScript RT Enzyme Mix1 and the total volume was transferred at 20 μL per tube with RNase Free dH2O. Reverse transcription was carried out in a ABI 2720 Thermal cycler (Applied Biosystems, CA, USA), programmed to run the reverse transcription for 15 min at 37 °C, and inactivation the reverse transcriptase for 5 s at 85 °C and finally incubated the cDNA products at 4 °C. Before the PCR step, the cDNA products were stored at −20 °C.
The mRNA levels of autophagy markers, LC3B, BECN 1, and ATG16L1, were measured by using Real-Time RT–PCR twostep multiplex technique in an ABI PRISMTM 7700 (Applied Biosystems, CA, USA). Probes and primers were obtained from Applied Biosystems. Real time PCR experiments were carried out by ABI 7500 Fast Real-Time PCR System (Applied Biosystems). The reagent used the Fast SYBR Green Master Mix (Thermo Fisher #4385612, Applied Biosystems, CA, USA) and followed the manufacturer’s instructions. The reaction mix contained 5 μL of sample mixed with 15 μL of PCR cocktail (200 nM for each primer). The reaction for each well was carried out as follows: 95 °C for 20 s, followed by 95 °C for 3 s and 60 °C for 30 s and repeated for 40 cycles. Dissociation stage was carried out as follows, 95 °C for 15 s, 60 °C for 60 s, 95 °C for 15 s and 60 °C for 15 s. The ABI7500 software (SDS V2.3) was used to obtain raw fluorescence data (Rn and DRn) for analysis. Many aspects of MIQE guidelines were taken into consideration for methods and analysis [1]. The primers used for amplification were listed in supplemental table (Table S1). The number of transcripts was calculated using the comparative threshold cycle (2ΔΔCt) formula, with the endogenous GAPDH expression as the internal control.
2.3. Statistical Analysis
Statistical analyses were performed using the paired Student’s t-test or Mann–Whitney U test for comparison of two parametric or nonparametric variables and Kruskal–Wallis one-way analysis of variance for the comparison of nonparametric three or more variables (GraphPad Prism 8; GraphPad software, San Diego, CA, USA). A p-value of <0.05 was considered statistically significant.
3. Results
3.1. Demographic Data
In all, 20 children with KD (aged 1.40 ± 0.69 years; 6 males and 14 females) were included in this study. Twenty patients with an acute febrile infectious disease (aged 1.49 ± 0.78 years; 12 males and 8 females) and 20 healthy children (aged 1.47 ± 0.61 years; 14 males and 6 females) were enrolled as the controls. There was no significant difference in age and sex between the children with KD and those in both the control groups (Table 1, Table 2, Table 3, Table 4 and Table 5).
3.2. Differential Counts of Leukocytes and Monocytes among the Control Groups and the Children with KD before and after Treatment
There was no significant difference in the differential counts of neutrophils (30.75 ± 11.1% vs. 39.05 ± 22.12%, p = 0.287) and monocytes (6.18 ± 2.29% vs. 8.15 ± 4.31%, p = 0.202) between the healthy and febrile control groups (Table 1). However, we observed a significant increase in the differential counts of leukocytes (59.79 ± 10.4%, p < 0.05) in children with KD before treatment (Table 2 and Table 3) and a decrease in the differential counts of monocytes (4.90 ± 1.13%, p < 0.05) in children with KD after treatment (Table 4 and Table 5) compared to both the control groups. Although the differential counts of lymphocytes were different between the KD1 and control groups, the total number of lymphocytes was not significantly different.
3.3. Analyses of Autophagy Markers mRNA in the Peripheral White Blood Cells of Kawasaki Disease (KD) Patients before and after IVIG Treatment
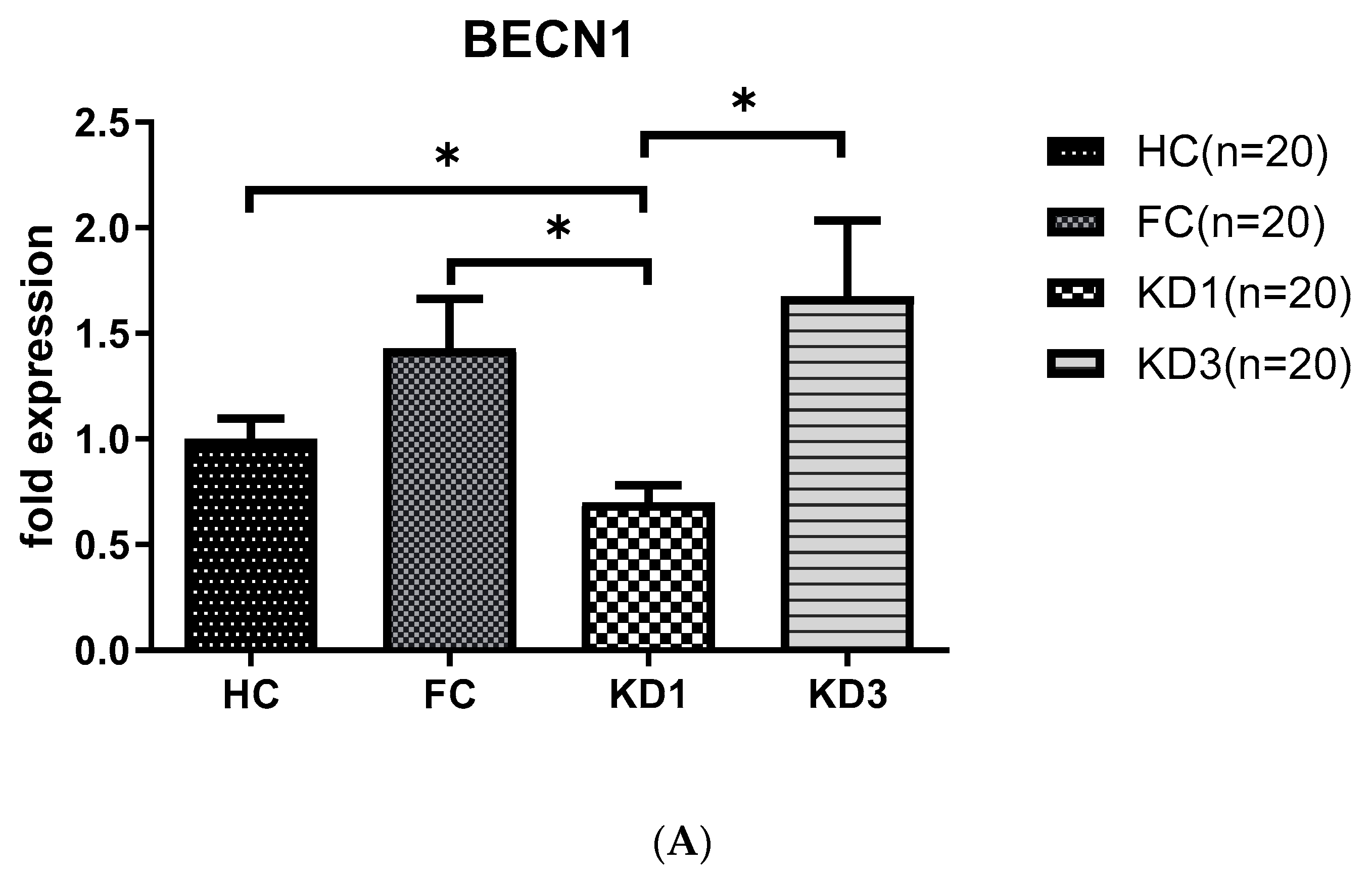
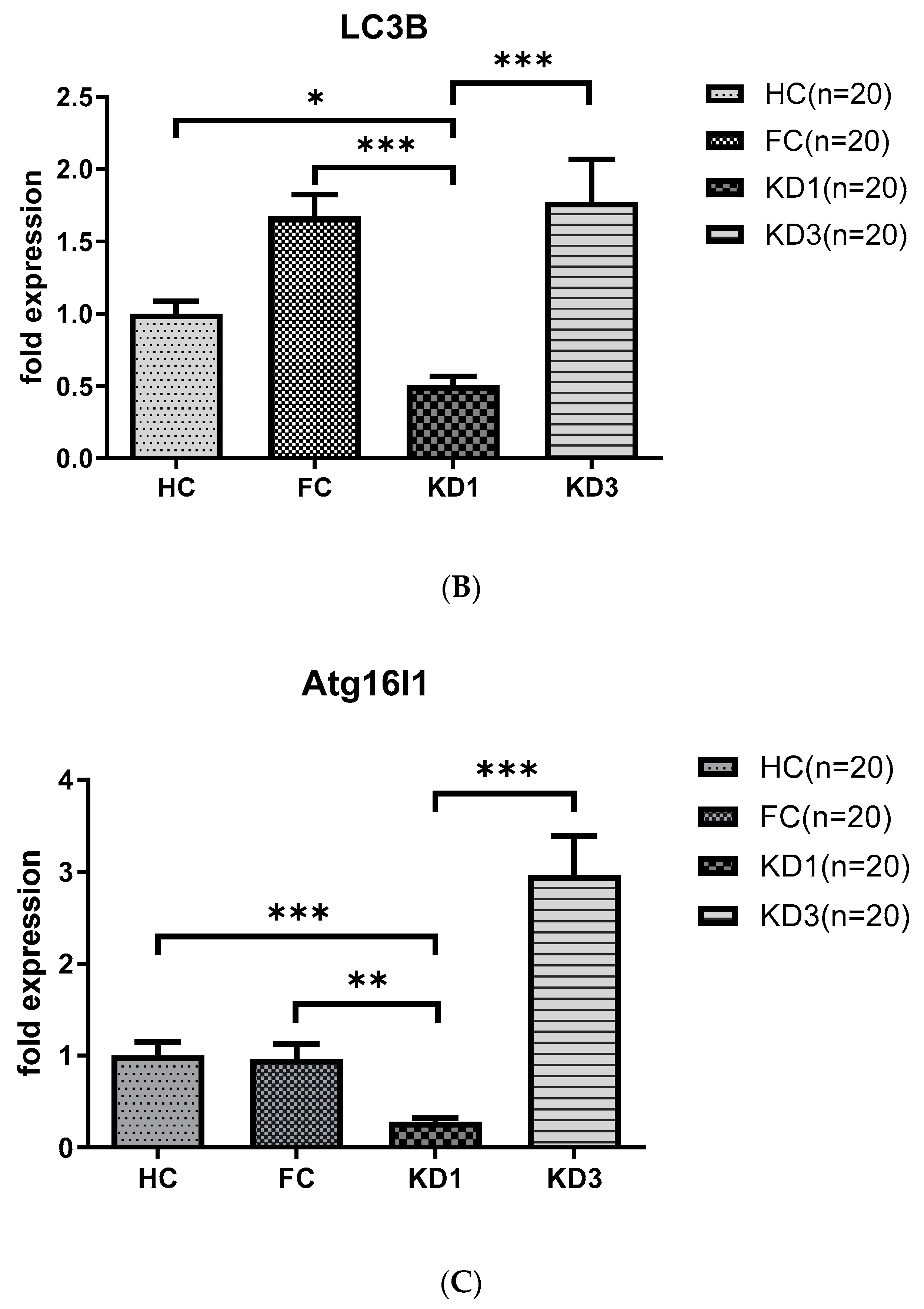
As shown in Figure 1, we observed that patients with KD before treatment (KD1) had downregulated levels of LC3B (0.50 ± 0.06 vs. 1.67 ± 0.15, p < 0.001), BECN 1 (0.70 ± 0.08 vs. 1.43 ± 0.23, p < 0.05) and ATG16L1 (0.28 ± 0.04 vs. 0.96 ± 0.16, p < 0.01) mRNA compared to the febrile control and healthy control groups. The produced mRNA of all these markers increased significantly 21 days after IVIG therapy (KD3)(LC3B, 1.77 ± 0.29 vs. 0.50 ± 0.06, p < 0.001; BECN 1, 1.67 ± 0.36 vs. 0.70 ± 0.08, p < 0.05; and ATG16L1, 2.96 ± 0.43 vs. 0.28 ± 0.04, p < 0.001) compared to KD1.
3.4. The Lower Levels of ATG16L1 mRNA Persist in KD Patients with CAL Compared to Those without CAL 21 Days after IVIG Therapy
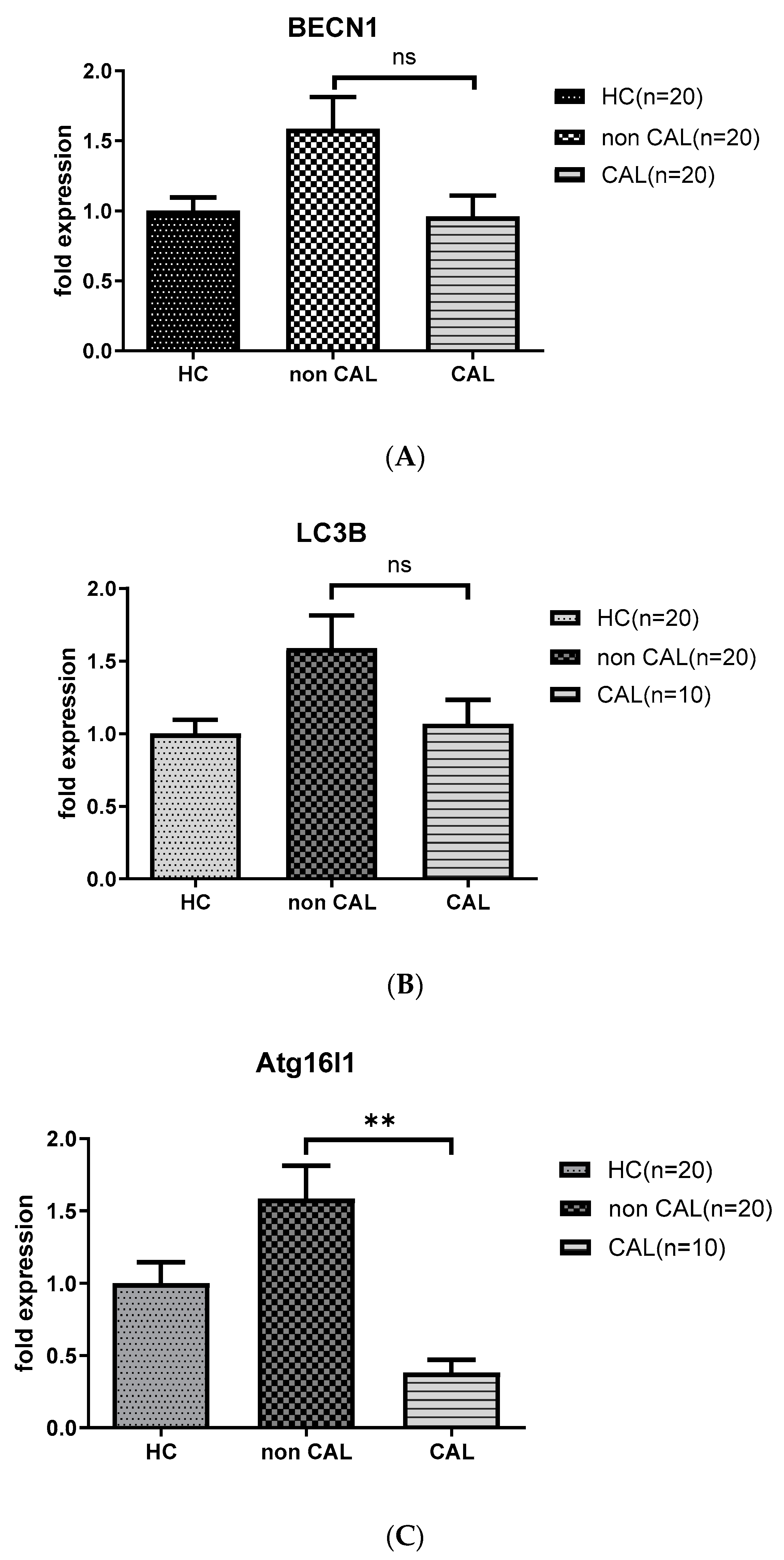
As shown in Figure 2, KD patients with CAL (CAL; n = 10) had persistent downregulated levels of ATG16L1 mRNA (0.38 ± 0.04 vs. 2.85 ± 0.46, p < 0.01) compared to the age-matched KD patients without CAL (non CAL; n = 20) 21 days after IVIG therapy; while the levels of BECN 1 and LC3B mRNA not significantly different between the aforementioned groups.
4. Discussion
In the normal heart, basal levels of autophagy is observed, and autophagy is characterized by maintaining a homeostatic status of heart. However, the functional studies on autophagy in the diseased heart have reported conflicting results [8,10,11,12,13,16,17,18]. In some cases, increased autophagy is beneficial and promotes functional recovery, and the myocardium may be salvaged after ischemia/reperfusion injury [8,13,17,18,19]; however, in other cases, autophagy is associated with cell death and promotion of heart failure [8,10,11,12]. Some studies suggest the role of autophagy proteins, Beclin 1 and LC3B, in the cardioprotection of the ischemic heart. In the current study, we observed that patients with KD had lower mRNA levels of autophagy markers, including LC3, Beclin 1, and ATG16L1 compared to the febrile control group. Additionally, the expression of these markers significantly increased 21 days after IVIG therapy.
Although the etiology of KD remains unknown, it is believed a combination of microbial infection, immune response or genetic susceptibility contribute to the development of KD. Both clinical and epidemiological characteristics of KD strongly suggest the involvement of infectious agents in the onset of KD [20]. Most studies report that an infectious agent triggers the illness [21,22,23,24], although the infectious agent remains unidentified at present. Heterogeneous infectious etiologies may account for the development of KD in different countries and during different seasons. In our study, neutrophils, which were predominantly noted in the patients with KD before treatment, may indicate the triggering role of bacterial infection in KD. The lower levels of autophagy in children KD in our study suggests that unknown microorganisms may play an important role in triggering KD [25,26], with poor autophagic clearance of these pathogens. The expression of ATG16L1 T300A variant decreases selective autophagy, leading to altered cytokine signaling and decreased antibacterial defense [27]. Neutrophils are the most abundant leukocytes and play a central role in the primary mechanisms underlying host defense. IVIG enhances the killing and autophagy activities of neutrophils isolated from either immunocompetent [28] or immunocompromised [29] patients exposed to multidrug-resistant bacteria. We revealed that the mRNA expression of autophagy markers was lower in the leukocytes of patients with KD who predominantly had neutrophils (Table 2 and Table 4). This can explain the therapeutic effect of IVIG on patients with KD [30] and the higher incidence of CAL in patients with IVIG-resistant KD [31]. Neutrophil-mediated damage of endothelial cells [32] contributes towards the generation of CAL in patients with KD. The infiltration of neutrophils in CAL during the acute phase of KD contributes towards the initial damage to coronary arteries, while sustained activity of neutrophils [33] may be involved in the pathogenesis of KD vasculitis [34]. Effective elimination of neutrophils is a resolution of detrimental inflammatory response. Most of the current data suggest that autophagy assists death of the neutrophils [35]. The absence of autophagy may contribute to sustained activity of neutrophils, leading to the injury of endothelial cells, while IVIG enhances neutrophil autophagy [28] and death of neutrophils and prevents the development of CAL in patients with KD.
The increase in autophagy in macrophages after ingesting pathogens contributes towards pathogen defense mechanisms, while dysregulated autophagy may allow pathogen survival in macrophages and stimulation of inflammatory responses. Macrophage autophagy limits acute toxic liver injury in mice through downregulation of interleukin-1β expression [36]. Several clinical studies on KD suggest that serum levels of IL-1β or IL-1β, gene expression may play an essential role in KD [37,38] and development of CAL [39]. Decreased expression of the autophagy protein, ATG16L1, enhances endotoxin-induced IL-1β production [40]. ATG16L1 polymorphisms together with the excessive production of IL-1β and IL-6 in human peripheral mononuclear cells [41] may account for the chronic inflammatory process in Crohn’s disease. It is compatible with our observation that the downregulated levels of ATG16L1 mRNA persists in KD children with CAL even after IVIG therapy (Figure 2) and may provide a mechanistic explanation for the involvement of this protein in the pathogenesis of CAL in children with KD. The persistent downregulated ATG16L1 mRNA level in KD patients may contribute towards the marked chronic inflammation in arteritis. The levels of ATG16L1 mRNA may be used as an indicator for CAL development in children with KD, and ATG16L1 polymorphism or miRNA controlling the expression of ATG16L1 may play an essential role in the pathogenesis of CAL in children with KD. Moreover, our preliminary data (data not shown), based on Illumina Next generation sequencing platform and miRSeq analysis for determining miRNA expression profiles in our Lab [42], revealed a significant increase in miR142-3p and miR93-3p expression at disease onset in children with KD before administration of IVIG treatment compared to the febrile controls. Targeting of autophagy protein, ATG16L1, by miR142-3p suggests that this miRNA may play a role in intestinal inflammation and Crohn’s disease [43]. Disruption of ATG16L1-mediated autophagy by miR-93 hinders the elimination of bacteria from the epithelial cells [44]. This finding is similar to that reported in a recent study that miR-93 may participate in regulating gene expression in circulating peripheral blood mononuclear cells and contribute towards the pathogenesis of CAL in acute KD [45]. In our study, the differential counts of peripheral monocytes are decreased in KD patients 21 days after IVIG therapy, which may lead to the activation of macrophages. Macrophage autophagy downregulates IL-1β expression, leading to the repair of endothelial damage after IVIG therapy.
Additionally, autophagy has an essential regulatory function in macrophage polarization that downregulates inflammation [46]. The ingestion of dead neutrophils may also influence the macrophage phenotype [47]. In the absence of neutrophils, macrophages predominantly assume M2-like phenotypes [48]. Impaired macrophage autophagy influences macrophage polarization by increasing proinflammatory M1 and decreasing anti-inflammatory M2 polarization in obese mice [46] and leads to hepatic inflammation and liver injury. M2 macrophage polarization mediates the anti-inflammatory effects of endothelial nitric oxide signaling [49]. In Table 3 and Table 5, besides neutrophils, patients with KD presented with a decreased number of monocytes after treatment (KD3), which were supposed to be polarized into macrophages M1 or M2 in the coronary arteries. Furthermore, IVIG induces autophagy in M1 macrophages but not in M2 macrophages and reduced inflammatory cytokines in the circulation [50]. It explains in part the mechanism by which IVIG therapy benefits patients with autoimmune and inflammatory disease. However, it is difficult to differentially sort macrophages into M1 and M2 types for determining a precise count. Although the differential counts of peripheral monocytes were similar in our patient groups, it does not mean that the proportion of M1 or M2 was similar. On the other hand, a phosphopeptide P140 that directly acts on chaperone-mediated autophagy, which is hyperactivated in certain subsets of lymphocytes in lupus, can be a very promising therapeutic agent in autoinflammatory diseases [51].
This study has certain limitations. First, all of our KD patients belonged to the Taiwanese population, so our findings need to be investigated in other KD populations, and clinical samples of a second population should be considered to confirm our findings. Second, we studied the autophagy expression in peripheral WBC, but studying autophagy expression in infiltrated neutrophils of coronary arteries, monocytes or lymphocytes in circulation in early-stage KD patients could yield better results. Such research would provide important insights into the pathophysiology of autophagy in the CAL of KD, which may ultimately lead to developing novel approaches for treating the disease.
5. Conclusions
We observed that the downregulated mRNA levels of autophagy markers in children with KD return to normal or even exceed the normal level in the general population 21 days after IVIG therapy, and the downregulated ATG16L1 mRNA levels persists in KD children with CAL even after IVIG therapy. Thus, the data obtained support the notion that change of autophagy in both monocytes/macrophages and neutrophils contributes to elimination or development of CAL in KD children, focusing on ATG16L1; thus, raising the possibility that manipulation of autophagy can have far-reaching therapeutic benefits. However, well-controlled clinical trials are needed to determine the effects of the administration of autophagy-enhancing agents in KD children.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2227-9067/7/10/166/s1, Table S1: Primer sequences used for real time RT-PCR.
Author Contributions
F.-C.H. analyzed and interpreted the patient data, and was a major contributor in writing the manuscript. Y.-H.H. contributed to sample preparation. H.-C.K. supervised the findings of this work. S.-C.L. performed the analytic calculations. All authors discussed the results and contributed to the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by grants from the Ministry of Science and Technology of Taiwan (104-2314-B-182-057) and grants from the Chang Gung Memorial Hospital (CMRPG8F1911, CMRPG8F1921, CMRPG8F1931, and CMRPG8F1941). Although these institutes provided financial support, they had no influence on the collection, analysis, or interpretation of the data or the preparation of this manuscript.
Conflicts of Interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Approval and Consent to Participate
This study was approved by the Institutional Review Board of the Chang Gung Memorial Hospital (no. 201601024B0). All informed consents were obtained in writing from these people or in the case of children, their parent or legal guardian, prior to participation.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- Taubert, K.A.; Rowley, A.H.; Shulman, S.T. Nationwide survey of Kawasaki disease and acute rheumatic fever. J. Pediatr. 1991, 119, 279–282. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kosaki, F.; Okawa, S.; Shigematsu, I.; Yanagawa, H. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics 1974, 54, 271–276. [Google Scholar]
- Suzuki, A.; Kamiya, T.; Kuwahara, N.; Ono, Y.; Kohata, T.; Takahashi, O.; Kimura, K.; Takamiya, M. Coronary arterial lesions of Kawasaki disease: Cardiac catheterization findings of 1100 cases. Pediatr. Cardiol. 1986, 7, 3–9. [Google Scholar] [CrossRef]
- McCrindle, B.W.; Rowley, A.H.; Newburger, J.W.; Burns, J.C.; Bolger, A.F.; Gewitz, M.; Baker, A.L.; Jackson, M.A.; Takahashi, M.; Shah, P.B.; et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals from the American Heart Association. Circulation 2017, 135, e927–e999. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sugimura, T.; Akagi, T.; Sato, N.; Hashino, K.; Maeno, Y.; Kazue, T.; Eto, G.; Yamakawa, R. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation 1996, 94, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Newburger, J.W.; Takahashi, M.; Burns, J.C.; Beiser, A.S.; Chung, K.J.; Duffy, C.E.; Glode, M.P.; Mason, W.H.; Reddy, V.; Sanders, S.P.; et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N. Engl. J. Med. 1986, 315, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Han, R.K.; Silverman, E.D.; Newman, A.; McCrindle, B.W. Management and outcome of persistent or recurrent fever after initial intravenous gamma globulin therapy in acute Kawasaki disease. Arch. Pediatr. Adolesc. Med. 2000, 154, 694–699. [Google Scholar] [CrossRef]
- Hill, J.A. Autophagy in cardiac plasticity and disease. Pediatr. Cardiol. 2011, 32, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Sawada, K.; Shimomura, H.; Kawamura, K.; James, T.N. On the nature of cell death during remodeling of hypertrophied human myocardium. J. Mol. Cell. Cardiol. 2000, 32, 161–175. [Google Scholar] [CrossRef]
- Miyata, S.; Takemura, G.; Kawase, Y.; Li, Y.; Okada, H.; Maruyama, R.; Ushikoshi, H.; Esaki, M.; Kanamori, H.; Li, L.; et al. Autophagic cardiomyocyte death in cardiomyopathic hamsters and its prevention by granulocyte colony-stimulating factor. Am. J. Pathol. 2006, 168, 386–397. [Google Scholar] [CrossRef] [Green Version]
- Hein, S.; Arnon, E.; Kostin, S.; Schönburg, M.; Elsässer, A.; Polyakova, V.; Bauer, E.P.; Klövekorn, W.-P.; Schaper, J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: Structural deterioration and compensatory mechanisms. Circulation 2003, 107, 984–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostin, S.; Pool, L.; Elsasser, A.; Hein, S.; Drexler, H.C.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klövekorn, W.-P.; et al. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Saji, T.; Otani, T.; Takeuchi, K.; Nakamura, T.; Arakawa, H.; Kato, T.; Hara, T.; Hamaoka, K.; Ogawa, S.; et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): A randomised, open-label, blinded-endpoints trial. Lancet 2012, 379, 1613–1620. [Google Scholar] [CrossRef]
- Guo, M.M.; Tseng, W.N.; Ko, C.H.; Pan, H.-M.; Hsieh, K.-S.; Kuo, H.-C. Th17- and Treg-related cytokine and mRNA expression are associated with acute and resolving Kawasaki disease. Allergy 2015, 70, 310–318. [Google Scholar] [CrossRef]
- Knaapen, M.W.; Davies, M.J.; De Bie, M.; Haven, A.J.; Martinet, W.; Kockx, M.M. Apoptotic versus autophagic cell death in heart failure. Cardiovasc. Res. 2001, 51, 304–312. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Vatner, D.E.; Kim, S.J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Decker, R.S.; Wildenthal, K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am. J. Pathol. 1980, 98, 425–444. [Google Scholar]
- Takahashi, K.; Oharaseki, T.; Yokouchi, Y.; Naoe, S.; Saji, T. Kawasaki disease: Basic and pathological findings. Clin. Exp. Nephrol. 2013, 17, 690–693. [Google Scholar] [CrossRef]
- Rowley, A.H. The etiology of Kawasaki disease: Superantigen or conventional antigen? Pediatr. Infect. Dis. J. 1999, 18, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Meissner, H.C.; Leung, D.Y. Superantigens, conventional antigens and the etiology of Kawasaki syndrome. Pediatr. Infect. Dis. J. 2000, 19, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, T.; Matsutani, T.; Toyosaki-Maeda, T.; Suzuki, H.; Uemura, S.; Suzuki, R.; Koike, M.; Hinuma, Y. Relation of streptococcal pyrogenic exotoxin C as a causative superantigen for Kawasaki disease. Pediatr. Res. 2003, 53, 403–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.Y.; Chang, I.S.; Lu, C.Y.; Chiang, B.-L.; Lee, C.-Y.; Chen, P.-J.; Wang, J.-T.; Ho, H.-N.; Chen, D.; Huang, L.-M.; et al. Epidemiologic features of Kawasaki disease in Taiwan, 1996–2002. Pediatrics 2004, 114, e678–e682. [Google Scholar] [CrossRef] [Green Version]
- Cimaz, R.; Falcini, F. An update on Kawasaki disease. Autoimmun. Rev. 2003, 2, 258–263. [Google Scholar] [CrossRef]
- Leung, D.Y.; Meissner, H.C.; Shulman, S.T.; Mason, W.H.; Gerber, M.A.; Glodé, M.P.; Myones, B.L.; Wheeler, J.; Ruthazer, R.; Schlievert, P.M. Prevalence of superantigen-secreting bacteria in patients with Kawasaki disease. J. Pediatr. 2002, 140, 742–746. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.-C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef] [Green Version]
- Itoh, H.; Matsuo, H.; Kitamura, N.; Yamamoto, S.; Higuchi, T.; Takematsu, H.; Kamikubo, Y.; Kondo, T.; Yamashita, K.; Sasada, M.; et al. Enhancement of neutrophil autophagy by an IVIG preparation against multidrug-resistant bacteria as well as drug-sensitive strains. J. Leukoc. Biol. 2015, 98, 107–117. [Google Scholar] [CrossRef]
- Matsuo, H.; Itoh, H.; Kitamura, N.; Kamikubo, Y.; Higuchi, T.; Shiga, S.; Ichiyama, S.; Kondo, T.; Takaori-Kondo, A.; Adachi, S. Intravenous immunoglobulin enhances the killing activity and autophagy of neutrophils isolated from immunocompromised patients against multidrug-resistant bacteria. Biochem. Biophys. Res. Commun. 2015, 464, 94–99. [Google Scholar] [CrossRef]
- Newburger, J.W.; Takahashi, M.; Beiser, A.S.; Burns, J.C.; Bastian, J.; Chung, K.J.; Colan, S.D.; Duffy, C.E.; Fulton, D.R.; Glode, M.P.; et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N. Engl. J. Med. 1991, 324, 1633–1639. [Google Scholar] [CrossRef]
- Wallace, C.A.; French, J.W.; Kahn, S.J.; Sherry, D.D. Initial intravenous gammaglobulin treatment failure in Kawasaki disease. Pediatrics 2000, 105, E78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smedly, L.A.; Tonnesen, M.G.; Sandhaus, R.A.; Haslett, C.; Guthrie, L.A.; Johnston, R.B.; Henson, P.M.; Worthen, G.S. Neutrophil-mediated injury to endothelial cells. Enhancement by endotoxin and essential role of neutrophil elastase. J. Clin. Investig. 1986, 77, 1233–1243. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, H.; Takeshita, S.; Nakatani, K.; Kawamura, Y.; Tokutomi, T.; Sekine, I. Delayed apoptosis of circulating neutrophils in Kawasaki disease. Clin. Exp. Immunol. 2001, 126, 355–364. [Google Scholar] [CrossRef]
- Biezeveld, M.H.; van Mierlo, G.; Lutter, R.; Kuipers, I.M.; Dekker, T.; Hack, C.E.; Newburger, J.W.; Kuijpers, T.W. Sustained activation of neutrophils in the course of Kawasaki disease: An association with matrix metalloproteinases. Clin. Exp. Immunol. 2005, 141, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Mihalache, C.C.; Simon, H.U. Autophagy regulation in macrophages and neutrophils. Exp. Cell Res. 2012, 318, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, G.; Zhao, E.; Liu, K.; Lin, Y.; Tesfa, L.; Tanaka, K.E.; Czaja, M.J. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1beta. J. Hepatol. 2016, 64, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maury, C.P.; Salo, E.; Pelkonen, P. Circulating interleukin-1 beta in patients with Kawasaki disease. N. Engl. J. Med. 1988, 319, 1670–1671. [Google Scholar] [CrossRef] [PubMed]
- Popper, S.J.; Shimizu, C.; Shike, H.; Kanegaye, J.T.; Newburger, J.W.; Sundel, R.P.; Brown, P.O.; Burns, J.C.; Relman, D.A. Gene-expression patterns reveal underlying biological processes in Kawasaki disease. Genome Biol. 2007, 8, R261. [Google Scholar] [CrossRef] [Green Version]
- Weng, K.P.; Hsieh, K.S.; Ho, T.Y.; Huang, S.-H.; Lai, C.-R.; Chiu, Y.-T.; Lin, C.-C.; Hwang, Y.-T.; Ger, L.-P.; Huang, S.-C. IL-1B polymorphism in association with initial intravenous immunoglobulin treatment failure in Taiwanese children with Kawasaki disease. Circ. J. 2010, 74, 544–551. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Plantinga, T.S.; Crisan, T.O.; Oosting, M.; Van De Veerdonk, F.L.; De Jong, D.J.; Philpott, D.J.; Van Der Meer, J.W.M.; Girardin, S.E.; Joosten, L.A.B.; Netea, M.G. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 2011, 60, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Hsieh, K.S.; Ming-Huey Guo, M.; Weng, K.-P.; Ger, L.-P.; Chan, W.-C.; Li, S.-C. Next-generation sequencing identifies micro-RNA-based biomarker panel for Kawasaki disease. J. Allergy Clin. Immunol. 2016, 138, 1227–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, Z.; Wu, F.; Dong, F.; Chuang, A.Y.; Messer, J.S.; Boone, D.L.; Kwon, J.H. Human autophagy gene ATG16L1 is post-transcriptionally regulated by MIR142-3p. Autophagy 2014, 10, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Chen, J.; Xu, H.G.; Zhou, X.; He, Q.; Li, Y.; Jiang, G.; Shan, Y.; Xue, B.; Zhao, R.; et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology 2014, 146, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Nakaoka, H.; Takasaki, I.; Hirono, K.; Yamamoto, S.; Kinoshita, K.; Miyao, N.; Ibuki, K.; Ozawa, S.; Watanabe, K.; et al. MicroRNA-93 may control vascular endothelial growth factor A in circulating peripheral blood mononuclear cells in acute Kawasaki disease. Pediatr. Res. 2016, 80, 425–432. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Tateya, S.; Cheng, A.M.; Rizzo-DeLeon, N.; Wang, N.F.; Handa, P.; Wilson, C.L.; Clowes, A.W.; Sweet, I.R.; Bomsztyk, K.; et al. M2 Macrophage Polarization Mediates Anti-inflammatory Effects of Endothelial Nitric Oxide Signaling. Diabetes 2015, 64, 2836–2846. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Karnam, A.; Stephen-Victor, E.; Gilardin, L.; Bhatt, B.; Sharma, V.K.; Rambabu, N.; Patil, V.; Lecerf, M.; Käsermann, F.; et al. Intravenous immunoglobulin mediates anti-inflammatory effects in peripheral blood mononuclear cells by inducing autophagy. Cell Death Dis. 2020, 11, 50. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Autophagy: A new concept in autoimmunity regulation and a novel therapeutic option. J. Autoimmun. 2018, 94, 16–32. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Analyses of mRNA autophagy markers in the peripheral white blood cells of Kawasaki disease (KD) patients before and after IVIG treatment. Total RNA from the peripheral white blood cells of healthy controls (HC, n = 20), febrile controls (FC, n = 20) and KD patients before (KD1, n = 20) and after (KD3, n = 20) IVIG treatment were isolated using a total RNA purification kit. Reverse transcription was performed using a high-performance reverse transcriptase system as described in the Methods section. The levels of BECN 1 (A), LC3B (B) and ATG16L1 (C) mRNA was measured by a real-time quantitative polymerase chain reaction (RT-qPCR). The expression levels were normalized to those of GAPDH. The comparisons among these groups were analyzed. Data are expressed as mean ± standard error. * p < 0.05, ** p < 0.01, *** p < 0.001 (by Kruskal–Wallis one-way analysis of variance).
Figure 1.
Analyses of mRNA autophagy markers in the peripheral white blood cells of Kawasaki disease (KD) patients before and after IVIG treatment. Total RNA from the peripheral white blood cells of healthy controls (HC, n = 20), febrile controls (FC, n = 20) and KD patients before (KD1, n = 20) and after (KD3, n = 20) IVIG treatment were isolated using a total RNA purification kit. Reverse transcription was performed using a high-performance reverse transcriptase system as described in the Methods section. The levels of BECN 1 (A), LC3B (B) and ATG16L1 (C) mRNA was measured by a real-time quantitative polymerase chain reaction (RT-qPCR). The expression levels were normalized to those of GAPDH. The comparisons among these groups were analyzed. Data are expressed as mean ± standard error. * p < 0.05, ** p < 0.01, *** p < 0.001 (by Kruskal–Wallis one-way analysis of variance).


Figure 2.
Analyses of mRNA autophagy markers in the peripheral white blood cells of Kawasaki disease (KD) patients without and with coronary artery lesions. Total RNA from the peripheral white blood cells of healthy controls (HC, n = 20) and KD patients without (non CAL, n = 20) and with coronary artery lesions (CAL, n = 10) 21 days after (KD3) IVIG treatment was isolated using a total RNA purification kit. Reverse transcription was performed using a high-performance reverse transcriptase system, as described in the Methods section. The levels of BECN 1 (A), LC3B (B) and ATG16L1 (C) mRNA was measured by a real-time quantitative polymerase chain reaction (RT-qPCR). The expression levels were normalized to those of GAPDH. The comparisons between the two groups of KD patients were analyzed. Data are expressed as mean ± standard error. ** p < 0.01 (by Mann–Whitney U test).
Figure 2.
Analyses of mRNA autophagy markers in the peripheral white blood cells of Kawasaki disease (KD) patients without and with coronary artery lesions. Total RNA from the peripheral white blood cells of healthy controls (HC, n = 20) and KD patients without (non CAL, n = 20) and with coronary artery lesions (CAL, n = 10) 21 days after (KD3) IVIG treatment was isolated using a total RNA purification kit. Reverse transcription was performed using a high-performance reverse transcriptase system, as described in the Methods section. The levels of BECN 1 (A), LC3B (B) and ATG16L1 (C) mRNA was measured by a real-time quantitative polymerase chain reaction (RT-qPCR). The expression levels were normalized to those of GAPDH. The comparisons between the two groups of KD patients were analyzed. Data are expressed as mean ± standard error. ** p < 0.01 (by Mann–Whitney U test).


{kind=link}
{kind=link}
{kind=link}
Table 1.
Demographic and Clinical Characteristics of Enrolled Patients.
| HC (n = 20) | FC (n = 20) | p Value | |
|---|---|---|---|
| age | 1.47 ± 0.61 | 1.49 ± 0.78 | 0.932 |
| sex | ♂(14)/♀(6) | ♂(12)/♀(8) | 0.519 |
| WBC | 9.035 ± 2.06 | 9.95 ± 3.47 | 0.371 |
| RBC | 4.81 ± 0.55 | 4.48 ± 0.24 * | 0.033 |
| Hemoglobin | 12.32 ± 0.9 | 11.58 ± 0.94 * | 0.047 |
| Hematocrit | 35.87 ± 2.57 | 35.01 ± 2.59 | 0.397 |
| Platelets | 314.25 ± 74.35 | 361.6 ± 145.1 | 0.243 |
| Segment | 30.75 ± 11.1 | 39.05 ± 22.12 | 0.287 |
| Lymphocyte | 58.17 ± 12.12 | 48.2 ± 21.08 | 0.110 |
| Monocyte | 6.18 ± 2.29 | 8.15 ± 4.31 | 0.202 |
| Eosinophil | 3.41 ± 2.07 | 1.42 ± 1.68 * | 0.014 |
| Basophil | 0.49 ± 0.77 | 0.18 ± 0.34 | 0.239 |
All variable data are expressed as mean ± SD. FC: indicates febrile control; HC: healthy control. * p < 0.05.
Table 2.
Demographic and Clinical Characteristics of Enrolled Patients.
| HC (n = 20) | KD1 (n = 20) | p Value | |
|---|---|---|---|
| age | 1.47 ± 0.61 | 1.40 ± 0.69 | 0.700 |
| sex | ♂(14)/♀(6) | ♂(13)/♀(7) | 0.747 |
| WBC | 9.03 ± 2.06 | 13.91 ± 4.98 * | 0.002 |
| RBC | 4.81 ± 0.55 | 4.30 ± 0.60 * | 0.014 |
| Hemoglobin | 12.32 ± 0.9 | 10.76 ± 0.94 *** | 0.000 |
| Hematocrit | 35.87 ± 2.57 | 32.5 ± 2.50 *** | 0.000 |
| Platelets | 314.25 ± 74.35 | 317.93 ± 94.42 | 0.896 |
| Segment | 30.75 ± 11.19 | 59.79 ± 10.40 *** | 0.000 |
| Lymphocyte | 58.17 ± 12.12 | 30.00 ± 9.84 *** | 0.000 |
| Monocyte | 6.18 ± 2.29 | 6.28 ± 2.77 | 0.899 |
| Eosinophil | 3.41 ± 2.07 | 2.42 ± 1.76 | 0.139 |
| Basophil | 0.49 ± 0.77 | 0.15 ± 0.26 | 0.078 |
All variable data are expressed as mean ± SD. HC: indicates healthy control and KD1: Kawasaki before intravenous immunoglobulin G (IVIG). * p < 0.05. *** p < 0.001.
Table 3.
Demographic and Clinical Characteristics of Enrolled Patients.
| FC (n = 20) | KD1 (n = 20) | p Value | |
|---|---|---|---|
| age | 1.49 ± 0.78 | 1.40 ± 0.69 | 0.725 |
| sex | ♂(12)/♀(8) | ♂(13)/♀(7) | 0.747 |
| WBC | 9.95 ± 3.47 | 13.91 ± 4.98 * | 0.038 |
| RBC | 4.48 ± 0.24 | 4.30 ± 0.61 | 0.322 |
| Hemoglobin | 11.58 ± 0.94 | 10.83 ± 0.96 | 0.062 |
| Hematocrit | 35.01 ± 2.59 | 32.5 ± 2.50 * | 0.022 |
| Platelets | 361.60 ± 145.10 | 317.93 ± 94.42 | 0.360 |
| Segment | 39.05 ± 22.12 | 59.79 ± 10.4 * | 0.017 |
| Lymphocyte | 48.2 ± 21.08 | 30.00 ± 9.84 * | 0.026 |
| Monocyte | 8.15 ± 4.31 | 6.28 ± 2.77 | 0.191 |
| Eosinophil | 1.42 ± 1.68 | 2.42 ± 1.76 | 0.163 |
| Basophil | 0.18 ± 0.34 | 0.15 ± 0.26 | 0.804 |
All variable data are expressed as mean ± SD. FC: indicates febrile control and KD1: Kawasaki before intravenous immunoglobulin G (IVIG). * p < 0.05.
Table 4.
Demographic and Clinical Characteristics of Enrolled Patients.
| HC (n = 20) | KD3 (n = 20) | p Value | |
|---|---|---|---|
| age | 1.47 ± 0.61 | 1.40 ± 0.69 | 0.75 |
| sex | ♂(14)/♀(6) | ♂(13)/♀(7) | 0.747 |
| WBC | 9.03 ± 2.06 | 8.28 ± 2.13 | 0.291 |
| RBC | 4.81 ± 0.55 | 4.53 ± 0.42 | 0.107 |
| Hemoglobin | 12.32 ± 0.9 | 11.66 ± 0.89 | 0.037 |
| Hematocrit | 35.87 ± 2.57 | 34.86 ± 1.97 | 0.208 |
| Platelets | 314.25 ± 74.35 | 337.64 ± 72.57 | 0.342 |
| Segment | 30.75 ± 11.19 | 29.43 ± 7.57 | 0.691 |
| Lymphocyte | 58.17 ± 12.12 | 61.89 ± 7.52 | 0.267 |
| Monocyte | 6.18 ± 2.29 | 4.90 ± 1.13 * | 0.045 |
| Eosinophil | 3.41 ± 2.07 | 3.26 ± 1.15 | 0.797 |
| Basophil | 0.49 ± 0.77 | 0.32 ± 0.27 | 0.421 |
All variable data are expressed as mean ± SD. HC: indicates healthy control and KD3: Kawasaki disease after intravenous immunoglobulin G (IVIG). * p < 0.05.
Table 5.
Demographic and Clinical Characteristics of Enrolled Patients.
| FC (n = 20) | KD3 (n = 20) | p Value | |
|---|---|---|---|
| age | 1.49 ± 0.78 | 1.40 ± 0.69 | 0.775 |
| sex | ♂(12)/♀(8) | ♂(13)/♀(7) | 0.747 |
| WBC | 9.95 ± 3.47 | 8.28 ± 2.13 | 0.140 |
| RBC | 4.48 ± 0.24 | 4.53 ± 0.42 | 0.734 |
| Hemoglobin | 11.58 ± 0.94 | 11.66 ± 0.89 | 0.828 |
| Hematocrit | 35.01 ± 2.59 | 34.86 ± 1.97 | 0.876 |
| Platelets | 361.60 ± 145.10 | 338.87 ± 74.89 | 0.594 |
| Segment | 39.05 ± 22.12 | 29.43 ± 7.57 | 0.213 |
| Lymphocyte | 48.20 ± 21.08 | 61.89 ± 7.52 | 0.075 |
| Monocyte | 8.15 ± 4.31 | 4.94 ± 1.09 ** | 0.007 |
| Eosinophil | 1.42 ± 1.68 | 3.26 ± 1.15 ** | 0.003 |
| Basophil | 0.18 ± 0.34 | 0.32 ± 0.27 | 0.245 |
All variable data are expressed as mean ± SD. FC: indicates febrile control and KD3: Kawasaki disease after intravenous immunoglobulin G (IVIG). ** p < 0.01.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huang, F.-C.; Huang, Y.-H.; Kuo, H.-C.; Li, S.-C. Identifying Downregulation of Autophagy Markers in Kawasaki Disease. Children 2020, 7, 166. https://0-doi-org.brum.beds.ac.uk/10.3390/children7100166
AMA Style
Huang F-C, Huang Y-H, Kuo H-C, Li S-C. Identifying Downregulation of Autophagy Markers in Kawasaki Disease. Children. 2020; 7(10):166. https://0-doi-org.brum.beds.ac.uk/10.3390/children7100166
Chicago/Turabian StyleHuang, Fu-Chen, Ying-Hsien Huang, Ho-Chang Kuo, and Sung-Chou Li. 2020. "Identifying Downregulation of Autophagy Markers in Kawasaki Disease" Children 7, no. 10: 166. https://0-doi-org.brum.beds.ac.uk/10.3390/children7100166
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.