New and Emerging Targeted Therapies for Pediatric Acute Myeloid Leukemia (AML)


1
Division of Pediatric Hematology/Oncology, Hackensack University Medical Center, Hackensack, NJ 07601, USA
2
Division of Pediatric Hematology/Oncology, NYU Winthrop Hospital, Mineola, NY 11501, USA
*
Authors to whom correspondence should be addressed.
Children 2020, 7(2), 12; https://0-doi-org.brum.beds.ac.uk/10.3390/children7020012
Submission received: 16 December 2019
/
Revised: 30 January 2020
/
Accepted: 1 February 2020
/
Published: 10 February 2020
(This article belongs to the Special Issue Pediatric Acute Myeloid Leukemia: Current Management and Future Directions)
Abstract
:The relapse rate for children with acute myeloid leukemia (AML) remains high despite advancements in risk classification, multi-agent chemotherapy intensification, stem cell transplantation, and supportive care guidelines. Prognosis for this subgroup of children with relapsed/refractory AML remains poor. It is well known that the ceiling of chemotherapy intensification has been reached, limited by acute and chronic toxicity, necessitating alternative treatment approaches. In the last several years, our improved understanding of disease biology and critical molecular pathways in AML has yielded a variety of new drugs to target these specific pathways. This review provides a summary of antibody drug conjugates (ADCs), small molecule inhibitors, and tyrosine kinase inhibitors with an emphasis on those that are currently under clinical evaluation or soon to open in early phase trials for children with relapsed/refractory AML.
1. Introduction
Acute myeloid leukemia (AML), a hematopoietic stem cell disorder characterized by clonal expansion of abnormally differentiated myeloid lineage blasts, accounts for 20% of childhood leukemia [1]. AML continues to pose a significant therapeutic challenge due to disease heterogeneity, high relapse rate, and therapy toxicity [2]. As compared to a 90% overall survival (OS) in childhood acute lymphoblastic leukemia (ALL), the OS approaches only 69–75% in childhood AML at best [2,3,4]. Despite significant advancements in risk classification and frontline therapeutic approaches, about 50% of children with AML relapse and prognosis for these children remains poor [3]. Recently published data generated from the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) AML initiative, a collaborative Children’s Oncology Group (COG)–National Cancer Institute (NCI) project to characterize the mutational, transcriptional, and epigenetic landscape of childhood AML, have significantly broadened our understanding of the biology of AML in children and how it differs from adults. Large-scale retrospective whole genome, DNA, and RNA sequencing as well as methylation profiling has revealed novel fusion genes, focal deletions, and recurrent mutations unique to pediatrics, some of which are associated with particularly poor prognoses. It has become clear that pediatric AML is a collection of molecularly diverse diseases with similar phenotype which has led to a refinement of our molecular risk classification and increased efforts toward personalized targeted therapy approaches [5]. It is evident that the ceiling of chemotherapy intensification has been reached, limited by infectious and cardiac toxicity, urgently necessitating novel therapeutic strategies. Alternate approaches to cytotoxic chemotherapy including epigenetic therapy, immunotherapy, antibody drug conjugates (ADCs), and small molecule inhibitor therapies are revolutionizing cancer care. In this review, we focus on the role of ADCs, small molecule inhibitors, and tyrosine kinase inhibitors in the treatment of childhood AML, discussing existing and emerging drugs that hold great promise in improving outcomes.
2. Antibody Drug Conjugates
Antibody-based therapies have emerged as an effective and well-tolerated approach to cancer care, whereby attaching a cytotoxic drug to an antibody can lead to increased dose intensity with reduced toxicity [6] (Figure 1, Table 1). The challenge in AML has been to identify targetable cell surface proteins that are expressed in AML blasts but not in normal hematopoietic cells.
2.1. Targeting CD33
The cell surface antigen, CD33, is present in more than 80% of patients with AML but is absent from hematopoietic stem cells, rendering it an ideal immunoconjugate target [2]. From early on, agents that were pursued as therapeutics included unconjugated antibodies, radioimmunoconjugates, ADCs, and immunotoxins; however, success with CD33-targeted therapeutics has been limited with several agents failing at various stages of clinical development [7]. For example, Lintuzumab (Seattle Genetics, HuM195, SGN-33), an unconjugated humanized murine monoclonal antibody, showed great promise in vitro eliciting antibody-dependent cellular cytotoxicity (ADCC) and phagocytosis leading to AML cell killing. However, this activity did not translate successfully to the clinic in adult clinical trials, resulting in termination of drug development [7,8,9]. The newer generation, vadastuximab talirine (Seattle Genetics, SGN-CD33A), a CD33 directed antibody conjugated to pyrrolobenzodiazepine (PBD) dimers, has shown more promise in relapsed/refractory (r/r) adult patients as monotherapy and in combination with a hypomethylating agent, but concerns over hepatic and hematopoietic toxicity derailed drug development [7,10,11].
The most promising data for targeting CD33 in both children and adults have come from studies with Gemtuzumab ozogamicin (Mylotarg, GO, Pfizer, New York, USA), a humanized IgG4 anti-CD33 antibody conjugated to calicheamicin, a DNA-cleaving cytotoxic agent, leading to its FDA approval in 2000. A subsequent randomized controlled trial in adults, Southwest Oncology Group (SWOG) S0106, failed to meet its primary end points of improved remission induction and safety, resulting in withdrawal of approval [2,12]. However, the results of this study were controversial and other concurrent adult studies did show improved survival with the addition of GO [13,14]. COG demonstrated feasibility and safety in adding 2 doses of GO to standard intensive chemotherapy for children with previously untreated AML in their pilot study AAML03P1 [15]. In the subsequent randomized controlled trial, AAML0531, newly diagnosed pediatric AML patients were randomized to receive the standard upfront chemotherapy backbone with or without 2 doses of GO at 3 mg/m2/dose. This landmark study showed that 3-year event free survival (EFS) was significantly improved with the addition of GO (53% vs. 46.9%, p = 0.04) by a significant reduction in 3-year relapse risk (RR) (32.8% vs. 41.3%, p = 0.006), particularly in the low and intermediate risk groups. Furthermore, the study did not demonstrate an increased risk of sinusoidal obstructive syndrome (SOS) in the GO group as had been the case in early studies using higher doses of GO [2]. Based on the results of these studies, GO again earned Food and Drug Administration (FDA) approval in 2017 for the treatment of newly diagnosed CD33-positive AML in r/r adults and children ≥2 years of age [16]. This success story has led to the incorporation of GO into the backbone of the upcoming COG randomized controlled clinical trial, AAML1831, comparing CPX-351, a liposomal preparation of cytarabine and daunorubicin versus standard cytarabine and daunorubicin, expected to open for enrollment in the first quarter of 2020.
2.2. Targeting Mesothelin
Mesothelin is a cell-surface tumor differentiation antigen expressed on mesothelial cells of serosal lining. It has been associated with malignant transformation, cellular proliferation, and tumor aggressiveness in a variety of solid tumors, including lung, pancreatic, and ovarian origin. Mesothelin was recognized as an attractive candidate for targeted cancer therapy due to its limited expression in normal tissue and high expression in cancer tissue [17,18]. Anetumab ravtensine (AR) (Bayer, Leverkusen, Germany) is an ADC that contains a human anti-mesothelin antibody conjugated to the maytansinoid tubulin inhibitor DM4 via a reducible disulfide linker [19]. Preclinical studies have shown potent antitumor activity in adult solid tumor models [19,20], which has led to the development of a number of Phase I and II clinical trials for adults with aggressive mesothelin-expressing solid tumors alone and in combination therapy [17]. Mesothelin was also shown to be expressed in pediatric AML cells [21]. Building on this finding, as part of the NCI/TARGET AML initiative, transcriptome sequencing (RNA-seq) was performed on AML cell lines which demonstrated that mesothelin was one of the most highly expressed genes in ~30% of childhood AML cases, a higher prevalence than in adult AML cases (~11%). Therefore, they conducted in vitro and in vivo studies with mesothelin-overexpressing AML cell lines and xenografts, respectively, and found that treatment with AR resulted in significant mesothelin-dependent efficacy at clinically achievable doses [22,23]. Furthermore, they demonstrated in vivo synergy between mesothelin-targeted therapy and conventional chemotherapy in mesothelin+ AML xenografts [24]. Based on this promising data and emerging safety and efficacy data from adult solid tumor clinical trials, a new Phase I COG study, AAML2011, is currently in development to assess treatment with AR for patients with r/r mesothelin-expressing AML.
2.3. Targeting CD123
CD123, the alpha subunit of the IL-3 receptor, is overexpressed in multiple hematologic malignancies, including AML, ALL, and blastic plasmacytoid dendritic cell neoplasm (BPDCN). Because of its high expression on leukemic blasts as compared with normal hematopoietic stem cells, CD123 has emerged as an attractive candidate for molecularly targeted therapeutics [25]. Tagraxofusp-erzs (Elzonris, Stemline) and IMGN632 (immunogen) are two anti-CD123-directed immunotoxins which have been developed in recent years. Tagraxofusp-erzs is a novel biologic targeted therapy, comprised of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis directed at the interleukin-3 receptor [26]. In December 2018, Tagraxofusp-erzs gained FDA approval for treatment of BPDCN in adult and pediatric patients ≥2 years of age. The approval was based on results of a single arm study, STML-401-0114, in which the pivotal cohort of 13 treatment-naïve BPDCN patients, treated with Tagraxofusp-erzs monotherapy, showed a 54% composite complete remission (CRc) rate and safety was established in 94 patients with myeloid neoplasms [27,28].
IMGN632 is comprised of a novel humanized anti-CD123 antibody, G4723A, linked to a unique DNA-alkylating payload of the recently developed IGN (indolinobenzodiazepine pseudodimer) class of cytotoxic compounds [25,29]. Kovtun et al. showed that IMGN632 demonstrated potent activity in all AML samples at concentrations well below levels that impacted normal bone marrow progenitors and exhibited robust antitumor activity with a wide therapeutic index in multiple AML xenografts [25]. Subsequently, Daver et al. conducted the first Phase I study of IMGN632 in adult patients with r/r AML and other CD123-positive hematologic malignancies (National Clinical Trial (NCT) 03386513), which is still recruiting. At the time of analysis, they found that 20% of AML patients (n = 66) and 43% (n = 7) of BPDCN patients had an objective response (complete remission (CR), complete remission with incomplete hematologic recovery (CRi), and partial remission (PR)) to IMGN632 [30]. These encouraging results have led to the development of a Phase Ib/II trial (NCT04086264) planned to evaluate IMGN632 as monotherapy or in combination with Venetoclax and/or Azacytidine for adults with untreated CD123+ AML ineligible for standard upfront therapy, minimal residual disease (MRD) positive, or r/r disease [31]. Through the Pediatric Acute Leukemia (PedAL) Initiative, the COG is currently developing a study for children with r/r leukemias, with a Phase I dose finding monotherapy arm for ALL and AML followed by a Phase II safety arm in combination with chemotherapy, followed by a Phase III randomization of chemotherapy ± IMGN632.
3. Small Molecule Inhibitors
With the discovery of multiple pathways critical to the proliferation and survival of AML cells, small molecule inhibitors have emerged as a promising therapeutic strategy in AML (Figure 1, Table 1). Small molecule inhibitors have the advantage of being able to target cell surface ligand receptors and intracellular proteins that promote the downstream signaling pathway for cell growth and metastatic disease. Targeted therapy with small molecule inhibitors, such as tyrosine kinase inhibitors (TKIs) for t(9;22) BCR-ABL gene fusion for Chronic Myeloid Leukemia (CML) or all-trans retinoic acid (ATRA) for acute promyelocytic leukemia (APML), have already proven to be successful [32]. The challenge in identifying potential targets in AML has been its heterogeneous genetic and molecular make up, with frequent evolution of the driver mutations at disease onset and progression.
3.1. E-Selectin Inhibitors
Binding of E-selectin adhesion molecules to AML blasts enhances the adherence of AML cells within the vascular bone marrow niche via activation of the Wnt signaling pathway. Protection within the vascular marrow niche supports leukemia cell quiescence, limits their exposure to cell-cycle dependent chemotherapy, and prolongs their survival [33,34,35]. The majority of primary AML blasts express E-selectin ligand, with increased expression in relapsed patients [35]. In addition, the higher expression of E-selectin associated with relapsed disease correlates with poor survival and high-risk disease in AML [36]. Because of its high expression and the protective role in AML cells, E-selectin has emerged as an ideal candidate for targeted therapy.
Uproleselan (Glycomimetics (GMI)-1271) is a small molecule inhibitor designed to prevent the binding of E-selectin to ligands on leukemia blasts. Preclinical work demonstrated that GMI-1271 can overcome adhesion-mediated chemotherapy resistance in vitro and reduce leukemia burden in mice engrafted with primary AML cells [35,37]. Uproleselan was granted Breakthrough Therapy Designation by the US FDA in 2017 for adults with r/r AML based on the notable result produced from a Phase II clinical trial in combination with mitoxantrone/etoposide/cytarabine (MEC) chemotherapy. This resulted in a CR/CRi for 22 out of 54 patients (41%), MRD-negative remission for 11 out of 16 evaluable patients (69%) at the recommended Phase II dose, and a median overall survival of 8.8 months. In addition, those with high expression of E-selectin ligand demonstrated greater inhibition by uproleselan and significantly longer overall survival [38]. A Phase III trial is ongoing to investigate the efficacy of uproleselan with MEC chemotherapy compared to MEC alone in adults with r/r AML (NCT03616470). The results of this trial have not yet been published. The study of E-selectin and its inhibitors in pediatric AML has been minimal; however, a recent report demonstrated that E-selectin ligand expression is also associated with poor survival in a small cohort of AML patients treated on COG AAML1031 [39]. Plans are being made for an early phase trial of chemotherapy ± uproleselan in children with r/r AML through the PedAL Initiative.
3.2. Targeting KMT2A-Fusion
KMT2A (formerly known as MLL or mixed lineage leukemia)-rearranged acute leukemia, involving fusions of 11q23, is a particularly aggressive leukemia affecting approximately 15–20% of childhood AML. KMT2A rearrangements are found in several groups of leukemia patients: Infantile ALL, AML, and acute megakaryoblastic leukemia (AMKL), a rare subtype of AML that occurs mainly in children with Down syndrome [40,41,42]. Outcome remains poor for KMT2A-rearranged leukemia following current conventional chemotherapy, although prognosis for AML is dependent on its fusion partner, of which over 100 fusion partners are known to date [40,41]. Clearly, there is a need for new treatment modalities for this group of patients. Two classes of small molecule inhibitors have emerged as possible therapies for KMT2A-rearranged leukemia: Disruptor of telomeric silencing 1-like (DOT1L) inhibitors and menin inhibitors.
DOT1L (disruptor of telomeric silencing 1-like) is a histone methyltransferase at H3K79 that in its normal form transcriptionally modifies chromatin of downstream target genes to help maintain the integrity of chromosomes and genes. However, the aberrant recruitment of DOT1L by a majority of KMT2A-fusion partners results in an inappropriate hypermethylation and overexpression of downstream, targeted genes that promote leukemia. The oncogenic drive of KMT2A-rearranged leukemia is dependent on the activation of DOT1L [43,44]. Therefore, the inhibition of DOT1L activation is an attractive target for new drug development. Preclinical work on DOT1L inhibitors in both cell lines and mice transformed with KMT2A-rearranged leukemia demonstrated inhibition of tumor proliferation and leukemia regression [45]. Pinometostat (Epizyme (EPZ)-5676), a DOT1L inhibitor, was one of the first histone methyltransferase inhibitors to undergo Phase I trials for both children and adults with r/r KMT2A-rearranged acute leukemia. However, single agent therapy with Pinometostat demonstrated very modest anti-leukemic activity in a cohort of 51 adults with r/r acute leukemia, a majority with KMT2A-rearrangement, resulting in 2 CR, both with t (11,19), 3 patients with resolution of leukemia cutis, and 9 patients with morphological evidence of myeloid differentiation [46]. In children with r/r, KMT2A-rearranged acute leukemia, there was no objective response as defined by protocol, although 7 out of 18 children showed evidence of transiently reduced peripheral blood or marrow blasts without meeting thresholds for objective response during therapy [47]. Although not significantly efficacious as a single agent, Pinometostat combined with azacitidine (NCT03701295) in KMT2A-rearranged r/r AML is currently being investigated in adults.
Another area of therapeutic interest has been to inhibit the binding between MLL and menin, an essential co-factor of the oncogenic component of the KMT2A complex that also acts as a histone methyltransferase to transcriptionally regulate targeted genes that are critical to leukemogenesis. Several small molecule menin inhibitors have been developed to date [48,49]. One of the earliest hallmark preclinical studies of first generation menin inhibitors, MI-463 and MI-503, demonstrated high potency inhibition of menin in KMT2A-rearranged leukemia cell lines and a substantial survival benefit in mice models harboring KMT2A-rearranged leukemia [48]. The most promising preclinical work has resulted from Vitae Pharmaceuticals (VTP)-50469 (close analog of Syndax (SNDX)-5613), an orally available, potent, and highly selective menin inhibitor which significantly inhibited proliferation of tumor growth in vitro and in vivo [44]. The robustness and consistency of these preclinical results have recently gained the approval of SNDX-5613 to enter into a Phase I/II clinical trial for r/r acute leukemia in adults, including those with KMT2A-rearranged ALL or AML, in late 2019 (NCT04065399). In children, however, VTP-50469 has only been investigated in preclinical, patient-derived xenograft (PDX) models harboring KMT2A-rearranged acute lymphoblastic leukemia. A report at the annual American Association for Cancer Research (AACR) meeting in 2018 revealed that 6 out of 7 of these PDX models demonstrated significantly longer EFS (ranging 1.1 to 109 days) compared to a non-KMT2A-rearranged PDX model following treatment with VTP-50469. Plans are being made for an early phase trial of chemotherapy ± SNDX-5613 in children with r/r acute leukemia through the PedAL Initiative [50].
Finally, another menin-KMT2A inhibitor, Kura Oncology (KO)-539, has been shown to inhibit leukemia growth and prolong survival in KMT2A-rearranged cell lines and in vivo models, earning the FDA Orphan Drug Designation [51]. This compound also recently entered a Phase I/II trial in adults with r/r AML (NCT04067336).
Targeting more than one component of the KMT2A complex has great potential for targeted treatment. Although a DOT1L inhibitor by itself has not shown promising results in Phase I trials, Dafflon et al. showed that the combination of a DOT1L inhibitor and a menin inhibitor demonstrated robust synergy in its anti-leukemic growth inhibition effect in a mouse model injected with KMT2A-AF9 leukemic cells, compared to either drug alone [44,52]. Further studies will be needed to investigate whether the combination of Pinometostat with another anti-leukemic agent can improve its efficacy.
3.3. MEK Inhibitors
Mitogen-activated protein kinase (MAPK) cascade is a signaling pathway crucial for the regulation of cell differentiation, proliferation, and survival. The cascade is activated by ligand binding to a receptor tyrosine kinase (RTK) to activate Ras then Raf, followed by Mitogen-activated protein kinase (MAPK, also known as MEK), finally resulting in activation of the last component of the pathway: Extracellular signal-regulated kinase (ERK). Activated ERK can then activate a variety of downstream substrates, and so the suppression of MEK/ERK activation can have profound effects on controlling cell growth [53,54,55,56]. Ras pathway mutations are highly prevalent in relapsed acute leukemia, especially in pediatric AML where it can represent up to 30% of cases, as KRAS, NRAS, fms-like tyrosine kinase 3 (FLT3) or PTPN11 mutations [5]. RAS pathway mutations are particularly common with KMT2A fusions [5], core-binding factor AML [57], and normal karyotype. Although it is not formally defined as a poor prognostic marker, it also does not significantly impact overall or event-free survival. RAS pathway mutations are nevertheless associated with high risk features as stated above. Due to difficulty in targeting RAS itself, most of the effort has concentrated on inhibiting downstream effectors such as MEK [58]. The most studied of the MEK inhibitors has been trametinib, a MEK 1/2 inhibitor.
The preclinical efficacy of MEK inhibitors in reducing leukemia burden and prolonging survival in cell lines, primary patient cells, and mouse xenograft models harboring RAS-mutated acute leukemia cells has been demonstrated in a variety of studies [59,60]. Data are not as robust in childhood acute leukemia, but ex vivo drug sensitivity testing on RAS-mutated primary ALL and AML cells from children enrolled onto Leukemia Precision-based Therapy (LEAP) Consortium (NCT026770525) has demonstrated dose-response sensitivity of leukemia cells to trametinib [61]. To date, the COG has only investigated a MEK inhibitor for acute leukemia through one of its registered studies, which is an ongoing Phase II trial of trametinib in r/r juvenile myelomonocytic leukemia (JMML). The results have not yet been published. Clinically, trametinib showed an overall response rate of 20% as a single-agent therapy in a cohort of r/r NRAS/KRAS-mutated AML in adults [62]. Plans are being made for an early phase trial of chemotherapy ± trametinib in children with r/r, RAS-mutated ALL or AML through the PedAL Initiative.
3.4. MDM2 Antagonists
Although Tumor Protein p53 (TP53) mutation is an infrequent event in AML [63], the inactivation of wild-type p53 occurs in a majority of AML. Multiple mechanisms exist for the inactivation of p53. The best-studied mechanism is by an overexpression of MDM2 (murine double minute 2). MDM2, like TP53, is also expressed in a variety of human cancers. More importantly, its overexpression is reported in up to 50% of AML [63,64]. MDM2 is primarily a negative regulator of the activity of TP53, binding to the transcriptional domain of TP53 to prevent it from binding to specific DNA sequences and marking P53 for proteasomal degradation. In this way, an overexpression of MDM2 in AML allows for the amplification of the MDM2–TP53 interaction to further limit the tumor suppressor functions of wild-type TP53 [65,66,67]. In addition, the overexpression of MDM2 is also associated with shorter CR duration and event-free survival [64]. Therefore, MDM2 antagonists have emerged as a promising anti-cancer agent to block the MDM2–TP53 interaction and restore the tumor suppressor functions of TP53.
Among the first studied small-molecule MDM2 antagonists are the Nutlins [68], which bind to MDM2 in the p53-binding pocket to activate p53 signaling pathways. As early as 2004, Nutlins demonstrated cell-cycle arrest, apoptosis, and tumor growth inhibition in AML cell lines, primary AML samples, and xenograft mice models harboring wild-type p53. As expected, those harboring p53 mutations were resistant to Nutlins [68,69]. More recent preclinical studies continue to support the in vitro and in vivo anti-leukemic effects of Nutlins in wild-type p53-expressing AML cells [70]. Idasanutlin, a second-generation Nutlin, was developed to achieve more potency and less toxicity than early generation Nutlins. A Phase Ib/II trial of Idasanutlin with Cytarabine in adults with r/r AML achieved a CR rate of 25% and a CRc rate of 29% [71]. Idasanutlin is now being investigated in a Phase III trial in r/r adult AML in combination with Cytarabine compared to Cytarabine alone (NCT02545283). The results have not yet been published. Idasanutlin is also being investigated in a Phase Ib/II trial for r/r adult AML in combination with venetoclax, a B cell lymphoma-2 (BCL-2) inhibitor (NCT02670044) based on synergistic effect in preclinical studies [72]. Early clinical data have shown the combination to be safe with an anti-leukemic effect of 50% and a CR/CRi/CRp rate of 29% at the recommended Phase II dose [73]. A review on the efficacy of Idasanutlin in AML was recently published [74]. In pediatrics, Carol et al. also demonstrated high anti-leukemic activity of RG7112 (Idasanutlin) as a single agent in pediatric ALL xenografts [75]. In children with r/r AML, Aileron (ALRN)-6924 (a dual MDM2/MDMX inhibitor) in combination with Cytarabine is now being investigated for the first time through an ongoing Phase 1 trial (NCT03654716).
3.5. Targeting Mutant TP53
The most frequent genetic variation across all human cancers is mutation of the tumor suppressor gene TP53. It is present mainly as a missense point mutation in up to 50% of some of the most common adult cancers, including head and neck cancers, pancreatic, colorectal, and ovarian [76]. In comparison, it is an infrequent occurrence in acute leukemia. About 8% of young adults and older with AML have TP53 mutations [63,77], although it has been reported in up to 18% of adults with newly diagnosed AML [78]. It is even less frequent in children with AML [5]. Despite its infrequent occurrence, it is still associated with therapy-related myelodysplastic syndrome (MDS)/AML, relapsed AML, and poor survival compared to TP53 wild-type AML [63,78]. Mutations of TP53 lead to loss of wild-type functions, including intrinsic tumor suppressive properties such as cell apoptosis and senescence. This results in cancer cell proliferation and survival with promotion of metastatic disease and chemoresistance [63,79].
Unlike MDM2 antagonists which aim to target the overexpression of a TP53-interacting protein, targeting the mutant TP53 itself is also of therapeutic interest. The most successful trial to date in AML has been testing Aprea Therapeutics (APR)-246, a methylated, small-molecule derivative of PRIMA-1 (p53 re-activation and induction of massive apoptosis), which has been shown to bind and reactivate mutant and inactivated p53 by restoring its wild-type transcriptional activity to induce cell apoptosis [80,81]. An ongoing Phase Ib/II clinical trial investigating APR-246 combined with Azacitidine in adults with TP53-mutated MDS and oligoblastic AML (≤30% blasts) was recently presented at the 2019 American Society of Hematology annual meeting (NCT03072043). The Phase II component of the trial achieved a CR rate of 50% for AML and an overall response rate (ORR) of 88% for MDS/AML with a well-tolerated combination. Median time to response was 2.1 months and the median duration of response was 6.5 months. In addition, an isolated TP53 mutation was predictive of a higher CR rate following APR-246 therapy [82]. In April 2019, APR-246 was granted Fast Track and Orphan Drug designations for the treatment of adult MDS with TP53 mutation by the FDA. These promising results also supported the ongoing adult Phase III trial of Azacitidine ± APR-246 in TP53-mutated MDS (NCT03745716). Further work will be needed to elucidate which of the thousands of TP53 mutations may be better targeted by APR-246.
4. FLT3 Inhibitors
FLT3 (fms-like tyrosine kinase 3), a cytokine receptor (CD135) belonging to the receptor tyrosine kinase class III, is expressed mainly on hematopoietic cells and plays a pivotal role in myeloid and lymphoid cell proliferation and survival. FLT3 internal tandem duplications (FLT3-ITDs), the most common class of FLT3 gene mutations found in AML, lead to constitutive activation of the kinase, promoting cell growth, survival, and anti-apoptotic signaling. FLT3-ITDs are associated with a poor prognosis due to a high relapse rate [6]. There has been great excitement in developing targeted FLT3 kinase inhibitors over the past two decades and these agents have improved outcomes in this AML subtype (Figure 1, Table 1).
First generation FLT3 inhibitors including sorafenib and midostaurin are multi-kinase inhibitors that show activity against a host of growth factors, including FLT3, KIT, platelet-derived growth factor receptor (PDGFR), and vascular endothelial growth factor (VEGF), leading to more off-target toxicity. Sorafenib is FDA-approved for treatment of renal cell carcinoma. There have been mixed results in early phase clinical trials of Sorafenib in combination with chemotherapy for adults with de novo FLT3-ITD+ AML [83,84]. However, the COG AAML 1031 clinical trial, evaluating the addition of Sorafenib to standard chemotherapy as well as maintenance sorafenib following hematopoietic stem cell transplant (HSCT) for children with high allelic ratio FLT3-ITD+ AML showed improved CR rates as compared to historical controls following induction I (73% vs. 56%, p = 0.078) and following induction II (91% vs. 70%, p = 0.007). Furthermore, 3-year EFS was improved to 57.5% from the historical control of 34.3%, p = 0.007, and the RR was reduced with sorafenib treatment (18.2% vs. historical 52.5%, p = 0.006), although 3-year OS was not significantly different (63.9% vs. 54.1%, p = 0.375) [85]. Sorafenib is also being investigated in a Phase I trial in combination with Palbociclib in adolescents and adults with R/R leukemia (NCT03132454).
Midostaurin, another first-generation FLT3 inhibitor, is the first tyrosine kinase inhibitor to be FDA approved for AML therapy in the first line. Approval largely resulted from the Alliance 10603 trial, in which adults with newly diagnosed, FLT3-mutated AML, who received midostaurin in combination with standard chemotherapy had superior EFS and OS as compared to standard chemotherapy alone [86]. In children, midostaurin has been investigated as monotherapy in r/r pediatric leukemia, including KMT2A-rearranged ALL or FLT3-mutated AML (NCT0866281), which unfortunately demonstrated limited efficacy in the 22 enrolled participants (the trial terminated early due to lack of enrollment) [87]. Midostaurin is now being investigated in combination with chemotherapy in children with newly diagnosed, FLT3-mutated AML (NCT03591510), which is ongoing.
Second-generation FLT3 inhibitors are much more highly selective in targeting FLT3, thereby showing more limited off-target toxicity [6]. Quizartinib has completed investigation through a Phase I clinical trial in combination with salvage chemotherapy in r/r childhood leukemia, both harboring FLT3 wild-type and FLT3-ITD mutations, and demonstrated 4/17 CR and 10/17 stable disease (SD). Of the 7 FLT3-ITD mutated patients on the trial, there were 3 CR and 4 SD, demonstrating some clinical efficacy in all FLT3-mutated patients [88]. Quizartinib is currently being investigated in a Phase I/II clinical trial in combination with re-induction chemotherapy and as monotherapy for maintenance in r/r, FLT3-ITD mutated AML in children (NCT03793478).
Finally, gilteritinib is another potent and highly selective second generation FLT3 inhibitor that exerts dual activity against FLT3 and AXL, an additional receptor tyrosine kinase that promotes proliferation and survival of AML cells [6]. Due to promising results from a Phase I/II trial on giltertinib in adults with r/r AML harboring both wild-type and FLT3 mutations [89], a randomized Phase III trial was conducted comparing gilteritinib monotherapy versus salvage chemotherapy in adults with r/r, FLT3-mutated AML. Patients who received gilteritinib demonstrated a significantly higher CR/Cri rate (34% vs. 15%), longer median OS (9.3 months vs. 5.6 months), and longer post-transplant survival (16.2 months vs. 8.4 months) as compared to those who received salvage chemotherapy, resulting in FDA approval for adults with r/r, FLT3-mutated AML [90]. Gilteritinib is planned to be investigated in the upcoming COG AML trial for children with newly diagnosed AML (AAML1831).
5. Conclusions
In summary, the last several years have been pivotal for AML clinicians, researchers, and patients, representing a period of robust discovery and development of targeted drugs for AML. Many of these drugs, as described in this review, have advanced from bench to bedside and are being evaluated in adults with either ongoing or emerging studies in children. The impact of these ADCs, small molecule inhibitors, and FLT3 inhibitors on improving survival in children with leukemia and other cancers remains to be seen as pediatric clinical trials become increasingly available. While the evaluation of these novel drugs as monotherapy is a critical first step, it will be equally critical to test their safety and efficacy in strategic combination with cytotoxic chemotherapy and/or other novel therapies. The standard of care for upfront AML therapy in children remains a risky, stratified, multi-agent chemotherapy regimen, but as evidenced by the last several landmark COG AML trials, incorporation of targeted therapies, including Gemtuzumab ozogamicin and tyrosine kinase inhibitors targeting FLT3-ITD mutations, are improving outcomes. The AML molecular landscape has evolved along with our understanding of the biology and genetic makeup of AML, and the potential of ongoing research may dramatically inform future treatment options.
Author Contributions
J.C. and C.L.G. both conceptualized, prepared the original draft, reviewed, and edited the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This review received no external funding.
Acknowledgments
The authors are grateful to Matthew Hockenberry for assistance with Figure 1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gamis, A.S.; Alonzo, T.A.; Perentesis, J.P.; Meshinchi, S.; COG Acute Myeloid Leukemia Committee. Children’s oncology group’s 2013 blueprint for research: Acute myeloid leukemia. Pediatr. Cancer 2013, 60, 964–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.B.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: Results from the randomized phase III children’s oncology group trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.H.; Reinhardt, D. Successes and challenges in the treatment of pediatric acute myeloid leukemia: A retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Kuhlen, M.; Klusmann, J.H.; Hoell, J.I. Molecular approaches to treating pediatric leukemias. Front. Pediatr. 2019, 7, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohl, S.R.; Bullinger, L.; Rucker, F.G. New targeted agents in acute myeloid leukemia: New hope on the rise. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Walter, R.B. Investigational CD33-targeted therapeutics for acute myeloid leukemia. Expert Opin. Investig. Drugs 2018, 27, 339–348. [Google Scholar] [CrossRef]
- Raza, A.; Jurcic, J.G.; Roboz, G.J.; Maris, M.; Stephenson, J.J.; Wood, B.L.; Feldman, E.J.; Galili, N.; Grove, L.E.; Drachman, J.G.; et al. Complete remissions observed in acute myeloid leukemia following prolonged exposure to lintuzumab: A phase 1 trial. Leuk. Lymphoma 2009, 50, 1336–1344. [Google Scholar] [CrossRef]
- Feldman, E.J.; Brandwein, J.; Stone, R.; Kalaycio, M.; Moore, J.; O’Connor, J.; Wedel, N.; Roboz, G.J.; Miller, C.; Chopra, R.; et al. Phase iii randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J. Clin. Oncol. 2005, 23, 4110–4116. [Google Scholar] [CrossRef]
- Fathi, A.T.; Erba, H.P.; Lancet, J.E.; Stein, E.M.; Ravandi, F.; Faderl, S.; Walter, R.B.; Advani, A.S.; DeAngelo, D.J.; Kovacsovics, T.J.J.B. A phase 1 trial of vadastuximab talirine combined with hypomethylating agents in patients with CD33-positive AML. Blood 2018, 132, 1125–1133. [Google Scholar] [CrossRef]
- Stein, E.M.; Walter, R.B.; Erba, H.P.; Fathi, A.T.; Advani, A.S.; Lancet, J.E.; Ravandi, F.; Kovacsovics, T.; DeAngelo, D.J.; Bixby, D.J.B. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood 2018, 131, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garfin, P.M.; Feldman, E.J. Antibody-based treatment of acute myeloid leukemia. Curr. Hematol. Malig. Rep. 2016, 11, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terre, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Franklin, J.; Gerbing, R.B.; Alonzo, T.A.; Hurwitz, C.; Raimondi, S.C.; Hirsch, B.; Smith, F.O.; Mathew, P.; Arceci, R.J.; et al. AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: A report from the children’s oncology group. Cancer 2012, 118, 761–769. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. Fda approval summary: Mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- Ghafoor, A.; Thomas, A.; Hassan, R. Targeting mesothelin in ovarian cancer. Oncotarget 2018, 9, 36050–36051. [Google Scholar] [CrossRef]
- Tarlock, K.; Kaeding, A.J.; Alonzo, T.A.; Loken, M.R.; Ries, R.E.; Pardo, L.; Gerbing, R.; Farrar, J.E.; Auvil, J.M.G.; Gerhard, D.S. Discovery and validation of cell-surface protein mesothelin (MSLN) as a novel therapeutic target in AML: Results from the COG/NCI target AML initiative. Blood 2016, 128, 2873. [Google Scholar] [CrossRef]
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab ravtansine: A novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol. Cancer Ther. 2014, 13, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Quanz, M.; Hagemann, U.B.; Zitzmann-Kolbe, S.; Stelte-Ludwig, B.; Golfier, S.; Elbi, C.; Mumberg, D.; Ziegelbauer, K.; Schatz, C.A. Anetumab ravtansine inhibits tumor growth and shows additive effect in combination with targeted agents and chemotherapy in mesothelin-expressing human ovarian cancer models. Oncotarget 2018, 9, 34103–34121. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, D.; Onda, M.; Voigt, A.; Dawczynski, K.; Wittig, S.; Hassan, R.; Gruhn, B.; Pastan, I. Mesothelin, a possible target for immunotherapy, is expressed in primary aml cells. Eur. J. Haematol. 2007, 79, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kaeding, A.; Tarlock, K.; Barwe, S.; Gopalakrisnapillai, A.; Alonzo, T.A.; Gerbing, R.B.; Loken, M.R.; Ries, R.E.; Pardo, L.; Chou, C. Mesothelin is a novel disease marker and potential therapeutic target in pediatric acute myeloid leukemia. Blood 2017, 130, 2461. [Google Scholar]
- Kaeding, A.; Tarlock, K.; Kolb, E.A.; Meshinchi, S. Immunotherapeutic targeting of mesothelin in acute myeloid leukemia in vitro with anetumab ravtansine and a novel antibody-drug conjugate. Blood 2018, 132, 1448. [Google Scholar] [CrossRef]
- Gopalakrishnapillai, A.; Kaeding, A.; Schatz, C.; Sommer, A.; Meshinchi, S.; Kolb, E.A.; Barwe, S. In vivo evaluation of mesothelin as a therapeutic target in pediatric acute myeloid leukemia. Blood 2019, 134, 1370. [Google Scholar] [CrossRef]
- Kovtun, Y.; Jones, G.E.; Adams, S.; Harvey, L.; Audette, C.A.; Wilhelm, A.; Bai, C.; Rui, L.; Laleau, R.; Liu, F.; et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. 2018, 2, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Hogge, D.E.; Rizzieri, D.A.; Cirrito, T.P.; Kornblau, S.M.; Borthakur, G.; Bivins, C.; Garcia-Manero, G.; Kadia, T.M.; Ravandi, F. SL-401, a targeted therapy directed to the interleukin-3 receptor present on leukemia blasts and cancer stem cells, is active as a single agent in patients with advanced AML. Blood 2012, 21, 3625. [Google Scholar] [CrossRef]
- Jen, E.Y.; Gao, X.; Li, L.; Zhuang, L.; Simpson, N.E.; Aryal, B.; Wang, R.; Przepiorka, D.; Shen, Y.L.; Leong, R.; et al. FDA approval summary: Tagraxofusp-erzs for treatment of blastic plasmacytoid dendritic cell neoplasm. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Pemmaraju, N.; Sweet, K.L.; Lane, A.A.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Aung, P. Results of pivotal phase 2 trial of SL-401 in patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood 2018, 132, 765. [Google Scholar] [CrossRef]
- Daver, N.G.; Erba, H.P.; Papadantonakis, N.; DeAngelo, D.J.; Wang, E.S.; Konopleva, M.Y.; Sloss, C.M.; Culm-Merdek, K.; Zweidler-McKay, P.A.; Kantarjian, H.M. A phase I, first-in-human study evaluating the safety and preliminary antileukemia activity of IMGN632, a novel CD123-targeting antibody-drug conjugate, in patients with relapsed/refractory acute myeloid leukemia and other CD123-positive hematologic malignancies. Blood 2018, 132, 27. [Google Scholar]
- Daver, N.G.; Montesinos, P.; DeAngelo, D.J.; Wang, E.S.; Papadantonakis, N.; Deconinck, E.; Erba, H.P.; Pemmaraju, N.; Lane, A.A.; Rizzieri, D.A. Clinical Profile of IMGN632, a Novel CD123-Targeting Antibody-Drug Conjugate (ADC), in Patients with Relapsed/Refractory (r/r) Acute Myeloid Leukemia (AML) or Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN); American Society of Hematology: Washington, DC, USA, 2019. [Google Scholar]
- Daver, N.G.; Erba, H.P.; Papadantonakis, N.; DeAngelo, D.J.; Wang, E.S.; Konopleva, M.Y.; Sloss, C.M.; Wang, J.; Malcolm, K.E.; Zweidler-McKay, P.A. A phase 1b/2 study of the CD123-targeting antibody-drug conjugate imgn632 as monotherapy or in combination with venetoclax and/or azacitidine for patients with CD123-positive acute myeloid leukemia. Blood 2018, 134, 2601. [Google Scholar] [CrossRef]
- Al-Hussaini, M.; DiPersio, J.F. Small molecule inhibitors in acute myeloid leukemia: From the bench to the clinic. Expert Rev. Hematol. 2014, 7, 439–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashidi, A.; DiPersio, J.F. Targeting the leukemia-stroma interaction in acute myeloid leukemia: Rationale and latest evidence. Ther. Adv. Hematol. 2016, 7, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Barbier, V.; Pattabiraman, D.R.; Gonda, T.J.; Magnani, J.L.; Levesque, J.-P. Vascular niche e-selectin protects acute myeloid leukaemia stem cells from chemotherapy. Blood 2014, 124, 620. [Google Scholar] [CrossRef]
- Chien, S.; Haq, S.U.; Pawlus, M.; Moon, R.T.; Estey, E.H.; Appelbaum, F.R.; Othus, M.; Magnani, J.L. Adhesion of acute myeloid leukemia blasts to e-selectin in the vascular niche enhances their survival by mechanisms such as wnt activation. Blood 2013, 122, 61. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P.; O’Dwyer, M.E.; Fogler, W.E.; Magnani, J.L.; Chen, M.M.J.B. High e-selectin ligand expression contributes to chemotherapy-resistance in poor risk relapsed and refractory (r/r) acute myeloid leukemia (AML) patients and can be overcome with the addition of uproleselan. Blood 2019, 134, 2690. [Google Scholar] [CrossRef]
- Chien, S.; Zhao, X.; Brown, M.; Saxena, A.; Patton, J.T.; Magnani, J.L.; Becker, P.S. A novel small molecule e-selectin inhibitor GMI-1271 blocks adhesion of AML blasts to e-selectin and mobilizes cells in nodscid IL2RGC−/− mice engrafted with human AML. Blood 2012, 120, 4092. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P.; O’Dwyer, M.E.; Fogler, W.E.; Wolfgang, C.D.; Magnani, J.L. Uproleselan (GMI-1271), an e-selectin antagonist, improves the efficacy and safety of chemotherapy in relapsed/refractory (r/r) and newly diagnosed older patients with acute myeloid leukemia: Final, correlative, and subgroup analyses. Blood 2018, 132, 331. [Google Scholar] [CrossRef]
- Leonti, A.R.; Pardo, L.; Alonzo, T.A.; Gerbing, R.B.; Eidenschink Brodersen, L.; Ries, R.E.; Smith, J.L.; Le, Q.; Aplenc, R.; Kolb, E.A. Transcriptome profiling of glycosylation genes defines correlation with e-selectin ligand expression and clinical outcome in AML. Blood 2019, 134, 3772. [Google Scholar] [CrossRef]
- Winters, A.C.; Bernt, K.M. MLL-rearranged leukemias-an update on science and clinical approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Burmeister, T.; Groger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- De Rooij, J.D.; Masetti, R.; Van Den Heuvel-Eibrink, M.M.; Cayuela, J.-M.; Trka, J.; Reinhardt, D.; Rasche, M.; Sonneveld, E.; Alonzo, T.A.; Fornerod, M.J.B. Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: A retrospective intergroup study. Blood 2016, 127, 3424–3430. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. Mll-rearranged leukemia is dependent on aberrant H3k79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Eschle, B.K.; Witkin, M.; Gadrey, J.Y.; Uckelmann, H.J.; Kitajima, S.; McGeehan, G.M.; Armstrong, S.A. Vtp50469 is a novel, orally available MENIN-MLL1 inhibitor effective against MLL-rearranged and NPM1-mutant leukemia. Cancer Res. 2018, 78, 4958. [Google Scholar]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The dot1l inhibitor pinometostat reduces H3k79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.; Wetmore, C.; O’Brien, M.M.; Silverman, L.B.; Brown, P.; Cooper, T.M.; Thomson, B.; Blakemore, S.J.; Daigle, S.; Suttle, B. Final report of phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in children with relapsed or refractory MLL-R acute leukemia. Blood 2016, 128, 2780. [Google Scholar] [CrossRef]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the MENIN-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Cierpicki, T.; Grembecka, J. Challenges and opportunities in targeting the MENIN-MLL interaction. Future Med. Chem. 2014, 6, 447–462. [Google Scholar] [CrossRef] [Green Version]
- Richard, B.L.; Kathryn, E.; Tara, P.; Stephen, W.E.; Yuelong, G.; David, A.C.; Gerard, M.M.; Beverly, A.T.; Malcolm, A.S. Pediatric preclinical testing consortium evaluation of the menin inhibitor, VTP-50469, against xenograft models of MLL-rearranged infant acute lymphoblastic leukemia. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Burrows, F.; Wu, T.; Kessler, L.; Li, S.; Zhang, J.; Zarrinkar, P.; Li, L.; Cierpicki, T.; Grembecka, J.; Ren, P. Abstract lb-a27: A novel small molecule MENIN-MLL inhibitor for potential treatment of MLL-rearranged leukemias and NPM1/DNMT3A-mutant AML. Available online: https://mct.aacrjournals.org/content/17/1_Supplement/LB-A27 (accessed on 1 February 2020).
- Dafflon, C.; Craig, V.J.; Mereau, H.; Grasel, J.; Schacher Engstler, B.; Hoffman, G.; Nigsch, F.; Gaulis, S.; Barys, L.; Ito, M.; et al. Complementary activities of DOT1L and MENIN inhibitors in MLL-rearranged leukemia. Leukemia 2017, 31, 1269–1277. [Google Scholar] [CrossRef]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the RAS/RAF/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef] [Green Version]
- Knight, T.; Irving, J.A. RAS/Raf/MEK/ERK pathway activation in childhood acute lymphoblastic leukemia and its therapeutic targeting. Front. Oncol. 2014, 4, 160. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.S.; Shapiro, P.S.; Ahn, N.G. Signal transduction through MAP kinase cascades. Adv. Cancer Res. 1998, 74, 49–139. [Google Scholar] [PubMed]
- Towatari, M.; Iida, H.; Tanimoto, M.; Iwata, H.; Hamaguchi, M.; Saito, H. Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia 1997, 11, 479–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goemans, B.F.; Zwaan, C.M.; Miller, M.; Zimmermann, M.; Harlow, A.; Meshinchi, S.; Loonen, A.H.; Hahlen, K.; Reinhardt, D.; Creutzig, U.; et al. Mutations in kit and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 2005, 19, 1536–1542. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Smalley, K.S.; Sosman, J.A. Molecular pathways: Targeting NRAS in melanoma and acute myelogenous leukemia. Clin. Cancer Res. 2014, 20, 4186–4192. [Google Scholar] [CrossRef] [Green Version]
- Burgess, M.R.; Hwang, E.; Firestone, A.J.; Huang, T.; Xu, J.; Zuber, J.; Bohin, N.; Wen, T.; Kogan, S.C.; Haigis, K.M.; et al. Preclinical efficacy of MEK inhibition in NRAS-mutant AML. Blood 2014, 124, 3947–3955. [Google Scholar] [CrossRef]
- Kerstjens, M.; Pinhancos, S.S.; Castro, P.G.; Schneider, P.; Wander, P.; Pieters, R.; Stam, R.W. Trametinib inhibits RAS-mutant MLL-rearranged acute lymphoblastic leukemia at specific niche sites and reduces erk phosphorylation in vivo. Haematologica 2018, 103, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Tasian, S.K.; Sulis, M.L.; Cooper, T.M.; Pauly, M.; Maloney, K.W.; Burke, M.J.; Brown, P.; Gossai, N.; Cole, P. Matched targeted therapy for pediatric patients with relapsed, refractory or high-risk leukemias: A report from the leap consortium. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Borthakur, G.; Popplewell, L.; Boyiadzis, M.; Foran, J.; Platzbecker, U.; Vey, N.; Walter, R.B.; Olin, R.; Raza, A.; Giagounidis, A.; et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in ras-mutant relapsed or refractory myeloid malignancies. Cancer 2016, 122, 1871–1879. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, K.; Li, S.; Adams, P.D.; Deshpande, A.J. The role of tp53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosomes Cancer 2019, 58, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Faderl, S.; Kantarjian, H.M.; Estey, E.; Manshouri, T.; Chan, C.Y.; Rahman Elsaied, A.; Kornblau, S.M.; Cortes, J.; Thomas, D.A.; Pierce, S.; et al. The prognostic significance of p16(INK4A)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 2000, 89, 1976–1982. [Google Scholar] [CrossRef]
- Iwakuma, T.; Lozano, G. MDM2, an introduction. Mol. Cancer Res. 2003, 1, 993–1000. [Google Scholar] [PubMed]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.F.; Morano, W.F.; Lee, J.; Gleeson, E.; Babcock, B.D.; Michl, J.; Sarafraz-Yazdi, E.; Pincus, M.R.; Bowne, W.B. Emerging role of MDM2 as target for anti-cancer therapy: A review. Ann. Clin. Lab. Sci. 2016, 46, 627–634. [Google Scholar] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Shikami, M.; Cabreira-Hansen, M.; McQueen, T.; Ruvolo, V.; Tsao, T.; Zeng, Z.; Vassilev, L.T.; et al. MDM2 antagonists induce p53-dependent apoptosis in aml: Implications for leukemia therapy. Blood 2005, 106, 3150–3159. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.; Halilovic, E.; Cooke, V.G.; Nonami, A.; Ren, T.; Sanda, T.; Simkin, I.; Yuan, J.; Antonakos, B.; Barys, L.; et al. Inhibition of wild-type p53-expressing aml by the novel small molecule HDM2 inhibitor CGM097. Mol. Cancer Ther. 2015, 14, 2249–2259. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, G.; Pappayannidis, C.; Yee, K.; Vey, N.; Drummond, M.; Kelly, K.; Dickinson, M.; Lee, J.; Seiter, K.; Yoon, S.J.H. Phase 1b results of idasanutlin+ cytarabine (ARA-C) in acute myeloid leukemia (AML) patients (Pts). Available online: https://library.ehaweb.org/eha/2016/21st/135260/cristina.pappayannidis.phase.1b.results.of.idasanutlin.2B.cytarabine.28ara-c29.in.html (accessed on 1 February 2020).
- Dangl, M.; Chien, Y.; Lehmann, C.; Friess, T. Synergistic anticancer activity of clinical stage, non-genotoxic apoptosis inducing agents Rg7388 (MDM2 antagonist) and ABT-199 (GDC-0199, BCL2 inhibitor) in p53 wild-type aml tumor models. Cancer Res. 2014, 74, 5505. [Google Scholar]
- Daver, N.G.; Pollyea, D.A.; Garcia, J.S.; Jonas, B.A.; Yee, K.W.; Fenaux, P.; Assouline, S.; Vey, N.; Olin, R.; Roboz, G.J. Safety, efficacy, pharmacokinetic (pk) and biomarker analyses of BCL2 inhibitor venetoclax (Ven) plus MDM2 inhibitor Idasanutlin (IDASA) in patients (PTS) with relapsed or refractory (r/r) AML: A phase Ib, non-randomized, open-label study. Blood 2018. [Google Scholar] [CrossRef]
- Khurana, A.; Shafer, D.A. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: Perspectives on the therapeutic potential of idasanutlin (RG7388). Onco. Targets Ther. 2019, 12, 2903–2910. [Google Scholar] [CrossRef] [Green Version]
- Carol, H.; Reynolds, C.P.; Kang, M.H.; Keir, S.T.; Maris, J.M.; Gorlick, R.; Kolb, E.A.; Billups, C.A.; Geier, B.; Kurmasheva, R.T.; et al. Initial testing of the MDM2 inhibitor RG7112 by the pediatric preclinical testing program. Pediatr. Cancer 2013, 60, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. Tp53 variations in human cancers: New lessons from the IARC TP53 database and genomics data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Ivey, A.; Huntly, B.J. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 2016, 127, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. Tp53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.H.; Bally, C.; et al. Synergistic effects of PRIMA-1MET (APR-246) and azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2019. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front. Oncol. 2016, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.F.; McGraw, K. Phase 2 Results of APR-246 and Azacitidine (Aza) in Patients with Tp53 Mutant Myelodysplastic Syndromes (Mds) and Oligoblastic Acute Myeloid Leukemia (Aml); American Society of Hematology: Washington, DC, USA, 2019. [Google Scholar]
- Ravandi, F.; Cortes, J.E.; Jones, D.; Faderl, S.; Garcia-Manero, G.; Konopleva, M.Y.; O’Brien, S.; Estrov, Z.; Borthakur, G.; Thomas, D.J.J.o.c.o. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 1856. [Google Scholar] [CrossRef]
- Rollig, C.; Serve, H.; Huttmann, A.; Noppeney, R.; Muller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Brown, P.A.; Gerbing, R.B.; Fox, E.; Choi, J.K.; Fisher, B.T.; Hirsch, B.A.; Kahwash, S.; Levine, J.E. Sorafenib in Combination with Standard Chemotherapy for Children with High Allelic Ratio Flt3/Itd+ Aml Improves Event-Free Survival and Reduces Relapse Risk: A Report From the Children’s Oncology Group Protocol Aaml1031; American Society of Hematology: Washington, DC, USA, 2019. [Google Scholar]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Söderhäll, S.; Brethon, B.; Luciani, M.; Rizzari, C.; Stam, R.W.; Besse, E.; Dutreix, C.; Fagioli, F.; Ho, P.A.J.B.j.o.h. A phase 1/2, open-label, dose-escalation study of midostaurin in children with relapsed or refractory acute leukaemia. Br. J. Haematol. 2019, 185, 623. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Cassar, J.; Eckroth, E.; Malvar, J.; Sposto, R.; Gaynon, P.; Chang, B.H.; Gore, L.; August, K.; Pollard, J.A.; et al. A phase I study of quizartinib combined with chemotherapy in relapsed childhood leukemia: A therapeutic advances in childhood leukemia & lymphoma (TACL) study. Clin. Cancer Res. 2016, 22, 4014–4022. [Google Scholar] [PubMed] [Green Version]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
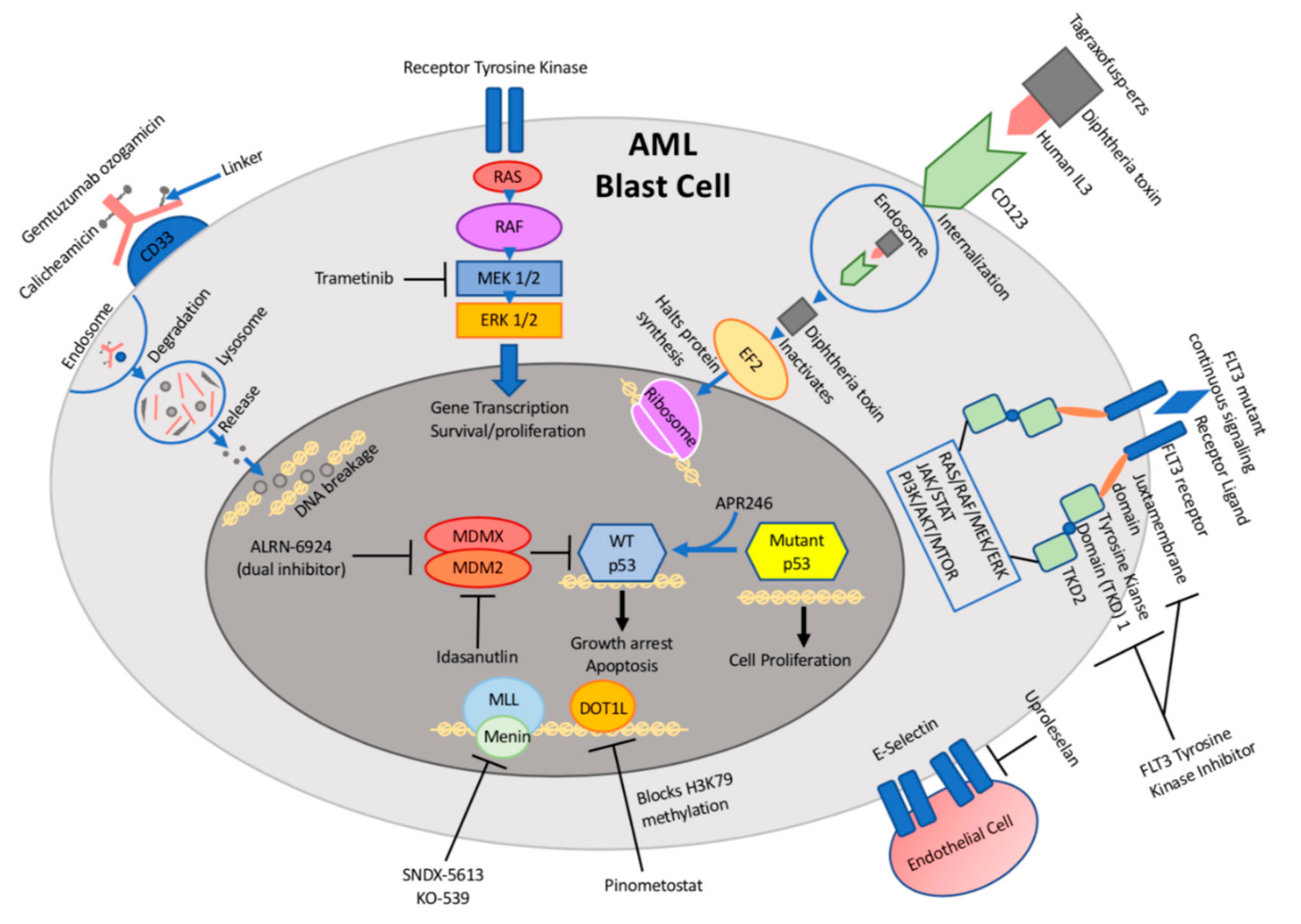
Figure 1.
Schematic illustration of targetable pathways and drug mechanisms in pediatric acute myeloid leukemia (AML).
Figure 1.
Schematic illustration of targetable pathways and drug mechanisms in pediatric acute myeloid leukemia (AML).


{kind=link}
Table 1.
Selected novel AML therapies currently in clinical trials in children and adults.
| Therapy | Type | Target | Clinical Trial | Patient Population |
|---|---|---|---|---|
| Gemtuzumab ozogamicin + chemotherapy | Antibody drug conjugate | CD33 | Phase III COG AAML1831 GO included in standard of care treatment backbone—in development | Newly diagnosed AML in children |
| Anetumab ravtensine | Antibody drug conjugate | Mesothelin | Phase I (COG AAML2011)—in development | ≥2nd relapse AML in children with mesothelin+ AML |
| IMGN632 | Antibody drug conjugate | CD123 | Phase I (NCT03386513) Phase I/II—in development | R/R AML Adults with CD123+ AML and other hematologic malignancies R/R AML in children |
| Uproleselan (GMI-1271) | Small molecule inhibitor | E-selectin | Randomized phase III (NCT03616470) | R/R AML in adults |
| Pinometostat + Azacitidine | Small molecular inhibitor | DOT1L | Phase I/II (NCT03701295) | Newly diagnosed or R/R AML with KMT2A rearrangement in adults |
| SNDX-5613 (VTP-50469) | Small molecule inhibitor | KMT2A rearrangement or NPM1 mutation | Phase I/II (NCT04065399) | Phase I: R/R acute leukemia Phase II in adults:
|
| KO = 539 | Small Molecule Inhibitor | KMT2A rearrangement | Phase I/II (NCT04067336) | R/R AML in adults |
| Trametinib | Small molecule inhibitor | RAS-pathway mutations | COG Phase II ADVL1521 (NCT03190915) | R/R juvenile myelomonocytic leukemia (JMML) in children |
| Idasanutlin+Cytarabine | Small molecule inhibitor | MDM2 antagonist | Randomized Phase III (NCT02545283) | R/R AML with WT and mutated TP53 in adults |
| ALRN-6924 (dual MDM2/MDMX inhibitor) | Small molecule inhibitor | MDM2 antagonist | Phase I (NCT03654716) | R/R AML, ALL, MPAL, or other undifferentiated acute leukemia (Cohort C) with WT TP53 in children |
| APR-246 | Small molecule inhibitor | TP53 | Randomized Phase III (NCT03745716) | TP53-mutated MDS in adults |
| Sorafenib + Palbociclib | Tyrosine kinase inhibitor | FLT3 | Phase I (NCT03132454) | R/R AML and ALL in adolescents and adults |
| Midostaurin + chemotherapy | Tyrosine kinase inhibitor | FLT3 | Phase II (NCT03591510) | Newly diangosed FLT3-mutated AML in children |
| Quizartinib + chemotherapy | Tyrosine kinase inhibitor | FLT3 | Phase I/II (NCT03793478) | R/R FLT3-mutated AML in children |
| Gilteritinib + chemotherapy | Tyrosine kinase inhibitor | FLT3 | Phase III (COG AAML1831)—in development | Newly diagnosed FLT3-mutated AML in children |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, J.; Glasser, C.L. New and Emerging Targeted Therapies for Pediatric Acute Myeloid Leukemia (AML). Children 2020, 7, 12. https://0-doi-org.brum.beds.ac.uk/10.3390/children7020012
AMA Style
Chen J, Glasser CL. New and Emerging Targeted Therapies for Pediatric Acute Myeloid Leukemia (AML). Children. 2020; 7(2):12. https://0-doi-org.brum.beds.ac.uk/10.3390/children7020012
Chicago/Turabian StyleChen, Jing, and Chana L. Glasser. 2020. "New and Emerging Targeted Therapies for Pediatric Acute Myeloid Leukemia (AML)" Children 7, no. 2: 12. https://0-doi-org.brum.beds.ac.uk/10.3390/children7020012
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.