Thermo-Responsive Hydrogels for Stimuli-Responsive Membranes

Department of Chemical Engineering, McMaster University, 1280 Main Street West, Hamilton, Ontario L8S 4L7, Canada
*
Author to whom correspondence should be addressed.
Processes 2013, 1(3), 238-262; https://0-doi-org.brum.beds.ac.uk/10.3390/pr1030238
Submission received: 26 August 2013
/
Revised: 17 September 2013
/
Accepted: 18 September 2013
/
Published: 30 September 2013
(This article belongs to the Special Issue Feature Papers)
Abstract
:Composite membranes with stimuli-responsive properties can be made by coating a thermo-responsive hydrogel onto a micro- or macroporous support. These hydrogels undergo a temperature induced volume-phase transition, which contributes towards the composite membrane’s stimuli-responsive properties. This paper reviews research done on complimentary forms of temperature responsive “thermophilic” hydrogels, those exhibiting positive volume-phase transitions in aqueous solvent. The influences of intermolecular forces on the mechanism of phase-transition are discussed along with case examples of typical thermophilic hydrogels.
1. Thermo-Responsive Hydrogels
By applying an adaptive property to a sedentary membrane, a membrane’s utility or productivity, or both, may be improved. Inspired by this concept, stimuli-responsive membranes are becoming increasingly prevalent in membrane research. For membranes employed in processes that make use of aqueous based solvents, hydrogels can be combined with a membrane substrate to create an environmentally adaptive technology. The hydrogel can be applied to the membrane substrate through simple submersion coating [1,2] or grafted through more complicated techniques, such as atom-transfer radical polymerization (ATRP) or surface initiation plasma treatment [3,4,5]. Hydrogels are water-soluble polymer gels that, instead of dissolving, entrap water in the interstitial spaces between polymer segments. Hydrophilic pendant groups like alcohols, carboxylic acids and amides are what enable the gels to be water-soluble. There are several forms of stimuli that will invoke a response from hydrogels such as temperature [6,7,8,9,10], electric fields [11,12,13], solvent composition [14,15], ionic strength [1,16,17], pH [18,19] and interaction with analytes like glucose [20,21]. Thermo-responsive hydrogels are of specific interest where chemical modification of the feed solvent is undesirable or impractical. This has implications for developments in membrane chromatography separations [10,22] and drug delivery [23].
There are a few well known models for gel structure such as those put forward by Flory for covalently bonded networks and Rees for non-covalent networks [24,25]. As we are considering hydrogels as a constituent of composite membranes, the focus of discussion in this review will predominantly be macrogels with a permanent covalently bonded network structure, avoiding specific microgel discussion and purposely excluding sol-gel systems. This will greatly affect the classification of thermo-responsive behavior observed in hydrogels. Typically sol-gel systems can be classified as having either an upper or lower critical solution temperature. On one side of this critical solution temperature, the system has two phases, as marked by a cloud point while on the other side, the system has one phase. For the permanent hydrogels that will be discussed in this review, this classification of temperature responsiveness is inaccurate as the polymer gel network is solvated but not dissolved meaning that there are always two physical phases. Instead of observing solution phase, volume-phase is a better basis of classification. Volume-phase transition is the response of a thermo-responsive hydrogel to heating and cooling. Tanaka et al. claim that volume phase transition is universal to all gels [26], however not all gels are able to have their volume affected by temperature in water. Additionally, temperature is typically an easy variable to control and predict in most practical setups, which makes these hydrogels an interesting and practical study.
Considering volume-phase transitions give us a volume-phase transition temperature (VPTT) instead of a critical solution temperature, there are still two main classifications of observed behavior, positive volume-phase transitions and negative volume-phase transitions. A positive volume-phase transition or “thermophilic” hydrogel will swell upon heating, increasing its volume. A negative volume-phase transition or “thermophobic” hydrogel will collapse upon heating, decreasing its volume. Thermophilic hydrogels typically maintain a collapsed conformation at temperatures below their VPTT while thermophobic hydrogels are able to swell at lower temperatures. The volume-phase transition may be reversible but not in all cases and hysteresis is frequently experienced [27,28,29,30,31].
Thermophilic behavior in water is uncommon for polymer gels, while the monomers of a polymer may be water soluble, the increased molecular weight from polymerization may cause the polymer not to be. Large hydrocarbon-based polymers tend to be hydrophobic, a property that favors negative volume-phase transition. Studies of both behavior types in aqueous based polymers have their roots in a similar time frame. Notable publications on thermophobic polymers were published in 1957 ((polyethylene glycol) [32] and 1968 (poly(N-isopropylacrylamide)) [33] while notable thermophilic polymers were published in 1964 (poly(N-Acryloylglycinamide)) [34]. Thermophobic hydrogels have been very popular since the early 1990’s reflected by the research and discussion concerning a single example, poly(N-isopropylacrylamide) (PNIPAm). Between 1984 and 1991, yearly research citations of PNIPAm quadrupled, as has been determined by other workers [11,22]. What made these hydrogels so popular was the novel concept that the thermophobic behavior of gels in aqueous solution is contrary to the majority of polymers in organic solution. Both single chain and crosslinked networks of PNIPAm exhibit negative volume-phase transition between 30 °C and 35 °C [33,35]. PNIPAm hydrogels and others that similarly exhibit thermophobic behavior are primarily enabled by water-polymer interactions, while polymers with thermophilic behavior are driven by both water-polymer and polymer-polymer interactions [36]. The heavy interest thermophobic hydrogels have garnered has made it difficult to find any information on thermophilic hydrogels, leading many researchers and reviewers to either quickly dismiss the positive volume-phase transition aspect of temperature responsive gels or ignore it altogether [37,38,39]. This review is a highlight of thermophilic hydrogels, the compliment thermo-responsive material to very popular thermophobic hydrogels, and will look at the specific mechanisms of classic thermophilic hydrogels as well as modern developments and applications.
2. Theory: Phase Transition of Thermophilic Hydrogels
There are several cooperative and competing forces active between polymer chains within hydrogels and from the solvent. Theoretically, thermophobic and thermophilic behavior should be possible for all hydrogels as represented by a closed loop coexistence curve on a phase diagram. This is providing that phase change of the solvent to gas or solid doesn’t occur [40]. Experimentally this may not be a practical observation due to the scarcity of substances with a small enough temperature range to display both solution behavior types without phase change, or due to the pressure conditions required to prevent phase change [14]. Much of the theoretical and experimental work for the phase change of gels has been transported from work done on single polymer chains in solution. Single polymer chains are easier to observe and characterize than gel networks and polymer gels are networks of single polymer chains. Literature concerning gels or single polymers in solution often reference theoretical work initiated by P.J. Flory [24]. Flory’s work explains that it can be expected that at sufficiently high temperatures or in good solvent conditions, a polymer system will be in an expanded state. At sufficiently low temperatures or in poor solvent conditions a polymer system will be in a collapsed state. Early work characterizing the volume-phase transition of gels found that transitions between collapsed and extended states of polymer chains were smooth and continuous or abrupt and discrete [15,41]. Other significant concepts were identified through this work such as collective diffusion theory [42]. A major factor governing volume-phase transition that is difficult to control and evaluate is the rubber elasticity of the gel as a result of the varying degrees of crosslinking that had been attained in the gels. Tanaka et al. (1979) identified that the rate of forming covalent crosslinks in a gel is logarithmic, once the initial fast gelation had occurred, the final curing process was slow due the movement of large molecule chains and low concentration of free chain ends. So depending on the time allotted to curing, two gel samples of the same composition may have considerably different solution properties. Due to the complexity of available models for polymer swelling and collapse, theoretical models that have been used to describe the volume-phase transition of hydrogels will not be examined. Instead, a brief overview of the forces that enable phase transition will be presented as background for later sections.
As briefly introduced, gels can be held together by either covalent bonds or non-covalent bonds [24,25]. Covalent bonds are chemical crosslinks between polymer chains and non-covalent bonds are physical crosslinks. The idea is that the covalent bonds are permanent while the non-covalent ones are not. The macrogels discussed in this review are held together by covalent bonds while the volume-phase transitions are dependent on non-covalent bonding interactions. Within the non-covalent physical bonds, there are those that can be classified as either strong physical bonds or weak physical bonds. Strong physical bonds are complexes like microcrystals and triple helices. They are stable over experimental time frames but can be broken by certain environmental conditions [43]. Weak physical bonds are driven by intermolecular forces that can be formed and broken over experimental time frames such as hydrogen bonding or van der Waals forces. Hydrogel polymer networks rely on chemical bonds for structure and physical bonds in order to undergo volume-phase transition. Weak physical bonds that are formed or broken during a phase transition are a combined effect of cooperative polymer-polymer interactions and polymer-solvent interactions [39,40,41]. A phase transition then becomes an equilibrium between repulsive forces that reduce polymer-polymer interactions and move to swell the gel and attractive forces that reduce the polymer-solvent interactions and move to collapse the gel. In terms of polymer-polymer interactions, attractive forces are hydrophobic interactions and van der Waals interactions while electrostatic interactions and hydrogen bonding may act as both attractive and repulsive forces depending on the environmental conditions. The following sections on intermolecular forces are not meant to be an exhaustive characterisation but instead to put the forces in context for this review.
2.1. Electrostatic Forces
Early work on hydrogels undergoing volume phase transitions surmised that the transition was driven by ionized pendant groups in the network and discrete or continuous change was determined by the degree of ionization [21,30,42]. Ionization of a gel was induced by copolymerization or hydrolysis. Toyoichi Tanaka had observed that “new PAm gels” that were tested after a shorter curing time showed continuous volume change while “old PAm gels” with a longer curing time showed discrete volume change [31]. Curing time was related to a natural hydrolysis process causing the “old gels” to have a higher degree of ionization than the “new gels”. From this work he determined that electrostatic forces due to ionization are the most effective repulsive force. Later works examining phase transition shows this to be an incomplete investigation since; other intermolecular forces must be considered, at least in the case of PAm gels, and electrostatic interactions are also an effective attractive force. Polyelectrolytes are the clearest example of a hydrogel where the phase transition is dominated by attractive and repulsive electrostatic forces. The phase transition is decided by the conformations of multiplets of ionomers induced by external stimuli. Electrostatic interactions are strongly affected by pH as well as the present of salts. Electrostatic interactions are capable of driving both thermophobic and thermophilic behaviors.
2.2. Van der Waals Interactions
Phase transitions driven by van der Waals interactions tend to be of thermophilic nature, where reference to van der Waals forces is in terms of permanent dipole interactions. Thermophilic behavior requires relatively strong associative intra and interpolymer-chain interactions at ambient conditions, causing the solvent (water) to be poor. Dipole interactions decrease upon heating effectively increasing the solvent quality [14]. Amide-amide interactions are a typical example in polymer gels where solubility in water is primarily controlled by dipole interactions. External stimuli can change the solution properties by affecting the strength and the alignment of these dipoles [44].
2.3. Hydrogen Bonding
Hydrogen bonding can be considered significant for phase transition in two ways; solvent interactions and solute interactions [14]. Researchers that consider solute interactions, polymer-polymer hydrogen bonding, observe hydrogen bonding to be characteristic of associative polymers and thermophilic behavior [26,43]. Consideration of polymer-solvent hydrogen bonding is typical of work concerning thermophobic hydrogels, as it is the dominant force allowing these gels to be water soluble at lower temperatures [35]. For hydrogels where phase transition is primarily driven by hydrogen bonding, thermophobic volume-phase transition is considered typical by many workers since hydrogen bonds are broken at elevated temperatures and therein should not be an influencing force [45]. Hydrogen bonding between the polymer and water is what permits water solubility. Formation of h-bonds between the polymer and water is both exothermic and forms higher structure order in the system. Adversely, where there is strong polymer-polymer hydrogen bonding causing gel collapse and preventing swelling, increasing temperature breaks the associative bonds effectively increasing the solvent quality to a similar effect as with dipole interactions. Interactions between carboxylic acid and amide functional groups can yield these strong polymer-polymer hydrogen bonds [26,46]. This was shown by Garay et al. through potentiometry in protic solvent. As carboxylic acid-amide bonds were formed, pH of the solvent increased reflecting a decrease in dissociated acid group concentration.
2.4. Hydrophobic Interactions
Hydrophobic interactions are an entropic effect induced mainly by the solvent. Aggregation of hydrophobic particles is driven by water to reduce the amount of interfacial area exposed. As mentioned above, higher temperatures typically raise the entropy of a system, which favors homogenous mixtures and better solvent quality, however, the solubility of small hydrophobic molecules increases with decreasing temperature. Heating reduces solubility and can lead to phase separation or precipitation. After reaching a minimum, the solubility sharply increases upon further heating. If phase separation occurred, one phase is again achieved on the condition boiling point has not been reached [47]. Hydrophobic interactions occur with polymers composed of primarily non-polar groups, such as phenyls, and tend to cause thermophobic behavior. As has already been suggested, volume phase transition is a result of a sum of several forces. Hydrophobic interactions tend to be thought of as being between water and non-polar solutes; however, they can also influence polar particles. Inclusion of hydrophobic functional groups in the pendant chains or as a co-monomer of polymer gels reduces the strength of the dipole interactions, increasing the significance of polymer-solvent hydrogen bonding. Enthalpy of dissolution becomes negative and enhances the water solubility of the polymer [44].
3. Ionic Polymer Hydrogels
Hydrogels composed of charged polymers are a main group of thermo-responsive hydrogels. There are several classifications of ionic polymer hydrogels, each one more specific in description than the last. A polyelectrolyte polymer has charged pendant groups, consisting of either cationic groups, anionic groups or both. Thus, they can have a net positive, net negative or neutral charge depending on environmental pH and ionic strength. Polyzwitterions refer specifically to those polymers that contain both positively and negatively charged pendant groups. Polyampholytes are those polyzwitterions where the charged groups are on different repeating monomer units while polybetaines are those where the charged groups are on nonadjacent sites of the same monomer unit and the cation doesn’t have an associated hydrogen (Figure 1). There isn’t consensus between researchers for the terminology of polyampholytes and polyzwitterions. Often their use can be the reverse of what is written above or polyzwitterion is used for what was described as a polybetaine with polyampholyte remaining the same [27,28,48]. There are some fundamental differences in their aqueous solution behaviors that can aid in identifying the uses of the above terms. As it is described above, electrostatic repulsions between charged groups on polyelectrolyte chains force an extended chain formation in deionized water at low concentrations. These repulsions can be disrupted by the addition of small electrolytes or changes to the system pH causing the chain to collapse. This is the polyelectrolyte effect. There is a complimentary antipolyelectrolyte effect where the polymer chain maintains a collapsed chain formation in deionized water at low concentrations and expands upon the addition of small electrolytes or change to the system pH. The antipolyelectrolyte effect is characteristic of polyampholytes and polybetaines. Polyampholytes can also exhibit polyelectrolyte behavior depending on solution conditions such as pH and tonicity. Polyampholytes are typically composed of weak acid and base monomers, causing them to have isoelectric points, further aiding in their identification [48]. Polybetaines are the most significant class of polyelectrolyte for this review as some are temperature responsive, a unique quality not typical of other polyzwitterions. Specifically, polysulfobetaines are of great interest to researchers of stimuli responsive materials.
Figure 1.
Sulfobetaine functional unit.

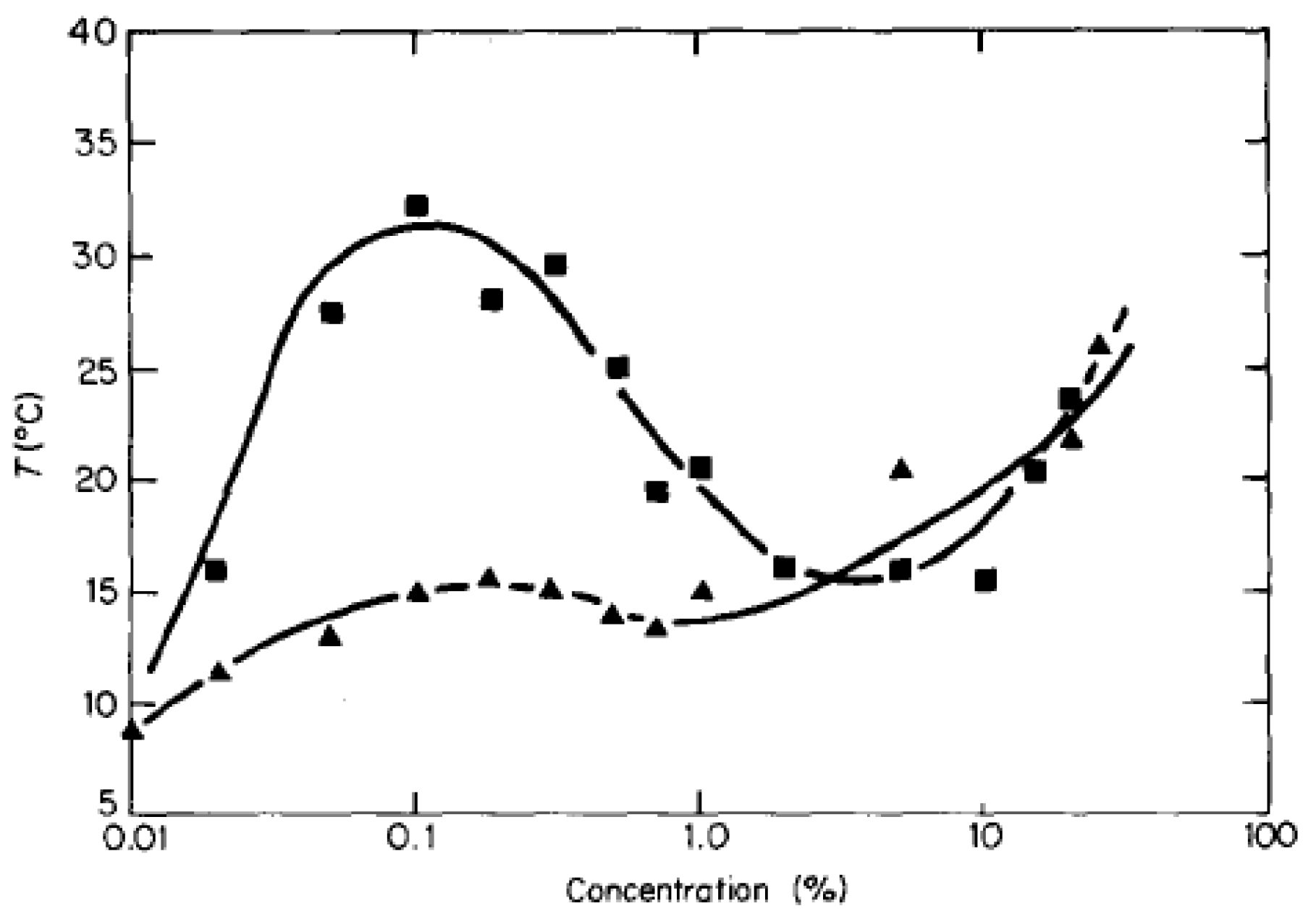
Early workers were required to synthesize their own sulfobetaines monomers, limiting the amount of research done for solution behavior. After polymerization, lengthy fractionation was also required to achieve adequately narrow polydispersity [49]. Soto and Galin worked with five unnamed polysulfobetaines determining their solubility in various solvents and the effect of adding salt. It was concluded that solubility for polysulfobetaines is limited to protic solvents and that addition of salt improves solubility in water specifically. It was suggested that this is because electrolytes disrupt the intra- and inter-molecular ionic network and that this effect is dependent on the polarizability of the anion. This is a clear example of antipolyelectrolyte effect. The commercial availability of sulfobetaine monomers enabled more research into solution properties of single polymers and gels in solution. N-(2-methacryloyloxyethyl)-N,N-dimethyl-N-(3-sulfopropyl) ammonium betaine was one of the first such monomers, offered under the name SPE by Raschig GmbH [17]. Along with the other IUPAC names for the compound (CAS# monomer: 3637-26-1, polymer: 41488-70-4), this monomer has been called sulfobetaine methacrylate (SBMA), DMMAPS and MEDSAH by other workers [27,28,50,51,52]. It was noted by Soto and Galin that although polysulfobetaine solubility is good in protic solvents, solubility in water above 20oC was unusual and was reaffirmed later [48]. However, it was found that solubility of polySPE in water was achieved at room temperature or upon heating. This signified that polySPE and possibly other polysulfobetaines have temperature dependent solution properties. Schulz was able to confirm this using cloud point titrations in both deionized water and salt water and map out the first full phase diagram for a sulfobetaine polymer in solution (Figure 2). The region above a coexistence curve represents one phase while the region below represents separation in two phases.
Figure 2.
Effect of polymer concentration and salt on polySPE phase behavior with respect to temperature. Polymer Mw = 4.35 × 105; ■, Water; ▲, 0.3% Aqueous NaCl (0.51 M) [17].
Figure 2.
Effect of polymer concentration and salt on polySPE phase behavior with respect to temperature. Polymer Mw = 4.35 × 105; ■, Water; ▲, 0.3% Aqueous NaCl (0.51 M) [17].

Both lower and upper critical solution temperature behaviors were observed at 16 °C and 33 °C respectively. These behaviors correspond to thermophobic and thermophilic VPTT in hydrogels of polySPE. It was mentioned in the previous section both behaviors should be possible for a single species and the claim is exemplified by this early work of Schulz. In agreement with Soto, it was also observed that salt solutions improve the solubility of polysulfobetaines. Thus when applied to temperature responsive solution properties, increasing salt concentrations decreases the thermophilic VPTT. Schulz was able to identify that the solution properties of polySPE are due to intra-chain and intra-group associations. Intra-chain associations are hydrogen bonds and dipole attractions on the same chain and the intra-group association is the ion pairing of the betaine. Strong intra-chain associations are what cause insolubility of the polymer. With the mention of inter-chain associations, the concept of thermophilic hydrogels was suggested but not discussed. Schulz was able to propose a model for the effect of salt on solution properties but was unable to account for the temperature responsiveness of the polySPE. Along with work on unperturbed dimension and θ-conditions, characterisation of single polySPE chains in solution allows better understanding of the behavior of the gels [53].
A study of polySPE chains grafted to a surface, by surface initiated atom-transfer radical polymerization, gives great insight into the mechanics of a hydrogel with positive volume-phase transition properties [27]. Azzaroni exhibits two main cases in this research: a surface with low molecular weight (short) chains, and a surface with high molecular weight (long) chains. The two cases are comparable to gels with high and low degrees of crosslinking respectively. Low molecular weight chains represent short polymer sections between crosslink nodes while high molecular weight chains are similar to long polymer sections between crosslink nodes. Azzaroni expands upon the description given by Schulz for the forces of thermophilic behavior of polybetaines. He invokes discussion of the dielectric properties of water, solvation effects and excluded volume effects from hydration of the charged sites in breaking chain associations for gel swelling. The energy of the electrostatic forces must exceed the energy required for dehydration of the charged sites in order to form high degrees of inter and intra-chain associations. The degree of chain association was measured by water contact angle in this study and qualified in terms of hydrophobicity and hydrophilicity of the hydrogel surfaces. Azzaroni claims that in the case of short polymer chains, there are no chain association as the density of electrostatic interactions is too low, allowing complete hydration. Conversely, long polySPE chains present a high number of charged sites increasing the probability of electrostatic interaction and chain association, hindering hydration. The long grafted chains were poorly soluble in water at 22 °C, however, upon heating to 52 °C the gel coating shifted from a hydrophobic surface to a hydrophilic one. The shift was found to be reversible and the polySPE coatings maintained a stable composition for several months, able to undergo thermal cycling with similar results. Although this work is comparable to a true gel, it doesn’t clearly qualitatively examine the equilibrium of interactions as the polybetaine is heated through its VPTT nor does it present the extent of hydration quantified by the volume increase of the polymer network.
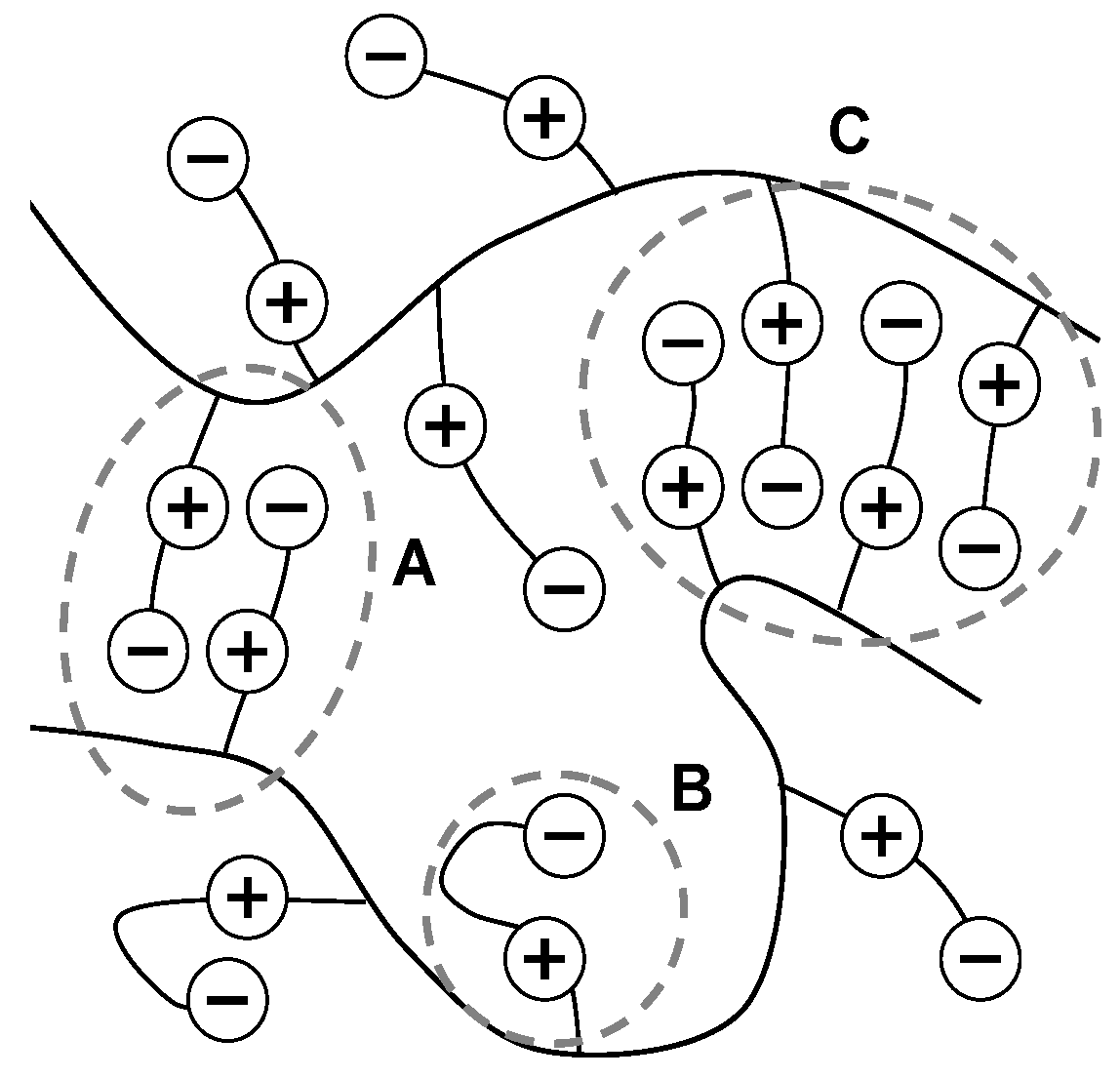
The work done by Azzaroni borrows the model of chain and group associations from research by Georgiev, presenting one of the first direct investigations of a polySPE hydrogel as it undergoes positive volume-phase transition [28]. Georgiev describes the reversible swelling of the hydrogel as an equilibrium between inter and intra associations further developing the theory of Schulz and providing a basis for Azzaroni. The associations are described as intragroup ion pairs and entropy driven zwitterionic clusters (Figure 3).
Figure 3.
Short zwitterionic cluster (A), intragroup ion pair (B), long zwitterionic cluster (C), nucleated from a short cluster with entropy driven propagation outwards. Figure adopted from [28].
Figure 3.
Short zwitterionic cluster (A), intragroup ion pair (B), long zwitterionic cluster (C), nucleated from a short cluster with entropy driven propagation outwards. Figure adopted from [28].

Georgiev is able to describe the shift of the equilibrium of the chain associations during the volume-phase transition. At low temperatures, zwitterionic clusters form as the enthalpy of the water decreases, making dehydration of the charged sites easier. The entropy of the solution also decreases, promoting higher order and a collapsed gel conformation. At high temperatures, increasing entropy and hydration energy favors ionic pairs allowing osmotic pressures to swell the gel. Dehydration of the charged sites is not mentioned by Georgiev as a factor in the volume transition of the hydrogel. The involvement of a dehydration activation energy is explored by Kamenova et al. [54], extending the model to include solvation factors in consolidation with the model of Azzaroni. This work assumes the model of associations established by Georgiev and uses an “isoconversional” method of thermogravimetric analysis to approximate the activation energy of dehydration for the gels [55]. While attempting to correlate the results of the analysis on the dehydration activation energy to the equilibrium swelling ratio of the gels, the data and results are poorly presented restricting the usefulness of the work by Kamenova et al. to affirming the theory of Georgiev. Missing from all three works was whether any pendant chains remain unpaired as the charged sites are solvated by the water.
The discussion above on the effect of polymer segment length that was extrapolated from the grafted polySPE chains of Azzaroni is also addressed, confirming the contentions presented. Hydrogels synthesized with low amounts of crosslinker provide long polymer segments between covalent nodes. It is observed that this leads to the abrupt positive change in swelling ratio observed across the VPTT. This transition in swelling ratio become smoother and then disappears altogether at higher incorporation of crosslinker in the hydrogel.
In the quantification of the swelling ratio transition, Georgiev presents the partial Flory-Huggins equation of osmotic pressure used by Tanaka [15,41] with a modification to account for charge distribution in the polyzwitterionic gel. It is found that the equation is adequate for describing the critical values of observable parameters but not the temperature dependence of interactions within the hydrogel through the transition. Additionally hysteresis is experienced upon cooling which is typical of dipole interactions seen in proteins undergoing coil-globule transition. These observations are used by Georgiev to infer that dipole-dipole associations as well as electrostatic interactions between ionic groups account for the large volume phase transition in the polybetaine hydrogels.
The progression of research with thermo-responsive polysulfobetaines has continued towards characterization and optimization of gel properties. Ethylene glycol dimethacrylate (EDMA) and N,N'-methylene bisacrylamide (MBAm) are typical crosslinkers when synthesizing polysulfobetaine gels. Attempts to optimize the hydrogel properties have used other crosslinking agents, with similar structure to SPE monomer, to improve the mechanical properties of polySPE gels [56]. This work shows that the concentration of crosslinker used in the synthesis of the hydrogel is more important for solution behavior than selection of crosslinker. However, it is shown that different crosslink distances due to linker selection will have an impact of mechanical properties such as Young’s modulus. Polysulfobetaines have also gained much interest as a biocompatible material with high protein fouling resistance [50]. Copolymerizations with other biocompatible thermo-responsive polymers have formed temperature triggered cell-detachment surfaces [57].
4. Acrylamide Based Hydrogels
It has been seen that polysulfobetaine-based hydrogels are the most studied ionic polymer hydrogel. Electrostatic interaction between the charged sites on the polymer side chains is the primary driving force for volume-phase transition. As mentioned prior, the phase transitions are in fact an equilibrium between several forces. Although electrostatic forces driven by the ionic nature of the gel composition is the primary force, dipole-dipole interactions from the permanent, non-adjacent and opposite charges as well as minor hydrogen bonding play a part. Unmentioned are hydrophobic interactions between the long hydrocarbon based polymer chains of the hydrogel. They were unaddressed by most workers as the hydrophobic interactions were not significant compared to the others. Another significant class of thermophilic hydrogel is that of acrylamide (Am)-based hydrogels. Many thermo-responsive hydrogels are Am-based as seen by work with PNIPAm and work on polysulfobetaines with an Am backbone [28,35]. As will be shown, this class of hydrogel is dominated by hydrogen bonding. However, it is not as dominating as the electrostatic forces in the polysulfobetaine gels. Dipole interactions, electrostatic interactions and hydrophobic interactions play a larger part in balancing the forces of phase-transition for Am-based hydrogels. These gels are water soluble above 0 °C and able to swell several times their dry-weight to an extent highly dependent on their synthesis methods [58,59,60,61]. Although Am itself can be polymerized to form hydrogels, they do not undergo volume-phase transition. To form thermophilic hydrogels, Am is typically combined with acrylic acid (AA) either as a random copolymer or as an interpenetrating network of the homopolymers. The isolated homopolymers of Am and AA in solution behave like a polymer in good solvent with near constant solubility upon heating while a mixture of the two in solution behaves like a polymer in poor solvent with improving solubility upon heating [62]. It is known that hydrogen bonded complexes are formed between the two monomers and that these complexes break down upon heating [63,64].
4.1. Interacting Forces
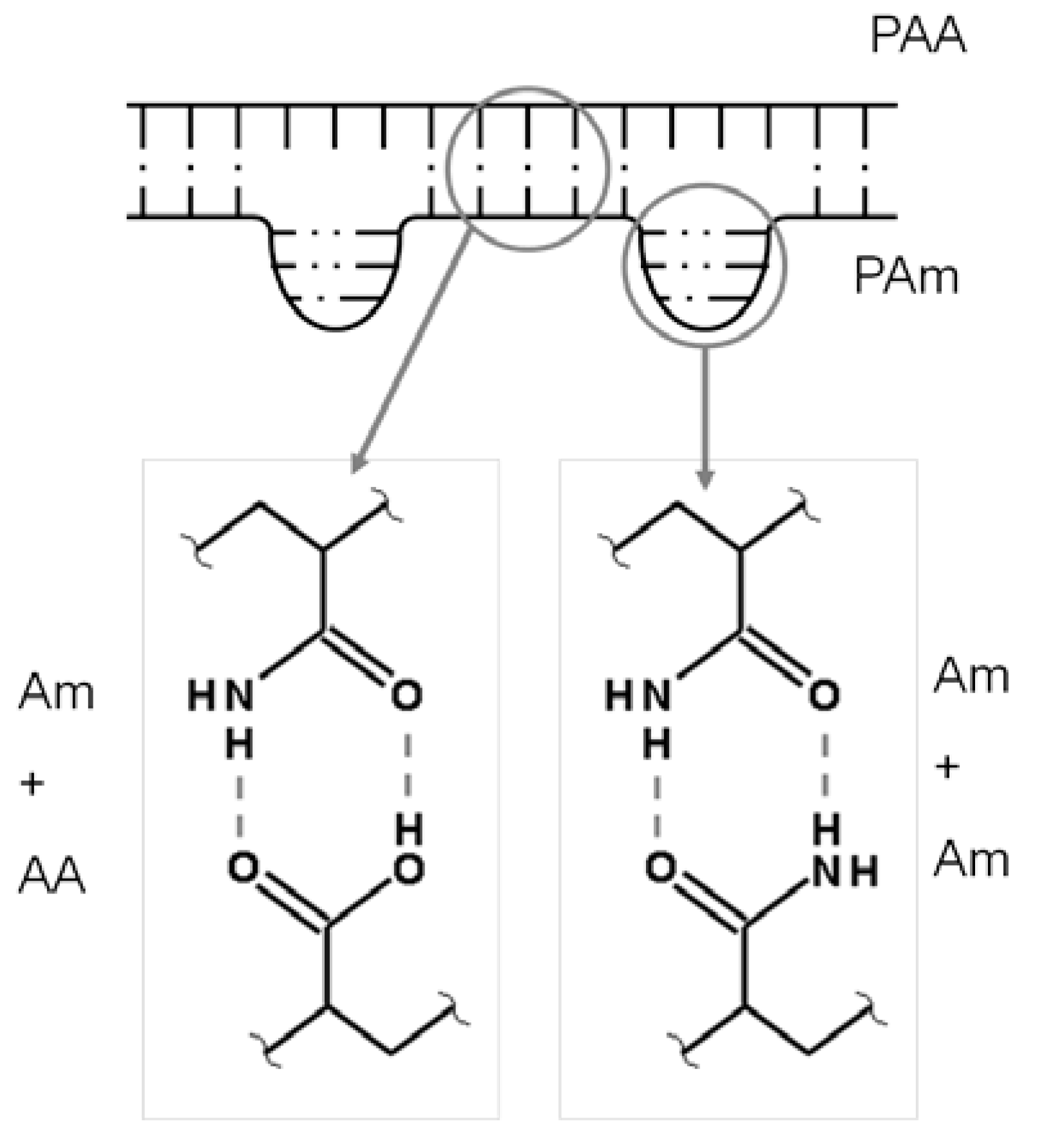
Although volume-phase transition had been shown for Am and AA polymer gels, complexation of the polymers by hydrogen bonding between the amide and carboxyl functional groups was first shown by Klenina [62]. Earlier work had examined interactions between the functional groups of Am and AA in acidic aqueous conditions [31,65]. It was believed that the inclusion of AA accounted for ionization of the gel network and was responsible for discontinuous volume phase transition. However, it was unsure as to whether complexation occurred when AA was not ionized. Stable complexes between polyacrylamide (PAm) and polyacrylic acid (PAA) in solution were developed without dissociating the carboxyl groups. In hydrogel structure, the hydrogen bonding forms a ladder structure between polymer segments (Figure 4). The ladder structure is formed and broken by a “zipping” action nucleated from an individual hydrogen bond similar to the long zwitterionic clusters of the polysulfobetaine gels. Dissolution of the structure occurs upon heating, hydration forces are generated as hydrogen bonds are broken by thermal energy and break adjacent hydrogen bonds resulting in rapid dissolution as there are now two functional destructive forces. Similarly, upon cooling, polymer-polymer hydrogen bonded structure is favored as thermal energy decreases and is aided in forming bonds by propagating dehydration between adjacent side groups.
Figure 4.
Competing hydrogen bond complexations in PAm and PAA hydrogels. Adapted from [36].
Figure 4.
Competing hydrogen bond complexations in PAm and PAA hydrogels. Adapted from [36].

Dipole-dipole interactions between the amide groups present another strong influence over the volume-phase transitions. Water solubility of PAm is primarily controlled by the dipole interactions of the amide groups. In the homopolymer, it is expected that the energy produced by the amide dipole interactions is larger than that provided by solvent interactions and other solute interactions [44]. In the gel network, hydrogen bonding becomes dominating weakening the effect of the dipole interactions but not making them negligible. The amide dipoles may be affected by temperature just as the hydrogen bonds are. External stimuli can change the strength and alignment of the dipoles modifying the solution properties [14]. For Am-AA based hydrogels, there are two main competing intermolecular complexation mechanisms. Am can form complexes with itself, the discussion to whether this is due to hydrogen bonding or dipole interactions is not complete, meaning that complete equimolar complexation with AA shouldn’t possible as seen in Figure 4 [36].
Confirmation of hydrogen bonding in Am-AA based hydrogels is often provided using urea [35,66], a known hydrogen bond and hydrophobic interaction disrupting analyte [67].One worker reported complete dissolution of Am-AA complexation at 10 wt% urea in water as confirmed by light transmittance [68]. The effectiveness of urea does not clearly identify hydrogen bonding as the sole force causing dissolution. Hydrophobic interactions are enhanced at higher temperature and are a strong attractive force significant in negative volume-phase transitions. However, considering that a thermophilic hydrogel swells upon heating as well as with the addition of aqueous urea at low temperatures suggests that Am-AA based hydrogel networks experience negligible associative hydrophobic interactions. In addition to work with urea, varying the Am-AA monomer ratio can show that the hydrogen bond between amide and carboxyl functional groups is essential to positive volume-phase transition. Both PAm and PAA homopolymers show negligible volume change with temperature change in pure water [14,15,41]. It has been shown that in a gel network, the monomer ratio (Am:AA) must be between 1:9 and 8:2 for any volume phase transition to occur [68].
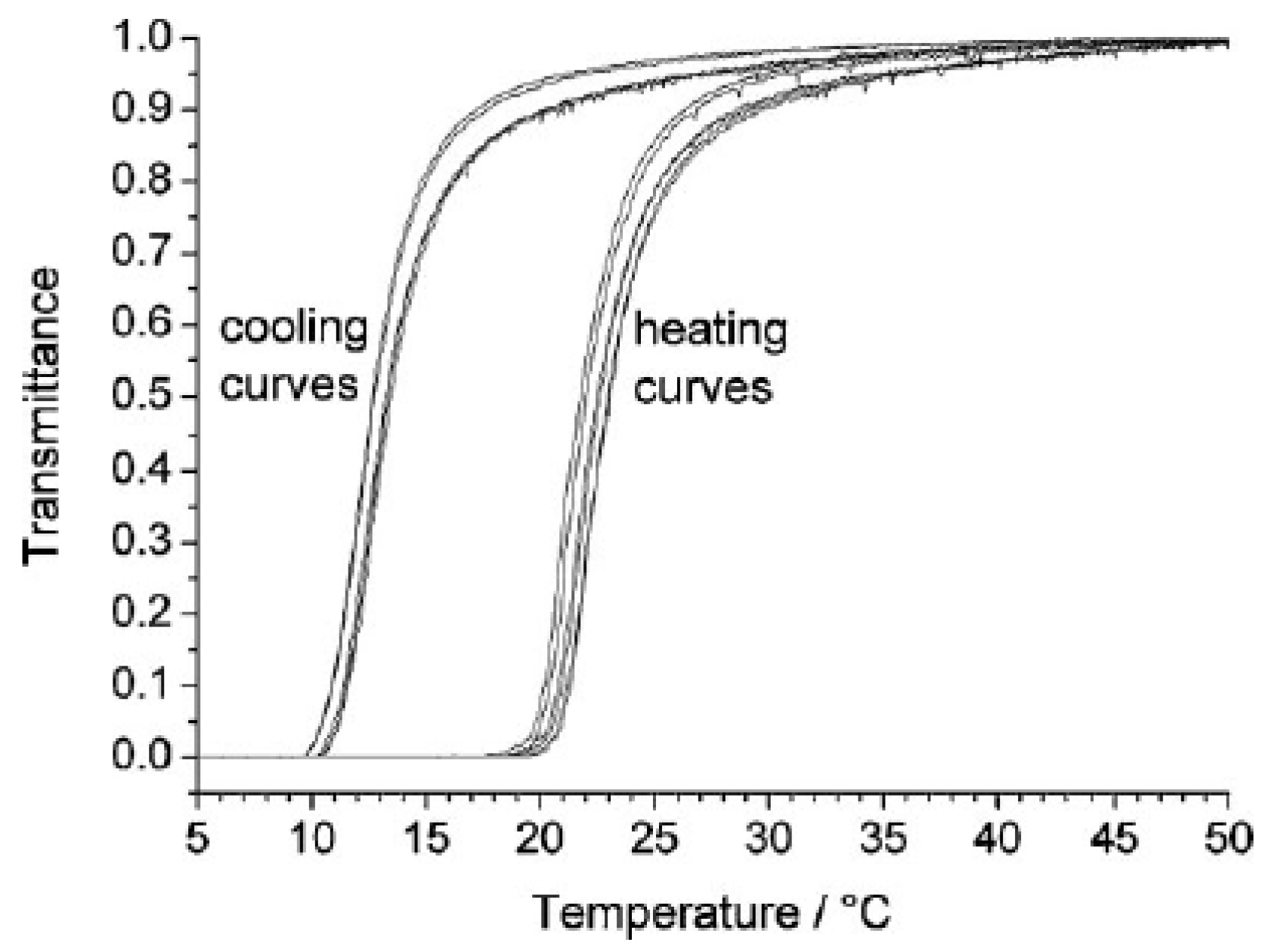
The swelling and collapse of a covalently crosslinked Am-AA based hydrogel is reversible and appears complete under light transmittance observation [68], however when viewed by mass or volume, reversibility is not complete with the absolute swelling volume becoming larger after each temperature cycle. Along with this, structure formation is kinetically slower than dissolution and hysteresis is often observed between heating and cooling cycles (Figure 5) [14,36,67,68]. Both these effects have similar root causes. The typically high swelling ratios of Am-AA based hydrogels above their VPTT impose great distances between polymer chains. These large interstitial spaces allow reduction in chain entanglement through the swelling and collapsing cycles, and, the polymer complexes formed below the VPTT are not reformed as they once were, their conformation changed as some chain segment may not reform complex at all [29]. Other aspects to consider which will be discussed in following sections are ionic dissociation of carboxyl groups and hydrophobic composition of the polymer chains.
Figure 5.
Hysteresis as observed by light transmittance characteristic of many Am-AA based hydrogels [30].
Figure 5.
Hysteresis as observed by light transmittance characteristic of many Am-AA based hydrogels [30].

Stabilizing the polymer complex of thermophilic hydrogels has become a great topic of research as workers attempt to optimize the gels’ responsive qualities. There are several strategies to do so: Increasing the amide density by using monomers with pendant groups with multiple amide functionality [69]; Increasing the hydrophobic content by copolymerization with a hydrophobic monomer [44,68,70,71]; Suppress amide-amide interactions to promote more efficient amide-carboxyl complex formation [36,72].
4.2. Synthesis
There are different volume-phase transition profiles that are attainable dependent on the microstructure of the Am-AA based hydrogel. The two most common structures are a random copolymer gel as well as an interpenetrating polymer network (IPN). The copolymers are typically prepared in a single polymerization step while the IPNs are prepared by sequential polymerization with the PAm network prepared first and then the PAA network throughout it. Workers have found that the random copolymerized hydrogels undergo smooth continuous volume transition through temperature change. The same workers have discovered IPNs to exhibit an abrupt volume change as the VPTT of the hydrogel is attained. This abrupt change is often described as a discontinuous or discrete transition profile. It has been asserted that both the continuous and discrete Am-AA hydrogel transition behaviors are reversible in the sense described above [29,31,70].
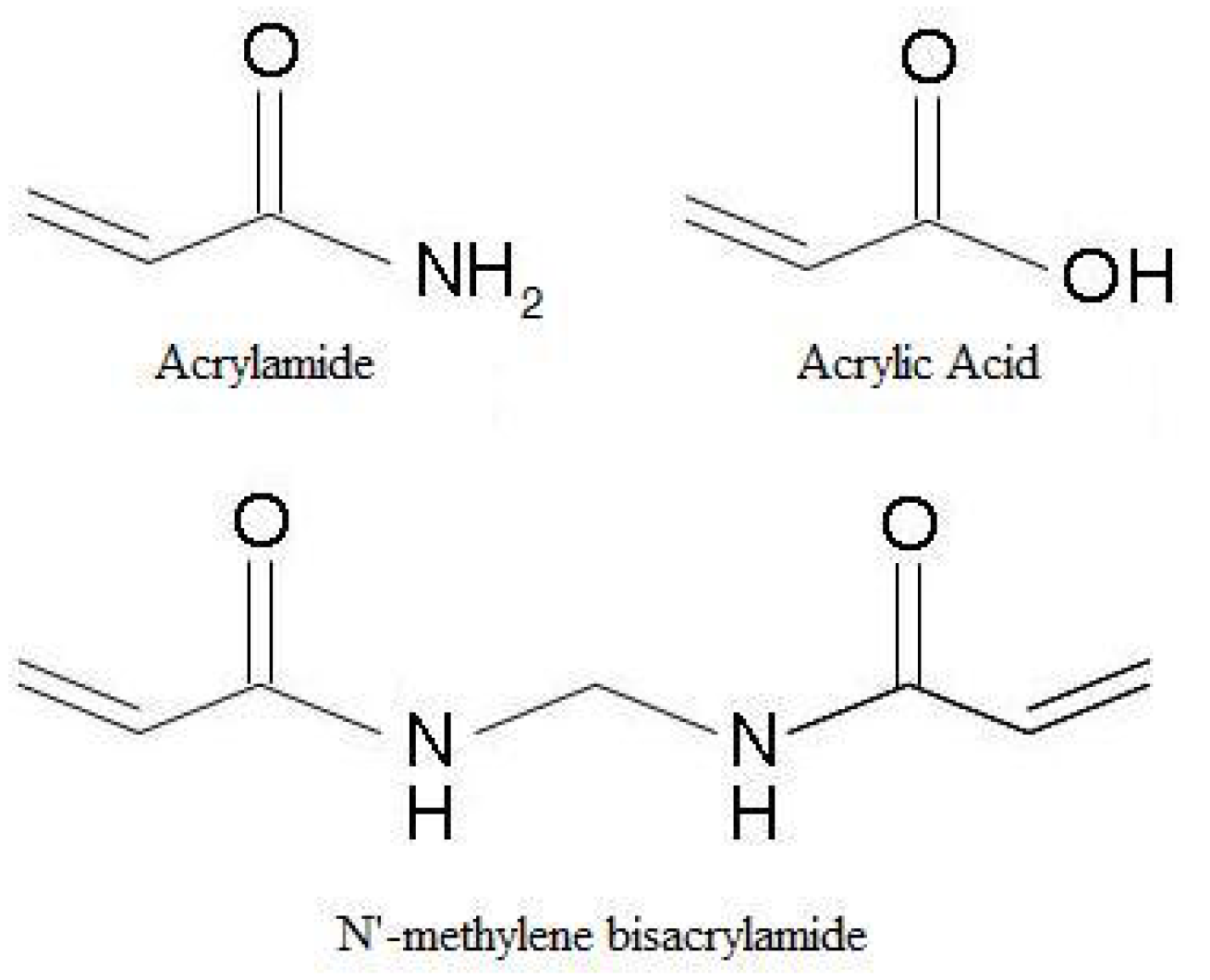
There are common methods and recipes for synthesis of Am-AA based hydrogels. N'-methylene bisacrylamide is a double functional amide monomer that is frequently used as a crosslinking agent for these gels due to its similar structure (Figure 6).
Figure 6.
Chemical similarities between acrylamide, acrylic acid and N'-methylene bisacrylamide.

Initiation for Am-AA based hydrogels is typically by free radical initiation. Thermally activated persulfate initiators like ammonium persulfate (AP) or potassium persulfate and often combined with an accelerator like tetraethylmethylenediamine (TEMED) to reduce the temperature of initiation. The crosslinker and choice of initiator are also common to other thermo-responsive hydrogels using N-alkyl substituted amide monomers [35]. Additional methods of initiation that have been used are irradiation and plasma treatment (see Table 1) as well as reversible addition-fragmentation chain transfer (RAFT) [73]. Hydrogel polymerization is usually carried out in glass pipettes, between glass plates with spacers or in glass dishes. Earlier it was mentioned that a range of monomer ratios were acceptable for a Am-AA hydrogel to posses thermophilic behavior, however, as shown in single polymer chain solutions of PAm and PAA, precipitate that forms below the VPTT has a stoichiometric ratio of 1:1 by refractive index analysis [62]. Results of this nature lead researchers who are developing hydrogels for functional applicability do design gels to have a 1:1 Am-AA monomer ratio in the final gel. Consistent of all workers, Am quantity is used to determine the amount of AA to satisfy the ratio.

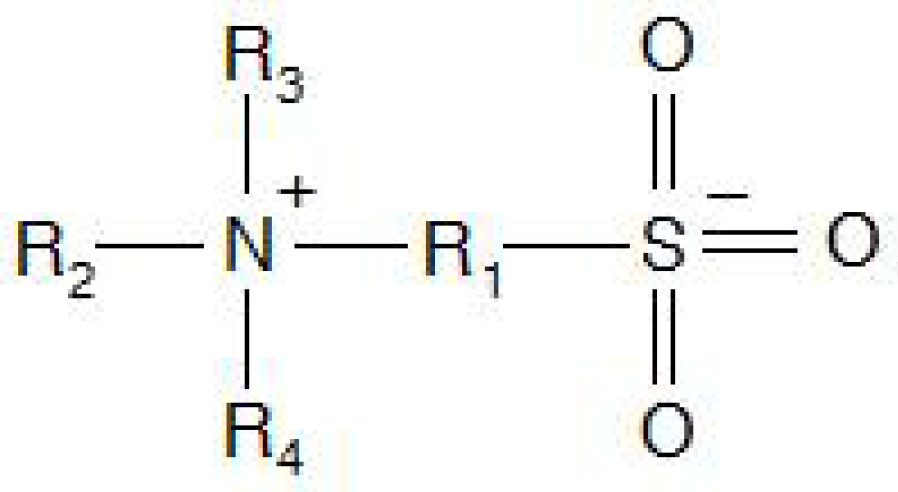{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | AA | Crosslinker | Initiator | Type | Source |
|---|---|---|---|---|---|
| 5 g | - | MBAm | AP & TEMED | Homopolymer | [15,31,41] |
| 5g | 5g | MBAm | AP & TEMED | Sequential IPN | [66] |
| 7.2 g | 7.3 g | MBAm | AP | Sequential IPN | [36] |
| 15–30 mol% | Various molar ratios | - | 60Co-γ irradiation | Random copolymer | [74] |
| 1:1 molar ratio | MBAm | Potassium peroxodisulfate | Random copolymer | [75] | |
| 1.775 g | 1.800 g | MBAm | α-Ketoglutaric acid | Random copolymer | [19] |
| 1–3 wt% | 0.2–2 wt% | MBAm | Plasma treatment, potassium persulfate | Sequential IPN | [76] |
Tanaka was one of the first researchers to work with thermophilic hydrogels. Initial work solely observed PAm gels and found discrete swelling behavior with respect to temperature in partial organic solvent [15]. As asserted above, PAm does not exhibit temperature responsiveness in purely aqueous solvent. Work to discover conditions that would allow PAm hydrogels a volume phase transition in water was extensive, even leading Tanaka to develop the spinodal curve for PAm [77]. It was discovered serendipitously that the gels needed to cure or “ionize” in order for thermophilic behavior to occur without partial organic solvent [41]. Older gels underwent volume collapse upon cooling while more freshly synthesized gels did not. Tanaka’s “ionization” turned out to be inclusion of carboxyl groups in the gel network [31]. This was possible by either hydrolysis of the amide groups or incorporation of AA as a co-monomer. Tanaka only synthesized random copolymers in early works positive volume-phase transition was induced by partial hydrolysis of the amide groups in a basic reaction medium (pH 12). It was discovered that while only a small fraction of AA units were necessary for volume-phase transition to occur in water, increasing fractions promoted discrete volume phase transitions. Tanaka surmised that the swelling transitions in aqueous media could be accounted for by osmotic pressure driven by dissociation of hydrogen ions from the carboxyl groups, leading to his use of Flory-Higgins solution theory as previously mentioned [15,41].
The results of Tanaka showing discrete swelling transition was primarily based on solvent composition, never truly able to show a direct correlation between temperature and volume-phase transition, deriving a “reduced temperature” instead from the Flory-Higgins’ osmotic pressure formula. The failure to show a direct relationship between temperature and hydrogel volume phase transition made Tanaka’s work difficult to reproduce. First works developing Am-AA IPNs discovered this as only smooth, continuous swelling profiles were observed for random copolymer gels while the IPNs displayed an abrupt discrete transition [29,70]. It was determined that rapid polymer complex formation and dissolution was responsible for the sharp volume transition observed for the IPN gels. The dull swelling of the copolymer was assumed to be due to structural discontinuities causing isolation of any Am-AA complexes that did form. As discussed above, complex formation and dissolution are both nucleated at specific sites and then aided in outward propagation by hydration forces. The random ordering of amide and carboxyl groups as well as the isolated Am-AA complexes in the copolymer gel prevents large range influence of nucleation sites. The homopolymer chain segments between crosslink nodes in the IPN promote more efficient complex formation and therefore dissolution. Equimolar composition of the IPN was perpetrated as the optimum condition for complete complexation of the hydrogel below the VPTT. This was not confirmed by the researchers as it was assumed that there was no concentration gradient through the initial PAm gel as it swelled with AA monomer solution prior to the secondary polymerization.
Application of Am-AA IPNs is well exemplified by work done grafting the hydrogels onto a porous nylon-6 substrate to create function gates to control permeability [76]. Consistent with the sequential polymerization method, PAm gels were surface initiated by plasma treatment to graft the initial gel onto the inside of the substrate pores. The initial gel was then swelled in an aqueous AA, MBAm, and potassium persulfate mixture before the secondary gel was polymerized. The functional gates showed that water permeability of the nylon substrate could be increased at temperatures below the VPTT as the hydrogel collapsed, opening the pores. The pores would close above the VPTT as the hydrogel gates swelled. Scanning electron microscopy and x-ray photoelectron spectroscopy were used to verify the structure of the membrane and the synthesis of the IPN. It was found that although the entire membrane was soaked in the secondary gel reaction mixture, it was only retained in the PAm gel (Figure 7). The thermophilic behavior of the IPN hydrogel gates was observed to be reversible through several temperature cycles varying between 10 °C and 40 °C.
Figure 7.
Scanning electron microscope image of nylon-6 membrane after sequential polymerization of a Am-AA IPN [76].
Figure 7.
Scanning electron microscope image of nylon-6 membrane after sequential polymerization of a Am-AA IPN [76].

4.3. Increasing Hydrophobic Content
Hydrophobic co-monomer is incorporated either randomly copolymerized along with the Am and AA monomers or randomly copolymerized with Am into the initial gel of a sequential IPN. Hydrophobic functional groups affect the complexation of amide and carboxyl groups in two main ways. Firstly, the hydrophobic groups increase the density of hydrophobic interactions. Hydrophobic interactions are strengthened at higher temperatures, so as hydrogen bonds break upon heating, hydrophobic interactions become more significant. Secondly, the hydrophobic functional groups in the polymer segments reduce the proximity of the amide groups from each other as well as from carboxyl groups, increasing the significance of polymer-solvent hydrogen bonding. This enhances the overall water solubility of the polymer as interchain associations are reduced [44]. Table 2 shows the quantities of hydrophobic monomer used in several studies. Butyl methacrylate (BMA) is the most commonly used hydrophobic monomer.
It is the consensus of these workers that incorporating a hydrophobic monomer into the hydrogel network increases the stability of the associative forces preventing swelling. This stability is observed as an increase in the VPTT for the complex. Further increases in hydrophobic content raises the VPTT even more, however a critical point where the gel is no longer water soluble is not reached by any worker. This is observed by several workers [29,36,70,71]. However, the VPTT may not be made to be lower than the same hydrogel without incorporation of the hydrophobic monomer. As well, at high BMA loadings, the volume-phase transition ceased to be significant and was only attainable at temperatures above 60 °C. One researcher suggested that the reduced swelling of hydrogels with higher BMA content was that the porosity of the gel network decreased, however this explanation was flawed due to the setup of the experiment [71]. This presented model failed to discuss the effect of higher polymer loading potentially causing a denser network.
| Am | AA | Hydrophobic monomer | Initiator | Type | Source |
|---|---|---|---|---|---|
| 3.80 g * | 3.85 g * | BMA (0.2 g) | t-Butyl peroctanoate, AP | Sequential IPN | [29,70] |
| 3.60 g * | 3.65 g * | BMA (0.4 g) | AP | Random copolymer | |
| 6.408 g | 6.497 g | BMA (1.584 g) | AP | Sequential IPN | [36] |
| 1.6 g | 1.6 mL | BMA (1.6 mL) | AP | Random copolymer | [71] |
| 1:1 molar ratio | Octylphenol polyoxylethylene ether (3 mol%) | Potassium persulfate | Random copolymer | [68] | |
* various monomer ratios and quantities were used in these studies. BMA, butyl methacrylate.
The work using octylphenyl polyoxyethylene ether (OP7-AC) as the hydrophobic monomer was interesting as a crosslinking agent was not used [68]. First, it should be noted that the monomer needed to be modified by adding an acryloyl chloride group in order for it to be polymerized into the Am chain. The hydrogel synthesized incorporating OP7-AC was crosslinked by interconnecting micelles of the OP7-AC pendant chains stabilized by sodium dodecyl sulfate (SDS) (Figure 8). These micelles acted as the physical crosslinks.
Figure 8.
Micellular crosslinks of OP7-AC and SDS [68].
Figure 8.
Micellular crosslinks of OP7-AC and SDS [68].

The effects of different concentrations of SDS and OP7-AC were investigated. SDS, as the micelle stabilizing molecule, becomes more effective at higher concentrations raising the VPTT with increasing concentration. Increasing OP7-AC content in the hydrogel initially decreased the VPTT sharply to a minimum of ~12 °C at 1.5 wt% of the monomer as the hydrophobic group disrupted amide-amide interactions and competed with the carboxyl groups for hydrogen bonding with the amide groups. Hydrophobic interactions became more dominant at higher OP7-AC inclusion, gradually increasing the VPTT. In the earlier work of Katono et al. it had been observed that inclusion of BMA in the hydrogel composition improved the mechanical strength of the gel and although this was a qualitative observation, suggested this to be typical effect for the inclusion of any hydrophobic monomer [29]. This was investigated for hydrogels containing OP7-AC as well. Stress-strain curves were prepared for several hydrogel recipes showing higher Am content promoted better stress properties while higher AA content promoted better elongation properties. Gels with the hydrophobic monomer were not compared to gels without OP7-AC making the results quantitatively inconclusive as to whether the mechanical properties were indeed improved.
4.4. Stabilizing Am-AA Complexation
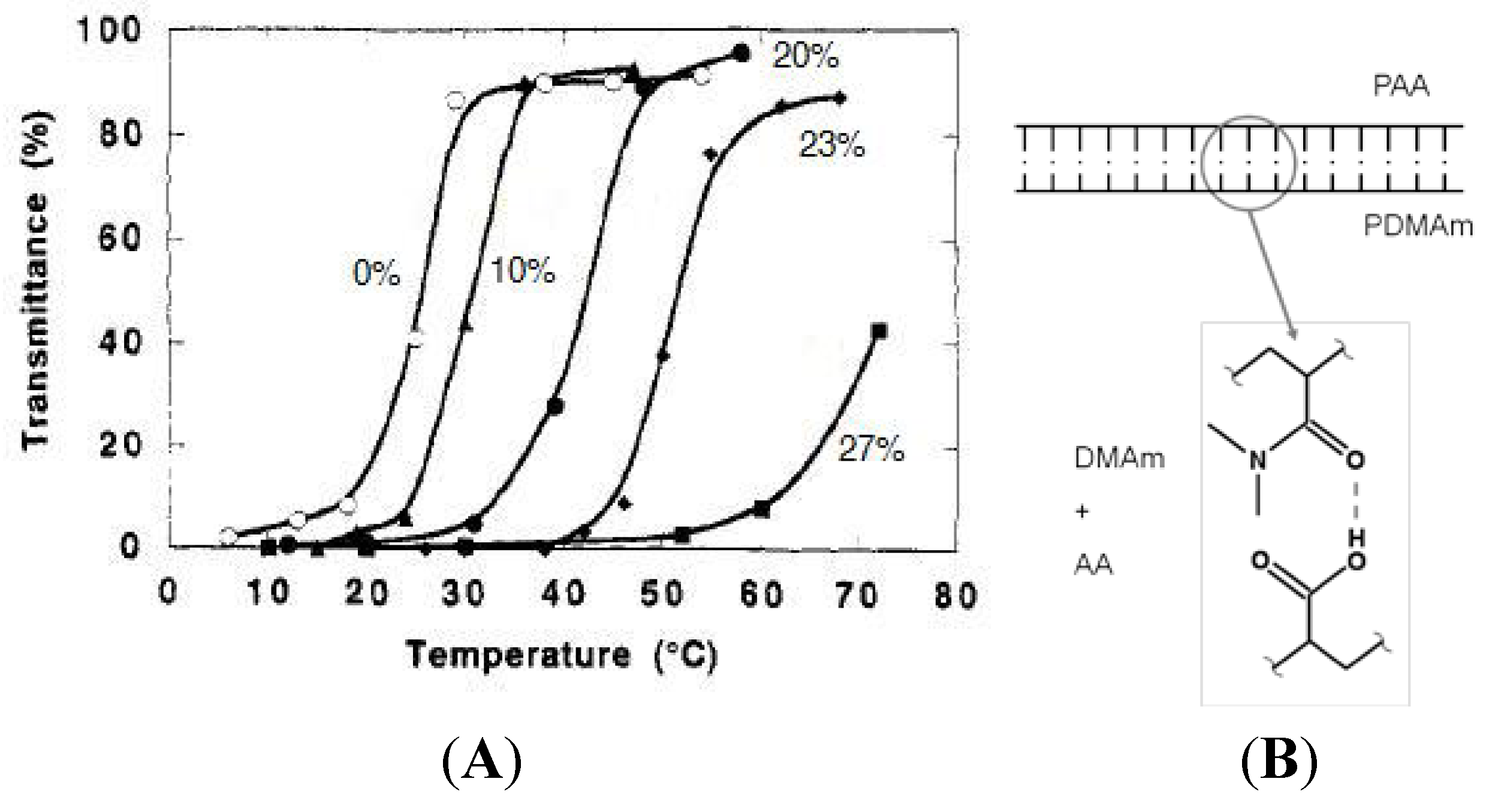
The suppression of amide-amide interactions is one of the effects of incorporating a hydrophobic co-monomer into the hydrogel network. However copolymerization of the hydrophobic monomer doesn’t always lead to more efficient hydrogen bonding between the amide and carboxyl groups as was seen with OP7-AC which had its own competing hydrogen bonding. There is another way to include hydrophobic functional groups which disrupt the dipole interactions between amide groups but do not hydrogen bond or create a steric hindrance which disables volume-phase transition. Substituting alkyl groups on the nitrogen of the amide introduces a hydrophobic property without another monomer. There is a balance however between the size of the substituted alkyl group and the strength of the amide-carboxyl hydrogen bond. NIPAm is one such N-substituted Am where the isopropyl group fully disrupts the interchain hydrogen bonding and hydrophobic interactions become dominant, promoting thermophobic behavior. N,N'-dimethylacryamide (DMAm) is a N-substituted Am that has been used with AA in IPN hydrogels to improve the amide-carboxyl group hydrogen bonding efficiency [36]. Like other IPNs, the DMAm monomer is copolymerized with Am to form the initial gel for a sequential IPN synthesis. MBAm and AP are used, respectively, as the crosslinking agent and initiator. Concentrations of DMAm used in the synthesis of the initial poly(DMAm-co-Am) gel varied between 0 mol% and 27 mol%. Similar to the other hydrogels incorporating a hydrophobic co-monomer, increasing DMAm content in the composition of poly(DMAm-co-Am)/PAA IPNs caused the VPTT to rise. This was confirmed by UV-transmittance, the VPTT rose from 25 °C at 0 mol% DMAm to 40 °C at 20 mol% (Figure 9A).
As briefly discussed, PAm can form hydrogen bonded complexes with itself which means that it shouldn’t be able to form a completely equimolar complex with PAA since there will be competing intermolecular complexations. PAm is a hydrogen donor and acceptor. PAA is a hydrogen donor. PDMAm is a hydrogen acceptor. This work argues that DMAm cannot form hydrogen bonded complexes with itself, which allows equimolar complex formation with PAA (See Figure 9B). This assertion by Aoki et al. is not wholly correct. If PAm can form hydrogen bonds with itself as both a donor and acceptor, it should be able to form hydrogen bonds with PDMAm where the Am unit acts a donor. Instead, it is possible that the Am self associations due to dipole-dipole interaction of the amide are a stronger associative force than the amide-amide hydrogen bonding. Am dipole-dipole interactions are disrupted by the non-polar n-alkyl substituted group. The substituted alkyl groups on the pendant chains of PDMAm prevent the amide from undergoing dipole-dipole interactions hence it cannot associate with PAm.
DMAm promotes more efficient amide-carboxyl in Am-AA based hydrogels only when incorporated as a comonomer with Am in the initial gel of an IPN. While PDMAm and PAA homopolymers can form strong complexes with each other in solution, poly(DMAm-co-AA) does not form a complex nor does PDMAm-graft-PAm [72]. Neither does the DMAm monomer unit impart temperature dependent solution properties, Am is required.
Figure 9.
Effect of DMAm content on poly(DMAm-co-Am)/PAA IPN (A). Percentages are by mol. Complexation of PDAm with PAA (B).
Figure 9.
Effect of DMAm content on poly(DMAm-co-Am)/PAA IPN (A). Percentages are by mol. Complexation of PDAm with PAA (B).

5. Novel Thermophilic Hydrogels
N-Acryloylglycinamide (NAGA), or N-(carbamoylmethyl)prop-2-enamide (Figure 10), is a monomer compound whose homopolymer (PNAGA) is capable of forming a thermally reversible, non-covalently bonded gel network in water. This temperature dependent hydrogel was first observed and extensively studied by Haas et al. [34,78,79,80,81,82]. Since this first observation and study there have been very few works further investigating the characteristics and mechanisms of complexation of NAGA as either homopolymer or co-monomer [83].
Figure 10.
N-Acryloylglycinamide.

A renewed interest in 2007 in patent literature [84,85] and again in 2010 in the solution properties of single polymer chains and synthesis methods for PNAGA [30,86,87] suggests that more investigations of the characteristics and properties of NAGA-based gels may be expected in the future. The only previous work discovered to observe the thermophilic behavior of NAGA-based gels was with IPNs by Sasase et al. [69]. Based on the observation of a VPTT of 35 °C for hydrogen bonded complexes of PNAGA and PAA in solution, sequential IPNs were synthesized incorporating BMA. Poly (NAGA-co-BMA)/PAA IPNs were synthesized and showed increased swelling with heating. Similar to other gels, the swelling ratio decreased at higher BMA content. These IPNs were compared to poly(Am-co-BMA)/PAA IPNs. When NAGA was substituted for Am in the hydrogel, the extent of swelling was lesser and the VPTT was higher. These results suggested that NAGA groups were capable of forming stronger hydrogen bonds than Am groups with AA groups.
The early works by Haas et al. did not actually determine volume-phase transition for PNAGA polymers. Thermo-reversible sol-gel gelation driven by randomly distributed hydrogen bonding was all that was observed. PNAGA is one of the rare temperature responsive polymers where thermophilic behavior was observed with the hydrogel prior to being characterized as single chains in solution. The more recent works are inspired by the work of Seuring et al. exhibiting the positive volume-phase transition of PNAGA in water [30]. The significant difference between a polymer in water undergoing sol-gel thermo-gelation versus thermophilic behavior in a hydrogel is that phase separation occurs below the VPTT for the sol-gel. The novel aspect of PNAGA having a VPTT in water is that it does so as a homopolymer and it is non-ionic. All the temperature responsive hydrogels that have been discussed so far are either ionic, which complicates their functionality in physiological and environmental milieu, or multiple monomer specie networks. Similar to the polysulfobetaines previously discussed, the pendant chains of PNAGA are capable of forming both inter and intra complexes without requirement of an additional hydrogen acceptor monomer species. The primary amide acts as the hydrogen donor and the carbonyl acts as the hydrogen acceptor. The VPTT was determined to be ~22.5 °C upon heating and ~12.3 °C upon cooling by a turbidity photometer at 670 nm at a heating rate of 1 °C min−1. A later publication by Seuring et al. investigated why thermophilic behavior for PNAGA had not been observed while it had been know to undergo thermo-reversible gelation for near half a century [87]. It was determined that any ionized groups in the polymer prevent phase separation, ionic groups can be introduced unintentionally by either “acrylate impurities in the monomer, hydrolysis of the polymer side chains, and/or usage of ionic initiators or chain transfer agents”. This work presents explanation for alternate synthesis attempts to polymerize PNAGA [86]. RAFT polymerization is attractive to many polymer scientists as is can be used to control the chain length of the polymers synthesized. It was observed that PNAGA synthesized in this method did not show a VPTT in water but instead reflected the work of Haas et al. The PNAGA that did show a VPTT in water was synthesized by free radical initiation with azo-bis-isobutyronitrile (AIBN).
The work by Seuring et al. was extended to examine NAGA in copolymerization with N-Acetalacrylamide (NAcAm) [30], an hydrogen acceptor, to investigate the nature of the hydrogen bonds proliferated by NAGA. NAcAm was chosen as it is similar in structure to NAGA but without the terminal primary amide. This structure difference, although showing both hydrogen donating and accepting functionalities, did not display VPTT behavior in water. Copolymers with a NAGA mol fraction of 0.645 or lower did not exhibit a VPTT either, although at higher NAGA mol fractions in the copolymer, the transition of solution to gel became smoother, less abrupt. This shows that the inter and intrachain associations of the NAGA pendant groups are stronger than the interchain associations with other species since VPTT increases with higher NAGA content in the copolymer and the transition becomes slower.
Following the example of characterizing previously discovered polymers, Glatzel et al., attempted RAFT polymerization of N-Acryloylasparaginamide (NAAm), a monomer unit again similar to NAGA but with two primary amides instead of one (Figure 11).
It was thought that by increasing the amide density, thermophilic polymers would result from a RAFT polymerization process. In addition, the water solubility of the NAAm monomer suggested strongly that the polymer would undergo volume-phase transition. Although this work presented another novel characterization of a thermophilic polymer, it is a rudimentary first look into the volume-phase transition of polyNAAm in water and there is much work between this and development of a functional hydrogel.
Figure 11.
N-Acryloylasparaginamide.

6. Conclusions
Volume-phase transition, in any case, is a balance between attractive and repulsive intermolecular forces. For a definitive transition, one set of forces must be dominant and in the case of thermo-responsive hydrogels, the forces must be sensitive to changes in temperature. As discussed in the preceding sections, hydrogen bonding, certain electrostatic interactions and certain van der Waals forces can be influenced by temperature change. Water-soluble thermophilic polymer gels require that any attractive polymer-polymer interactions formed at ambient temperature are be stronger than any polymer-solvent interactions. These polymer-polymer interactions must also be weak enough to be disrupted by temperature. The design constraints of thermophilic hydrogels have limited research to exploration on only a few unique hydrogel chemistries. Early in the development of thermo-responsive hydrogels, both positive and negative volume-phase transition materials received similar research interest. Negative volume-phase transition hydrogels became the mainstream thermo-responsive hydrogel, as more substance variation was possible. Although less numerous, work involving positive volume-phase transition hydrogels has mirrored that of their vastly more popular counterpart finding similar application and utility. While researchers attempt to modify the composition or microstructure of thermophobic hydrogels to fit special applications, many issues may be resolved by substituting for thermophilic hydrogels. New studies identifying thermophilic behavior in polymers, such as the works of Seuring et al., shown in previous sections mostly feature polymers from works on solution properties. Control of the VPTT is important for any application of thermo-responsive hydrogel. Creating membranes with temperature sensitive properties often requires that the coated or grafted hydrogel maintain a stable chemistry and physiology before and after the volume-phase transition. Future work on thermophilic hydrogels will most likely follow the development path already set by work on thermophobic hydrogels. Optimization of VPTT by chemical composition, gel microstructure, polymer microstructure and monomer sequence are all aspects that should be considered.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mah, K.Z.; Ghosh, R. Paper-based composite lyotropic salt-responsive membranes for chromatographic separation of proteins. J. Membr. Sci. 2010, 360, 149–154. [Google Scholar] [CrossRef]
- Yu, D.; Chen, X.; Pelton, R.; Ghosh, R. Paper-PEG-based membranes for hydrophobic interaction chromatography: Purification of monoclonal antibody. Biotechnol. Bioeng. 2008, 99, 1434–1442. [Google Scholar] [CrossRef]
- Kuroki, H.; Ito, T.; Ohashi, H.; Tamaki, T.; Yamaguchi, T. Biomolecule-recognition gating membrane using biomolecular cross-linking and polymer phase transition. Anal. Chem. 2011, 83, 9226–9229. [Google Scholar] [CrossRef]
- Rattan, S.; Sehgal, T. Stimuli-responsive membranes through peroxidation radiation-induced grafting of 2-hydroxyethyl methacrylate (2-HEMA) onto isotactic polypropylene film (IPP). J. Radioanal. Nuclear Chem. 2012, 293, 107–118. [Google Scholar] [CrossRef]
- Zhao, Y.-H.; Wee, K.-H.; Bai, R. A novel electrolyte-responsive membrane with tunable permeation selectivity for protein purification. ACS Appl. Mater. Interfaces 2009, 2, 203–211. [Google Scholar] [CrossRef]
- Chu, L.-Y.; Niitsuma, T.; Yamaguchi, T.; Nakao, S.-i. Thermoresponsive transport through porous membranes with grafted PNIPAM gates. AICHE J. 2003, 49, 896–909. [Google Scholar] [CrossRef]
- Liang, L.; Feng, X.; Peurrung, L.; Viswanathan, V. Temperature-sensitive membranes prepared by UV photopolymerization of N-isopropylacrylamide on a surface of porous hydrophilic polypropylene membranes. J. Membr. Sci. 1999, 162, 235–246. [Google Scholar] [CrossRef]
- Park, Y.S.; Ito, Y.; Imanishi, Y. Permeation control through porous membranes immobilized with thermosensitive polymer. Langmuir 1998, 14, 910–914. [Google Scholar] [CrossRef]
- Ying, L.; Kang, E.T.; Neoh, K.G. Synthesis and characterization of poly(N-isopropylacrylamide)-graft-poly(vinylidene fluoride) copolymers and temperature-sensitive membranes. Langmuir 2002, 18, 6416–6423. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, X.Y. Temperature and pH-responsive polymeric composite membranes for controlled delivery of proteins and peptides. Biomaterials 2004, 25, 5281–5291. [Google Scholar] [CrossRef]
- Kwon, I.C.; Bae, Y.H.; Kim, S.W. Electrically credible polymer gel for controlled release of drugs. Nature 1991, 354, 291–293. [Google Scholar] [CrossRef]
- Ly, Y.; Cheng, Y.-L. Electrically-modulated variable permeability liquid crystalline polymeric membrane. J. Membr. Sci. 1993, 77, 99–112. [Google Scholar] [CrossRef]
- Tanaka, T.; Nishio, I.; Sun, S.-T.; Ueno-Nishio, S. Collapse of gels in an electric field. Science 1982, 218, 467–469. [Google Scholar]
- Briscoe, B.; Luckham, P.; Zhu, S. On the effects of water solvency towards non–ionic polymers. Proc. Math. Phys. Eng. Sci. 1999, 455, 737–756. [Google Scholar] [CrossRef]
- Tanaka, T. Collapse of gels and the critical endpoint. Phys. Rev. Lett. 1978, 40, 820–823. [Google Scholar] [CrossRef]
- Kontturi, K.; Mafé, S.; Manzanares, J.A.; Svarfvar, B.L.; Viinikka, P. Modeling of the salt and pH effects on the permeability of grafted porous membranes. Macromolecules 1996, 29, 5740–5746. [Google Scholar] [CrossRef]
- Schulz, D.N.; Peiffer, D.G.; Agarwal, P.K.; Larabee, J.; Kaladas, J.J.; Soni, L.; Handwerker, B.; Garner, R.T. Phase behavior and solution properties of sulphobetaine polymers. Polymer 1986, 27, 1734–1742. [Google Scholar] [CrossRef]
- Siegel, R.A.; Firestone, C.A. pH-dependent equilibrium swelling properties of hydrophobic polyelectrolyte copolymer gels. Macromolecules 1988, 21, 3254–3259. [Google Scholar] [CrossRef]
- Zhou, X.; Weng, L.; Chen, Q.; Zhang, J.; Shen, D.; Li, Z.; Shao, M.; Xu, J. Investigation of pH sensitivity of poly(acrylic acid-co-acrylamide) hydrogel. Polym. Int. 2003, 52, 1153–1157. [Google Scholar] [CrossRef]
- Cartier, S.; Horbett, T.A.; Ratner, B.D. Glucose-sensitive membrane coated porous filters for control of hydraulic permeability and insulin delivery from a pressurized reservoir. J. Membr. Sci. 1995, 106, 17–24. [Google Scholar] [CrossRef]
- Chu, L.-Y.; Li, Y.; Zhu, J.-H.; Wang, H.-D.; Liang, Y.-J. Control of pore size and permeability of a glucose-responsive gating membrane for insulin delivery. J. Control. Release 2004, 97, 43–53. [Google Scholar] [CrossRef]
- Chu, L.; Xie, R.; Ju, X. Stimuli-responsive membranes: Smart tools for controllable mass-transfer and separation processes. Chin. J. Chem. Eng. 2011, 19, 891–903. [Google Scholar] [CrossRef]
- Jeon, G.; Yang, S.Y.; Kim, J.K. Functional nanoporous membranes for drug delivery. J. Mater. Chem. 2012, 22, 14814. [Google Scholar] [CrossRef]
- Flory, P.J. Priciples of Polymer Chemistry; Cornell University Press: Ithica, NY, USA, 1953. [Google Scholar]
- Rees, D.A. Polysaccharide Shapes; Chapman and Hall: London, UK, 1977. [Google Scholar]
- Tanaka, T. Phase Transitions of Gels. In Polyelectrolyte Gels; American Chemical Society: Cambridge, MA, USA, 1992; pp. 1–21. [Google Scholar]
- Azzaroni, O.; Brown, A.A.; Huck, W.T.S. UCST wetting transitions of polyzwitterionic brushes driven by self-association. Angew. Chem. 2006, 118, 1802–1806. [Google Scholar] [CrossRef]
- Georgiev, G.S.; Mincheva, Z.P.; Georgieva, V.T. Temperature-sensitive polyzwitterionic gels. Macromol. Symp. 2001, 164, 301–312. [Google Scholar] [CrossRef]
- Katono, H.; Maruyama, A.; Sanui, K.; Ogata, N.; Okano, T.; Sakurai, Y. Thermo-responsive swelling and drug release switching of interpenetrating polymer networks composed of poly(acrylamide-co-butyl methacrylate) and poly (acrylic acid). J. Control. Release 1991, 16, 215–227. [Google Scholar] [CrossRef]
- Seuring, J.; Agarwal, S. Non-ionic homo- and copolymers with h-donor and h-acceptor units with an UCST in water. Macromol. Chem. Phys. 2010, 211, 2109–2117. [Google Scholar] [CrossRef]
- Tanaka, T.; Sun, S.-T.; Nishio, I.; Swislow, G.; Shah, A. Phase transitions in ionic gels. Phys. Rev. Lett. 1980, 45, 1636–1639. [Google Scholar] [CrossRef]
- Malcolm, G.N.; Rowlinson, J.S. Thermodynamic properties of aqueous solutions of polyethylene glycol, polypropylene glycol and dioxane. Trans. Faraday Soc. 1957, 53, 921–931. [Google Scholar] [CrossRef]
- Heskins, M.; Guillet, J.E. Solution properties of poly(N-isopropylacrylamide). J. Macromol. Sci. Part A 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Haas, H.C.; Schuler, N.W. Thermally reversible homopolymer gel systems. J. Polym. Sci. Part B 1964, 2, 1095–1096. [Google Scholar] [CrossRef]
- Schild, H.G. Poly(N-isopropylacrylamide): Experiment, theory and application. Progr. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Aoki, T.; Kawashima, M.; Katono, H.; Sanui, K.; Ogata, N.; Okano, T.; Sakurai, Y. Temperature-responsive interpenetrating polymer networks constructed with poly(acrylic acid) and poly(N,N-dimthylacrylamide). Macromolecules 1994, 27, 947–952. [Google Scholar] [CrossRef]
- Gil, E.S.; Hudson, S.M. Stimuli-reponsive polymers and their bioconjugates. Progr. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Nath, N.; Chilkoti, A. Creating “smart” surfaces using stimuli responsive polymers. Adv. Mater. 2002, 14, 1243–1247. [Google Scholar] [CrossRef]
- Urban, A.M.; Urban, M.W. Stimuli-Responsive Macromolecules and Polymeric Coatings. In Stimuli-ReponsivePolymeric Films and Coatings; Urban, M.W., Ed.; American Chemical Society: Washington, DC, USA, 2005; pp. 1–25. [Google Scholar]
- Yalkowsky, S.H. Solubility and Solubilization in Aqueous Media; American Chemical Society: Washington, DC, USA, 1999; Volume 1, p. 480. [Google Scholar]
- Tanaka, T. Phase transitions in gels and a single polymer. Polymer 1979, 20, 1404–1412. [Google Scholar] [CrossRef]
- Matsuo, E.S.; Tanaka, T. Kinetics of discontinuous volume-phase transition of gels. J. Chem. Phys. 1988, 89, 1695–1703. [Google Scholar] [CrossRef]
- Rubinstein, M.; Dobrynin, A.V. Associations leading to formation of reversible networks and gels. Curr. Opin. Colloid Interface Sci. 1999, 4, 83–87. [Google Scholar] [CrossRef]
- Day, J.; Robb, I. Thermodynamic parameters of polyacrylamides in water. Polymer 1981, 22, 1530–1533. [Google Scholar] [CrossRef]
- Eisenberg, D.S.; Kauzmann, W. The Structure and Properties of Water. In Oxford Classic Texts in the Physical Sciences; Clarendon Press: Oxford, UK, 2005. [Google Scholar]
- Garay, M.T.; Llamas, M.C.; Iglesias, E. Study of polymer-polymer complexes and blends of poly(N-isopropylacrylamide) with poly(carboxylic acid): 1. Poly(acrylic acid) and poly(methacrylic acid). Polymer 1997, 38, 5091–5096. [Google Scholar] [CrossRef]
- Moelbert, S.; de Los Rios, P. Hydrophobic interaction model for upper and lower critical solution temperatures. Macromolecules 2003, 36, 5845–5853. [Google Scholar] [CrossRef]
- Lowe, A.B.; McCormick, C.L. Synthesis and solution properties of zwitterionic polymers. Chem. Rev. 2002, 102, 4177–4190. [Google Scholar] [CrossRef]
- Soto, V.M.M.; Galin, J.C. Poly(sulphopropylbetaines): 2. Dilute solution properties. Polymer 1984, 25, 254–262. [Google Scholar] [CrossRef]
- Kuo, W.H.; Wang, M.J.; Chien, H.W.; Wei, T.C.; Lee, C.; Tsai, W.B. Surface modification with poly(sulfobetaine methacrylate-co-acrylic acid) to reduce fibrinogen adsorption, platelet adhesion, and plasma coagulation. Biomacromolecules 2011, 12, 4348–4356. [Google Scholar] [CrossRef]
- Weaver, J.V.M.; Armes, S.P.; Bütün, V. Synthesis and aqueous solution properties of a well-defined thermo-responsive schizophrenic diblock copolymer. Chem. Commun. 2002, 18, 2122–2123. [Google Scholar] [CrossRef]
- Zhang, Z.; Chao, T.; Chen, S.; Jiang, S. Superlow fouling sulfobetaine and carboxybetaine polymers on glass slides. Langmuir 2006, 22, 10072–10077. [Google Scholar] [CrossRef]
- Huglin, M.B.; Radwan, M.A. Unperturbed dimensions of a zwitterionic polymethacrylate. Polym. Int. 1991, 26, 97–104. [Google Scholar] [CrossRef]
- Kamenova, I.; Harrass, M.; Lehmann, B.; Friedrich, K.; Ivanov, I.; Georgiev, G. Swelling of the zwitterionic copolymer networks and dehydration of their hydrogels. Macromol. Symp. 2007, 254, 122–127. [Google Scholar]
- Friedman, H.L. Kinetics of thermal degradation of char-forming plastics from thermogravimetry-application to a phenolic resin. J. Polym. Sci. 1964, 6C, 183–195. [Google Scholar]
- Kasák, P.; Kroneková, Z.; Krupa, I.; Lacík, I. Zwitterionic hydrogels crosslinked with novel zwitterionic crosslinkers: Synthesis and characterization. Polymer 2011, 52, 3011–3020. [Google Scholar] [CrossRef]
- Chang, Y.; Yandi, W.; Chen, W.Y.; Shih, Y.J.; Yang, C.C.; Chang, Y.; Ling, Q.D.; Higuchi, A. Tunable bioadhesive copolymer hydrogels of thermoresponsive poly(N-isopropyl acrylamide) containing zwitterionic polysulfobetaine. Biomacromolecules 2010, 11, 1101–1110. [Google Scholar] [CrossRef]
- Singhal, R.; Tomar, R.; Nagpal, A. Effect of cross-linker and initiator concentration on the swelling behavior and network parameters of superabsorbent hydrogels based on acrylamide and acrylic acid. Int. J. Plast. Technol. 2009, 13, 22–37. [Google Scholar] [CrossRef]
- Xie, J.; Liu, X.; Liang, J.; Luo, Y. Swelling properties of superabsorbent poly(acrylic acid-co-acrylamide) with different crosslinkers. J. Appl. Polym. Sci. 2009, 112, 602–608. [Google Scholar] [CrossRef]
- Baker, B.A.; Murff, R.L.; Milam, V.T. Tailoring the mechanical properties of polyacrylamide-based hydrogels. Polymer 2010, 51, 2207–2214. [Google Scholar] [CrossRef]
- Zhu, X.F.; Yang, M.; Zhang, H.X.; Nie, Y.J. The synthesis and characteristic properties of poly (AAc-co-AAm). Adv. Mater. Res. 2011, 213, 534–538. [Google Scholar] [CrossRef]
- Klenina, O.V.; Fain, E.G. Phase separation in the system polyacrylic acid-polyacrylamide-water. Polym. Sci. USSR 1981, 23, 1439–1446. [Google Scholar] [CrossRef]
- Osada, Y. Equilibrium study of polymer–polymer complexation of poly(methacrylic acid) and poly(acrylic acid) with complementary polymers through cooperative hydrogen bonding. J. Polym. Sci. Polym. Chem. Ed. 1979, 17, 3485–3498. [Google Scholar] [CrossRef]
- Painter, P.C.; Graf, J.; Coleman, M.M. A lattice model describing hydrogen bonding in polymer mixtures. J. Chem. Phys. 1990, 92, 6166–6174. [Google Scholar] [CrossRef]
- Klenina, O.V.; Fain, E.G. Phase separation in the hydrolyzed polyacrylamide-water-hydrochloric acid system. Kolloidnyi Zhurnal 1980, 42, 558–561. [Google Scholar]
- Ilmain, F.; Tanaka, T.; Kokufuta, E. Volume transition in a gel driven by hydrogen bonding. Nature 1991, 349, 400–401. [Google Scholar] [CrossRef]
- Cecil, R. Model system for hydrophobic interactions. Nature 1967, 214, 369–370. [Google Scholar] [CrossRef]
- Yang, M.; Liu, C.; Li, Z.; Gao, G.; Liu, F. Temperature-responsive properties of poly(acrylic acid-co-acrylamide) hydrophobic association hydrogels with high mechanical strength. Macromolecules 2010, 43, 10645–10651. [Google Scholar] [CrossRef]
- Sasase, H.; Aoki, T.; Katono, H.; Sanui, K.; Ogata, N.; Ohta, R.; Kondo, T.; Okano, T.; Sakurai, Y. Regulation of temperature-response swelling behavior of interpenetrating polymer networks composed of hydrogen bonding polymers. Die Makromol. Chem. Rapid Commun. 1992, 13, 577–581. [Google Scholar] [CrossRef]
- Katono, H.; Sanui, K.; Ogata, N.; Okano, T.; Sakurai, Y. Drug release off behavior and deswelling kinetics of thermo-responsive IPNs composed of poly(acrylamide-co-butyl methacrylate) and poly(acrylic acid). Polym. J. 1991, 23, 1179–1189. [Google Scholar] [CrossRef]
- Singhal, R.; Gupta, I. A study on the effect of butyl methacrylate content on swelling and controlled-release behavior of poly (acrylamide-co-butyl-methacrylate-co-acrylic acid) environment-responsive hydrogels. Int. J. Polym. Mater. 2010, 59, 757–776. [Google Scholar] [CrossRef]
- Shibanuma, T.; Aoki, T.; Sanui, K.; Ogata, N.; Kikuchi, A.; Sakurai, Y.; Okano, T. Thermosensitive phase-separation behavior of poly(acrylic acid)-graft-poly(N,N-dimethylacrylamide) aqueous solution. Macromolecules 1999, 33, 444–450. [Google Scholar]
- McCormick, C.L.; Sumerlin, B.S.; Lokitz, B.S.; Stempka, J.E. RAFT-synthesized diblock and triblock copolymers: Thermally-induced supramolecular assembly in aqueous media. Soft Matter 2008, 8, 1760–1773. [Google Scholar]
- Duran, S.; Solpan, D.; Güven, O. Synthesis and characterization of acrylamide-acrylic acid hydrogels and adsorption of some textile dyes. Nuclear Instrum. Methods Phys. Res. Sect. B 1999, 151, 196–199. [Google Scholar] [CrossRef]
- Katime, I.; Novoa, R.; de Apodaca, E.D.; Mendizábal, E.; Puig, J. Theophylline release from poly(acrylic acid-co-acrylamide) hydrogels. Polym. Test. 1999, 18, 559–566. [Google Scholar] [CrossRef]
- Chu, L.-Y.; Li, Y.; Zhu, J.-H.; Chen, W.-M. Negatively thermoresponsive membranes with functional gates driven by zipper-type hydrogen-bonding interactions. Angew. Chem. Int. Ed. 2005, 44, 2124–2127. [Google Scholar] [CrossRef]
- Hochberg, A.; Tanaka, T.; Nicoli, D. Spinodal line and critical point of an acrylamide gel. Phys. Rev. Lett. 1979, 43, 217–219. [Google Scholar] [CrossRef]
- Haas, H.C.; Moreau, R.D.; Schuler, N.W. Synthetic thermally reversible gel systems. II. J. Polym. Sci. Polym. Phys. Ed. 1967, 5, 915–927. [Google Scholar]
- Haas, H.C.; Chiklis, C.K.; Moreau, R.D. Synthetic thermally reversible gel systems. III. J. Polym. Sci. Part A 1970, 8, 1131–1145. [Google Scholar] [CrossRef]
- Haas, H.C.; MacDonald, R.L.; Schuler, A.N. Synthetic thermally reversible gel systems. IV. J. Polym. Sci. Part A 1970, 8, 1213–1226. [Google Scholar] [CrossRef]
- Haas, H.C.; Manning, M.J.; Mach, M.H. Synthetic thermally reversible gel systems. V. J. Polym. Sci. Part A 1970, 8, 1725–1730. [Google Scholar] [CrossRef]
- Haas, H.C.; MacDonald, R.L.; Schuler, A.N. Synthetic thermally reversible gel systems. VI. J. Polym. Sci. Part A 1970, 8, 3405–3415. [Google Scholar] [CrossRef]
- Seuring, J.; Agarwal, S.; Harms, K. N-Acryloyl glycinamide. Acta Crystallogr. Sect. E 2011, 67, o2170. [Google Scholar] [CrossRef]
- Nagaoka, H.; Ohnishi, N.; Eguchi, M. Thermoresponsive Polymer and Production Method Thereof; Chisso Corporation: Alexandria, VA, USA, 2007. [Google Scholar]
- Ohnishi, N.; Furukawa, H.; Kataoka, K.; Ueno, K. Polymer Having an Upper Critical Solution Temperature; Nathional Institute of Advanced Industrial Science and Technology, Chisso Corporation: Alexandria, VA, USA, 2007. [Google Scholar]
- Glatzel, S.; Badi, N.; Päch, M.; Laschewsky, A.; Lutzm, J.-F. Well-defined synthetic polymers with a protein-like gelation behavior in water. Chem. Commun. 2010, 46, 4517–2519. [Google Scholar] [CrossRef]
- Seuring, J.; Bayer, F.M.; Huber, K.; Agarwal, S. Upper critical solution temperature of poly(N-acryloyl glycinamide) in water: A concealed property. Macromolecules 2011, 44, 8962–8971. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Mah, E.; Ghosh, R. Thermo-Responsive Hydrogels for Stimuli-Responsive Membranes. Processes 2013, 1, 238-262. https://0-doi-org.brum.beds.ac.uk/10.3390/pr1030238
AMA Style
Mah E, Ghosh R. Thermo-Responsive Hydrogels for Stimuli-Responsive Membranes. Processes. 2013; 1(3):238-262. https://0-doi-org.brum.beds.ac.uk/10.3390/pr1030238
Chicago/Turabian StyleMah, Evan, and Raja Ghosh. 2013. "Thermo-Responsive Hydrogels for Stimuli-Responsive Membranes" Processes 1, no. 3: 238-262. https://0-doi-org.brum.beds.ac.uk/10.3390/pr1030238