
Mechanisms and Insights for the Development of Heart Failure Associated with Cancer Therapy

 , and
, and
Abstract
:1. Epidemiology of Cancer Therapy-Related Cardiotoxicity
2. Pathophysiology of Cardiotoxic Cancer Therapies
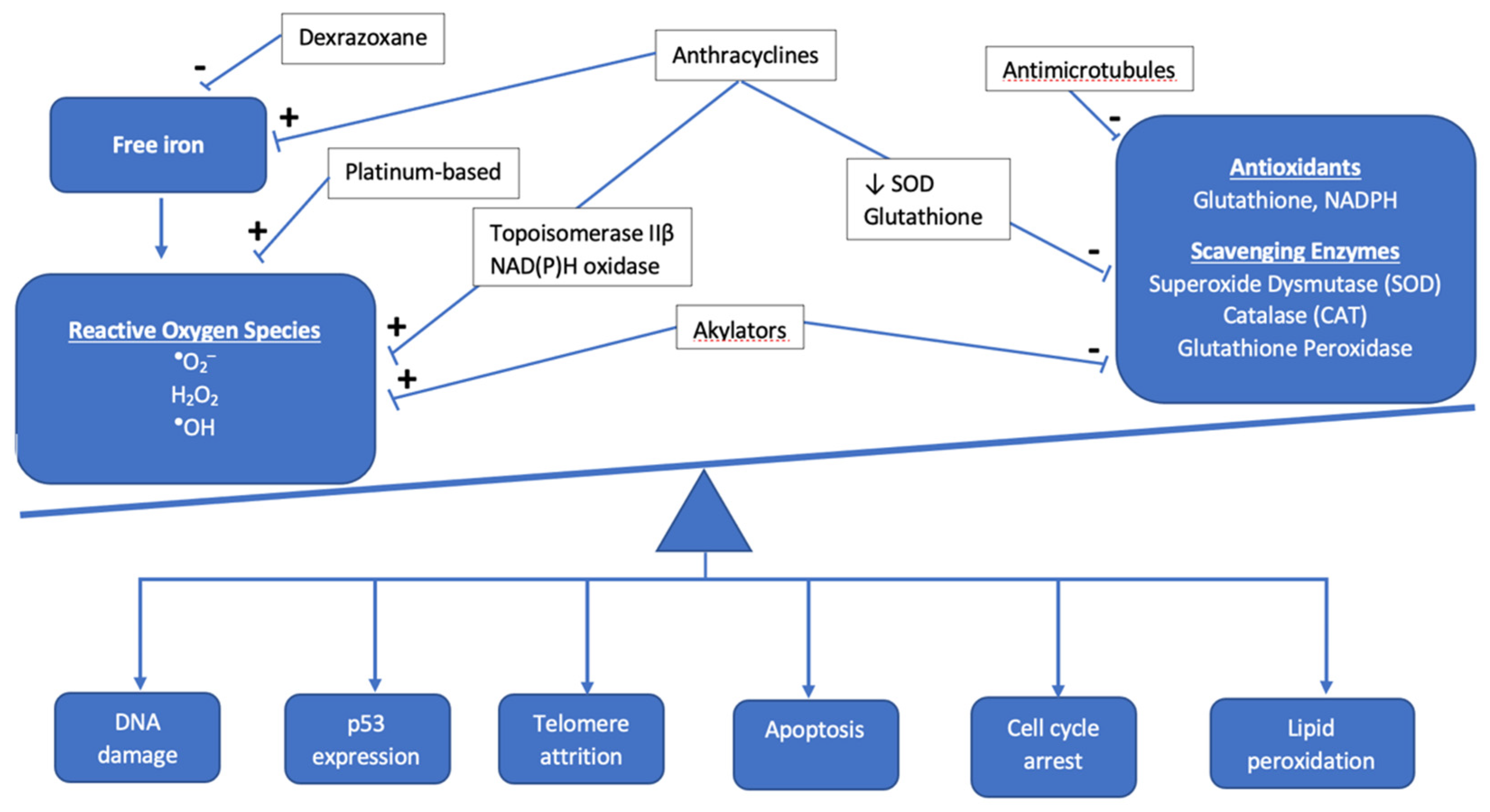
2.1. Anthracyclines
2.2. Non-Anthracycline Agents
2.3. Radiation-Induced Cardiotoxicity
2.3.1. Mechanisms for Toxicity
2.3.2. Dose-Toxicity Relationship
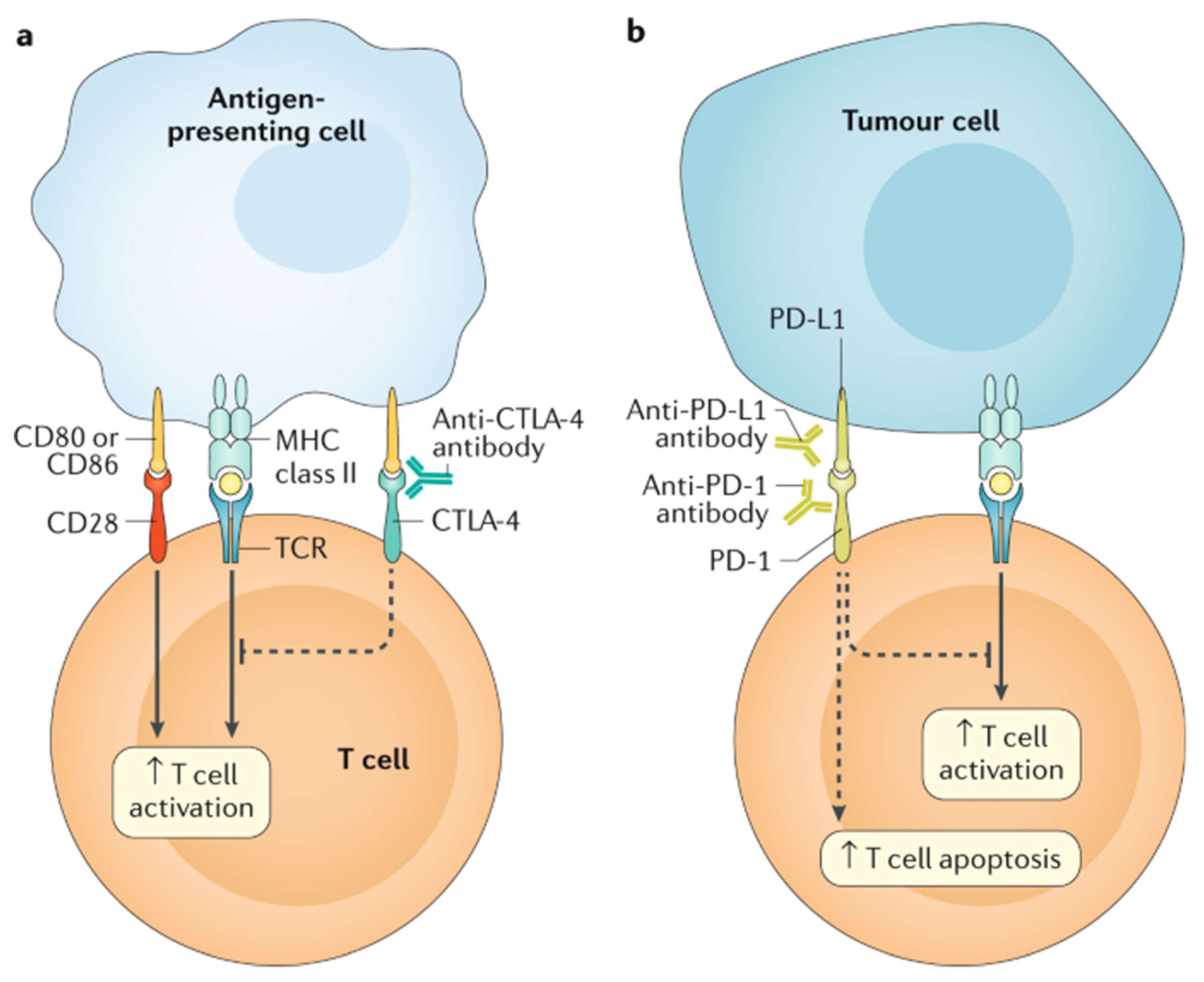
2.4. Targeted Cancer Therapies
2.5. Cellular Therapy and Hematopoietic Stem Cell Transplantation
3. Prevention
3.1. Drug Delivery
3.2. Dexrazoxane
3.3. Evolution in RT Treatment Planning to Reduce Cardiac Dose
4. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lipshultz, S.E.; Adams, M.J.; Colan, S.D.; Constine, L.S.; Herman, E.H.; Hsu, D.T.; Hudson, M.M.; Kremer, L.C.; Landy, D.C.; Miller, T.L.; et al. Long-term cardiovascular toxicity in children, adolescents, and young adults who receive cancer therapy: Pathophysiology, course, monitoring, management, prevention, and research directions: A scientific statement from the American Heart Association. Circulation 2013, 128, 1927–1995. [Google Scholar] [CrossRef] [Green Version]
- Getz, K.D.; Sung, L.; Ky, B.; Gerbing, R.B.; Leger, K.J.; Leahy, A.B.; Sack, L.; Woods, W.G.; Alonzo, T.; Gamis, A.; et al. Occurrence of Treatment-Related Cardiotoxicity and Its Impact on Outcomes Among Children Treated in the AAML0531 Clinical Trial: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 12–21. [Google Scholar] [CrossRef]
- Franzon, J.; Berry, N.M.; Ullah, S.; Versace, V.L.; McCarthy, A.L.; Atherton, J.; Roder, D.; Koczwara, B.; Coghlan, D.; Clark, R.A. Heart failure following blood cancer therapy in pediatric and adult populations. Asia Pac. J. Clin. Oncol. 2018, 14, 224–230. [Google Scholar] [CrossRef]
- Feijen, E.; Font-Gonzalez, A.; Van der Pal, H.J.H.; Kok, W.E.M.; Geskus, R.B.; Ronckers, C.M.; Bresters, D.; van Dalen, E.C.; van Dulmen-den Broeder, E.; van den Berg, M.H.; et al. Risk and Temporal Changes of Heart Failure Among 5-Year Childhood Cancer Survivors: A DCOG-LATER Study. J. Am. Heart Assoc. 2019, 8, e009122. [Google Scholar] [CrossRef] [Green Version]
- Mulrooney, D.A.; Yeazel, M.W.; Kawashima, T.; Mertens, A.C.; Mitby, P.; Stovall, M.; Donaldson, S.S.; Green, D.M.; Sklar, C.A.; Robison, L.L.; et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: Retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ 2009, 339, b4606. [Google Scholar] [CrossRef] [Green Version]
- Wolf, C.M.; Reiner, B.; Kuhn, A.; Hager, A.; Muller, J.; Meierhofer, C.; Oberhoffer, R.; Ewert, P.; Schmid, I.; Weil, J. Subclinical Cardiac Dysfunction in Childhood Cancer Survivors on 10-Years Follow-Up Correlates With Cumulative Anthracycline Dose and Is Best Detected by Cardiopulmonary Exercise Testing, Circulating Serum Biomarker, Speckle Tracking Echocardiography, and Tissue Doppler Imaging. Front. Pediatr. 2020, 8, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, E.J.; Leger, K.J.; Bhatt, N.S.; Mulrooney, D.A.; Ross, C.J.; Aggarwal, S.; Bansal, N.; Ehrhardt, M.J.; Armenian, S.H.; Scott, J.M.; et al. Paediatric cardio-oncology: Epidemiology, screening, prevention, and treatment. Cardiovasc. Res. 2019, 115, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Burstein, D.S.; Maude, S.; Grupp, S.; Griffis, H.; Rossano, J.; Lin, K. Cardiac Profile of Chimeric Antigen Receptor T Cell Therapy in Children: A Single-Institution Experience. Biol. Blood Marrow Transplant. 2018, 24, 1590–1595. [Google Scholar] [CrossRef] [Green Version]
- Shalabi, H.; Sachdev, V.; Kulshreshtha, A.; Cohen, J.W.; Yates, B.; Rosing, D.R.; Sidenko, S.; Delbrook, C.; Mackall, C.; Wiley, B.; et al. Impact of cytokine release syndrome on cardiac function following CD19 CAR-T cell therapy in children and young adults with hematological malignancies. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef] [Green Version]
- Deidda, M.; Madonna, R.; Mango, R.; Pagliaro, P.; Bassareo, P.P.; Cugusi, L.; Romano, S.; Penco, M.; Romeo, F.; Mercuro, G. Novel insights in pathophysiology of antiblastic drugs-induced cardiotoxicity and cardioprotection. J. Cardiovasc. Med. 2016, 17 (Suppl. 1), S76–S83. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Chatelain, P.; Caspers, J.; Ruysschaert, J.M. Evidence of a complex between adriamycin derivatives and cardiolipin: Possible role in cardiotoxicity. Biochem. Pharmacol. 1980, 29, 3003–3010. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Qiao, X.; van der Zanden, S.Y.; Wander, D.P.A.; Borràs, D.M.; Song, J.Y.; Li, X.; van Duikeren, S.; van Gils, N.; Rutten, A.; van Herwaarden, T.; et al. Uncoupling DNA damage from chromatin damage to detoxify doxorubicin. Proc. Natl. Acad. Sci. USA 2020, 117, 15182–15192. [Google Scholar] [CrossRef]
- Abramson, J.J.; Buck, E.; Salama, G.; Casida, J.E.; Pessah, I.N. Mechanism of anthraquinone-induced calcium release from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1988, 263, 18750–18758. [Google Scholar] [CrossRef]
- Sag, C.M.; Köhler, A.C.; Anderson, M.E.; Backs, J.; Maier, L.S. CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 51, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keltai, K.; Cervenak, L.; Makó, V.; Doleschall, Z.; Zsáry, A.; Karádi, I. Doxorubicin selectively suppresses mRNA expression and production of endothelin-1 in endothelial cells. Vascul. Pharmacol. 2010, 53, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Magdy, T.; Burmeister, B.T.; Burridge, P.W. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol. Ther. 2016, 168, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Aminkeng, F.; Bhavsar, A.P.; Visscher, H.; Rassekh, S.R.; Li, Y.; Lee, J.W.; Brunham, L.R.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; et al. A coding variant in RARG confers susceptibility to anthracyclineinduced cardiotoxicity in childhood cancer. Nat. Genet. 2015, 47, 1079–1084. [Google Scholar] [CrossRef]
- Wojnowski, L.; Kulle, B.; Schirmer, M.; Schluter, G.; Schmidt, A.; Rosenberger, A.; Vonhof, S.; Bickeboller, H.; Toliat, M.R.; Suk, E.K.; et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation 2005, 112, 3754–3762. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Rasi, S.; Franceschetti, S.; Capello, D.; Castelli, A.; De Paoli, L.; Ramponi, A.; Chiappella, A.; Pogliani, E.M.; Vitolo, U.; et al. Analysis of the host pharmacogenetic background for prediction of outcome and toxicity in diffuse large B-cell lymphoma treated with R-CHOP21. Leukemia 2009, 23, 1118–1126. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascales, A.; Pastor-Quirante, F.; Sanchez-Vega, B.; Luengo-Gil, G.; Corral, J.; Ortuno-Pacheco, G.; Vicente, V.; de la Pena, F.A. Association of anthracycline-related cardiac histological lesions with NADPH oxidase functional polymorphisms. Oncologist 2013, 18, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pavia, P.; Kim, Y.; Restrepo-Cordoba, M.A.; Lunde, I.G.; Wakimoto, H.; Smith, A.M.; Toepfer, C.N.; Getz, K.; Gorham, J.; Patel, P.; et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation 2019, 140, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Visscher, H.R.C.; Rassekh, S.R.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; van der Pal, H.J.; Rogers, P.C.; Rieder, M.J.; Carleton, B.C.; et al. Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatr. Blood Cancer 2013, 60, 1375–1381. [Google Scholar] [CrossRef]
- Leger, K.J.; Cushing-Haugen, K.; Hansen, J.A.; Fan, W.; Leisenring, W.M.; Martin, P.J.; Zhao, L.P.; Chow, E.J. Clinical and genetic determinants of cardiomyopathy risk among hematopoietic cell transplantation survivors. Biol. Blood Marrow Transplant. 2016, 26, 1094–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasielewski, M.; van Spaendonck-Zwarts, K.Y.; Westerink, N.D.; Jongbloed, J.D.; Postma, A.; Gietema, J.A.; van Tintelen, J.P.; van den Berg, M.P. Potential genetic predisposition for anthracycline-associated cardiomyopathy in families with dilated cardiomyopathy. Open Heart 2014, 1, e000116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.G.; Sharma, A.; Holmstrom, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhu, Y.; Dong, S.; Zhang, A.; Lu, Y.; Li, Y.; Lv, S.; Zhang, J. Role of oxidative stress in cardiotoxicity of antineoplastic drugs. Life Sci. 2019, 232, 116526. [Google Scholar] [CrossRef] [PubMed]
- Rtibi, K.; Grami, D.; Selmi, S.; Amri, M.; Sebai, H.; Marzouki, L. Vinblastine, an anticancer drug, causes constipation and oxidative stress as well as others disruptions in intestinal tract in rat. Toxicol. Rep. 2017, 4, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, C.; Deidda, M.; Piras, A.; Cadeddu, C.; Demurtas, L.; Puzzoni, M.; Piscopo, G.; Scartozzi, M.; Mercuro, G. Pathophysiology of cardiotoxicity induced by nonanthracycline chemotherapy. J. Cardiovasc. Med. 2016, 17 (Suppl. 1), e12–e18. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Cadeddu, C.; Di Lisi, D.; Femminò, S.; Madonna, R.; Mele, D.; Monte, I.; Novo, G.; Penna, C.; Pepe, A.; et al. From Molecular Mechanisms to Clinical Management of Antineoplastic Drug-Induced Cardiovascular Toxicity: A Translational Overview. Antioxid Redox Signal. 2019, 30, 2110–2153. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.; Porto, S.; Zappavigna, S.; Addeo, E.; Marra, M.; Miraglia, N.; Sannolo, N.; Vanacore, D.; Stiuso, P.; Caraglia, M. A mechanistic study on the cardiotoxicity of 5-fluorouracil in vitro and clinical and occupational perspectives. Toxicol. Lett. 2014, 227, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Koutroumpakis, E.; Palaskas, N.L.; Lin, S.H.; Abe, J.I.; Liao, Z.; Banchs, J.; Deswal, A.; Yusuf, S.W. Modern Radiotherapy and Risk of Cardiotoxicity. Chemotherapy 2020, 65, 65–76. [Google Scholar] [CrossRef]
- Nielsen, K.M.; Offersen, B.V.; Nielsen, H.M.; Vaage-Nilsen, M.; Yusuf, S.W. Short and long term radiation induced cardiovascular disease in patients with cancer. Clin. Cardiol. 2017, 40, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Aleman, B.M.; van den Belt-Dusebout, A.W.; De Bruin, M.L.; van ’t Veer, M.B.; Baaijens, M.H.; de Boer, J.P.; Hart, A.A.; Klokman, W.J.; Kuenen, M.A.; Ouwens, G.M.; et al. Late cardiotoxicity after treatment for Hodgkin lymphoma. Blood 2007, 109, 1878–1886. [Google Scholar] [CrossRef] [Green Version]
- van Nimwegen, F.A.; Schaapveld, M.; Janus, C.P.; Krol, A.D.; Petersen, E.J.; Raemaekers, J.M.; Kok, W.E.; Aleman, B.M.; van Leeuwen, F.E. Cardiovascular disease after Hodgkin lymphoma treatment: 40-year disease risk. JAMA Intern. Med. 2015, 175, 1007–1017. [Google Scholar] [CrossRef]
- Mulrooney, D.A.; Hyun, G.; Ness, K.K.; Ehrhardt, M.J.; Yasui, Y.; Duprez, D.; Howell, R.M.; Leisenring, W.M.; Constine, L.S.; Tonorezos, E.; et al. Major cardiac events for adult survivors of childhood cancer diagnosed between 1970 and 1999: Report from the Childhood Cancer Survivor Study cohort. BMJ 2020, 368, l6794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutter, D.J.; Schaapveld, M.; Darby, S.C.; Hauptmann, M.; van Nimwegen, F.A.; Krol, A.D.; Janus, C.P.; van Leeuwen, F.E.; Aleman, B.M. Risk of valvular heart disease after treatment for Hodgkin lymphoma. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Hancock, S.L.; Tucker, M.A.; Hoppe, R.T. Factors affecting late mortality from heart disease after treatment of Hodgkin’s disease. JAMA 1993, 270, 1949–1955. [Google Scholar] [CrossRef]
- van Nimwegen, F.A.; Schaapveld, M.; Cutter, D.J.; Janus, C.P.; Krol, A.D.; Hauptmann, M.; Kooijman, K.; Roesink, J.; van der Maazen, R.; Darby, S.C.; et al. Radiation Dose-Response Relationship for Risk of Coronary Heart Disease in Survivors of Hodgkin Lymphoma. J. Clin. Oncol. 2016, 34, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Armenian, S.H.; Hudson, M.M.; Mulder, R.L.; Chen, M.H.; Constine, L.S.; Dwyer, M.; Nathan, P.C.; Tissing, W.J.; Shankar, S.; Sieswerda, E.; et al. Recommendations for cardiomyopathy surveillance for survivors of childhood cancer: A report from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol. 2015, 16, e123–e136. [Google Scholar] [CrossRef] [Green Version]
- van Nimwegen, F.A.; Ntentas, G.; Darby, S.C.; Schaapveld, M.; Hauptmann, M.; Lugtenburg, P.J.; Janus, C.P.M.; Daniels, L.; van Leeuwen, F.E.; Cutter, D.J.; et al. Risk of heart failure in survivors of Hodgkin lymphoma: Effects of cardiac exposure to radiation and anthracyclines. Blood 2017, 129, 2257–2265. [Google Scholar] [CrossRef]
- Martel, M.K.; Sahijdak, W.M.; Ten Haken, R.K.; Kessler, M.L.; Turrisi, A.T. Fraction size and dose parameters related to the incidence of pericardial effusions. Int. J. Radiat. Oncol. Biol. Phys. 1998, 40, 155–161. [Google Scholar] [CrossRef]
- Haddy, N.; Diallo, S.; El-Fayech, C.; Schwartz, B.; Pein, F.; Hawkins, M.; Veres, C.; Oberlin, O.; Guibout, C.; Pacquement, H.; et al. Cardiac Diseases Following Childhood Cancer Treatment: Cohort Study. Circulation 2016, 133, 31–38. [Google Scholar] [CrossRef] [Green Version]
- van der Pal, H.J.; van Dalen, E.C.; van Delden, E.; van Dijk, I.W.; Kok, W.E.; Geskus, R.B.; Sieswerda, E.; Oldenburger, F.; Koning, C.C.; van Leeuwen, F.E.; et al. High risk of symptomatic cardiac events in childhood cancer survivors. J. Clin. Oncol. 2012, 30, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganatra, S.; Parikh, R.; Neilan, T.G. Cardiotoxicity of Immune Therapy. Cardiol. Clin. 2019, 37, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Love, V.A.; Grabie, N.; Duramad, P.; Stavrakis, G.; Sharpe, A.; Lichtman, A. CTLA-4 ablation and interleukin-12 driven differentiation synergistically augment cardiac pathogenicity of cytotoxic T lymphocytes. Circ. Res. 2007, 101, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Tanaka, Y.; Nishio, R.; Mitsuiye, T.; Mizoguchi, A.; Wang, J.; Ishida, M.; Hiai, H.; Matsumori, A.; Minato, N.; et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 2003, 9, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Grabie, N.; Gotsman, I.; DaCosta, R.; Pang, H.; Stavrakis, G.; Butte, M.J.; Keir, M.E.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation 2007, 116, 2062–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor-Associated Hypertension and Vascular Disease. Hypertension 2018, 71, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Ky, B.; Vejpongsa, P.; Yeh, E.T.; Force, T.; Moslehi, J.J. Emerging paradigms in cardiomyopathies associated with cancer therapies. Circ. Res. 2013, 113, 754–764. [Google Scholar] [CrossRef]
- Moslehi, J.; Minamishima, Y.A.; Shi, J.; Neuberg, D.; Charytan, D.M.; Padera, R.F.; Signoretti, S.; Liao, R.; Kaelin, W.G., Jr. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation 2010, 122, 1004–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, M.W.; Hamo, C.E.; Cardinale, D.; Ky, B.; Nohria, A.; Baer, L.; Skopicki, H.; Lenihan, D.J.; Gheorghiade, M.; Lyon, A.R.; et al. Cancer Therapy-Related Cardiac Dysfunction and Heart Failure: Part 1: Definitions, Pathophysiology, Risk Factors, and Imaging. Circ. Heart Fail. 2016, 9, e002661. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef]
- Ebb, D.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 2545–2551. [Google Scholar] [CrossRef] [Green Version]
- Rotz, S.J.; Ryan, T.D.; Hlavaty, J.; George, S.A.; El-Bietar, J.; Dandoy, C.E. Cardiotoxicity and cardiomyopathy in children and young adult survivors of hematopoietic stem cell transplant. Pediatr. Blood Cancer 2017, 64, e26600. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Ganatra, S.; Carver, J.R.; Hayek, S.S.; Ky, B.; Leja, M.J.; Lenihan, D.J.; Lenneman, C.; Mousavi, N.; Park, J.H.; Perales, M.A.; et al. Chimeric Antigen Receptor T-Cell Therapy for Cancer and Heart: JACC Council Perspectives. J. Am. Coll. Cardiol. 2019, 74, 3153–3163. [Google Scholar] [CrossRef]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947. [Google Scholar] [CrossRef] [Green Version]
- Pathan, N.; Hemingway, C.A.; Alizadeh, A.A.; Stephens, A.C.; Boldrick, J.C.; Oragui, E.E.; McCabe, C.; Welch, S.B.; Whitney, A.; O’Gara, P.; et al. Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet 2004, 363, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Loeffen, E.A.H.; van Dalen, E.C.; Mulder, R.L.; van de Wetering, M.D.; Kremer, L.C.M.; Tissing, W.J.E. Anthracycline Cardiotoxicity Working Group. The duration of anthracycline infusion should be at least one hour in children with cancer: A clinical practice guideline. Pediatr. Blood Cancer 2018, 65. [Google Scholar] [CrossRef]
- Tardi, P.G.; Boman, N.L.; Cullis, P.R. Liposomal doxorubicin. J. Drug Target. 1996, 4, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Absalon, M.; Alonzo, T.A.; Gerbing, R.B.; Leger, K.J.; Hirsch, B.A.; Pollard, J.A.; Razzouk, B.I.; Aplenc, R.; Kolb, E.A. AAML 1421, a phase I/II study of CPX-351 followed by fludarabine, cytarabine, and G-CSF (FLAG) for children with relapsed acute myeloid leukemia (AML): A Report from the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 10003. [Google Scholar] [CrossRef]
- Children’s Oncology Group and the National Cancer Institute. A Study to Compare Standard Chemotherapy to Therapy With CPX-351 and/or Gilteritinib for Patients With Newly Diagnosed AML With or Without FLT3 Mutations, ClinicalTrials.gov Identifier: NCT04293562. Available online: https://clinicaltrials.gov/ct2/show/NCT04293562 (accessed on 21 September 2021).
- Asselin, B.L.; Devidas, M.; Chen, L.; Franco, V.I.; Pullen, J.; Borowitz, M.J.; Hutchison, R.E.; Ravindranath, Y.; Armenian, S.H.; Camitta, B.M.; et al. Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children’s Oncology Group Randomized Trial Pediatric Oncology Group 9404. J. Clin. Oncol. 2016, 34, 854–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikh, F.; Dupuis, L.L.; Alexander, S.; Gupta, A.; Mertens, L.; Nathan, P.C. Cardioprotection and Second Malignant Neoplasms Associated With Dexrazoxane in Children Receiving Anthracycline Chemotherapy: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [Green Version]
- Getz, K.D.; Sung, L.; Alonzo, T.A.; Leger, K.J.; Gerbing, R.B.; Pollard, J.A.; Cooper, T.; Kolb, E.A.; Gamis, A.S.; Ky, B.; et al. Effect of Dexrazoxane on Left Ventricular Systolic Function and Treatment Outcomes in Patients With Acute Myeloid Leukemia: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2020, 38, 2398–2406. [Google Scholar] [CrossRef]
- Reichardt, P.; Tabone, M.D.; Mora, J.; Morland, B.; Jones, R.L. Risk-benefit of dexrazoxane for preventing anthracycline-related cardiotoxicity: Re-evaluating the European labeling. Future Oncol. 2018, 14, 2663–2676. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, H.J.; Park, K.D.; Koh, K.N.; Im, H.J.; Seo, J.J.; Lee, J.W.; Chung, N.G.; Cho, B.; Kim, H.K.; et al. Risk Factor Analysis for Secondary Malignancy in Dexrazoxane-Treated Pediatric Cancer Patients. Cancer Res. Treat. 2019, 51, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Kopp, L.M.; Womer, R.B.; Schwartz, C.L.; Ebb, D.H.; Franco, V.I.; Hall, D.; Barkauskas, D.A.; Krailo, M.D.; Grier, H.E.; Meyers, P.A.; et al. Effects of dexrazoxane on doxorubicin-related cardiotoxicity and second malignant neoplasms in children with osteosarcoma: A report from the Children’s Oncology Group. Cardiooncology 2019, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Bansal, N.; Adams, M.J.; Ganatra, S.; Colan, S.D.; Aggarwal, S.; Steiner, R.; Amdani, S.; Lipshultz, E.R.; Lipshultz, S.E. Strategies to prevent anthracycline-induced cardiotoxicity in cancer survivors. Cardiooncology 2019, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Liesse, K.; Harris, J.; Chan, M.; Schmidt, M.L.; Chiu, B. Dexrazoxane Significantly Reduces Anthracycline-induced Cardiotoxicity in Pediatric Solid Tumor Patients: A Systematic Review. J. Pediatr. Hematol. Oncol. 2018, 40, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Specht, L.; Yahalom, J.; Illidge, T.; Berthelsen, A.K.; Constine, L.S.; Eich, H.T.; Girinsky, T.; Hoppe, R.T.; Mauch, P.; Mikhaeel, N.G.; et al. Modern radiation therapy for Hodgkin lymphoma: Field and dose guidelines from the international lymphoma radiation oncology group (ILROG). Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.M.; Aznar, M.C.; Berthelsen, A.K.; Loft, A.; Schut, D.A.; Maraldo, M.; Josipovic, M.; Klausen, T.L.; Andersen, F.L.; Specht, L. Prospective phase II trial of image-guided radiotherapy in Hodgkin lymphoma: Benefit of deep inspiration breath-hold. Acta Oncol. 2015, 54, 60–66. [Google Scholar] [CrossRef]
- Pinnix, C.C.; Gunther, J.R.; Fang, P.; Bankston, M.E.; Milgrom, S.A.; Boyce, D.; Lee, H.J.; Nair, R.; Steiner, R.; Strati, P.; et al. Assessment of Radiation Doses Delivered to Organs at Risk Among Patients With Early-Stage Favorable Hodgkin Lymphoma Treated With Contemporary Radiation Therapy. JAMA Netw. Open 2020, 3, e2013935. [Google Scholar] [CrossRef]
- Seeger, T.; Porteus, M.; Wu, J.C. Genome Editing in Cardiovascular Biology. Circ. Res. 2017, 120, 778–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitani, T.; Ong, S.G.; Lam, C.K.; Rhee, J.W.; Zhang, J.Z.; Oikonomopoulos, A.; Ma, N.; Tian, L.; Lee, J.; Telli, M.L.; et al. Human-Induced Pluripotent Stem Cell Model of Trastuzumab-Induced Cardiac Dysfunction in Patients With Breast Cancer. Circulation 2019, 139, 2451–2465. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Genetic Modifiers of Cardiotoxicity Risk | |
|---|---|
| Deleterious Effect | |
| Gene/SNP | Mechanism of toxicity |
| RARG (S427L variant) | Increase topoisomerase IIβ expression |
| rs1883112 SNP, p40phox subunit NAD(P)H oxidase | Interstitial fibrosis |
| UGT1A6*A | Decreased drug glucuronidation and clearance |
| MYH7 | Variants documented in familial dilated cardiomyopathy Downregulated in hiPSC-CM treated with doxorubicin |
| Titin truncating variants (TTN) | Encode A and I bands in sarcomeres; associated with depressed LV function |
| Indeterminant Effect | |
| rs4673 SNP, p22phos subunit NAD(P)H | Protective against myocardial fibrosis Acute cardiotoxicity |
| First Author | n | Treatment Period | Age at Treatment, Years | Length of Follow-Up, Years (Median) | Risk of Grade ≥ 3 Cardiotoxicity (95% CI) |
|---|---|---|---|---|---|
| Haddy [48] | 3162 | 1942–1985 | <17 | 26 | No anthracycline:
|
| van der Pal [49] | 1362 | 1966–1996 | <18 | 22 | Radiation only: HR 13.0 (2.8–61) Anthracycline & radiation: HR 49.5 (10.7–230) HR 1.8 (1.4–2.2) per 10 Gy to heart |
| Mulrooney [41] | 23,462 | 1970–1999 | <20 (median 6) | 28 | HF:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraley, C.; Milgrom, S.A.; Kondapalli, L.; Taylor, M.R.G.; Mestroni, L.; Miyamoto, S.D. Mechanisms and Insights for the Development of Heart Failure Associated with Cancer Therapy. Children 2021, 8, 829. https://0-doi-org.brum.beds.ac.uk/10.3390/children8090829
Fraley C, Milgrom SA, Kondapalli L, Taylor MRG, Mestroni L, Miyamoto SD. Mechanisms and Insights for the Development of Heart Failure Associated with Cancer Therapy. Children. 2021; 8(9):829. https://0-doi-org.brum.beds.ac.uk/10.3390/children8090829
Chicago/Turabian StyleFraley, Claire, Sarah A. Milgrom, Lavanya Kondapalli, Matthew R. G. Taylor, Luisa Mestroni, and Shelley D. Miyamoto. 2021. "Mechanisms and Insights for the Development of Heart Failure Associated with Cancer Therapy" Children 8, no. 9: 829. https://0-doi-org.brum.beds.ac.uk/10.3390/children8090829