
A Comprehensive Review of Recent Advancements in Cancer Immunotherapy and Generation of CAR T Cell by CRISPR-Cas9

 ,
,  ,
,  ,
,  , ,
, ,  ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
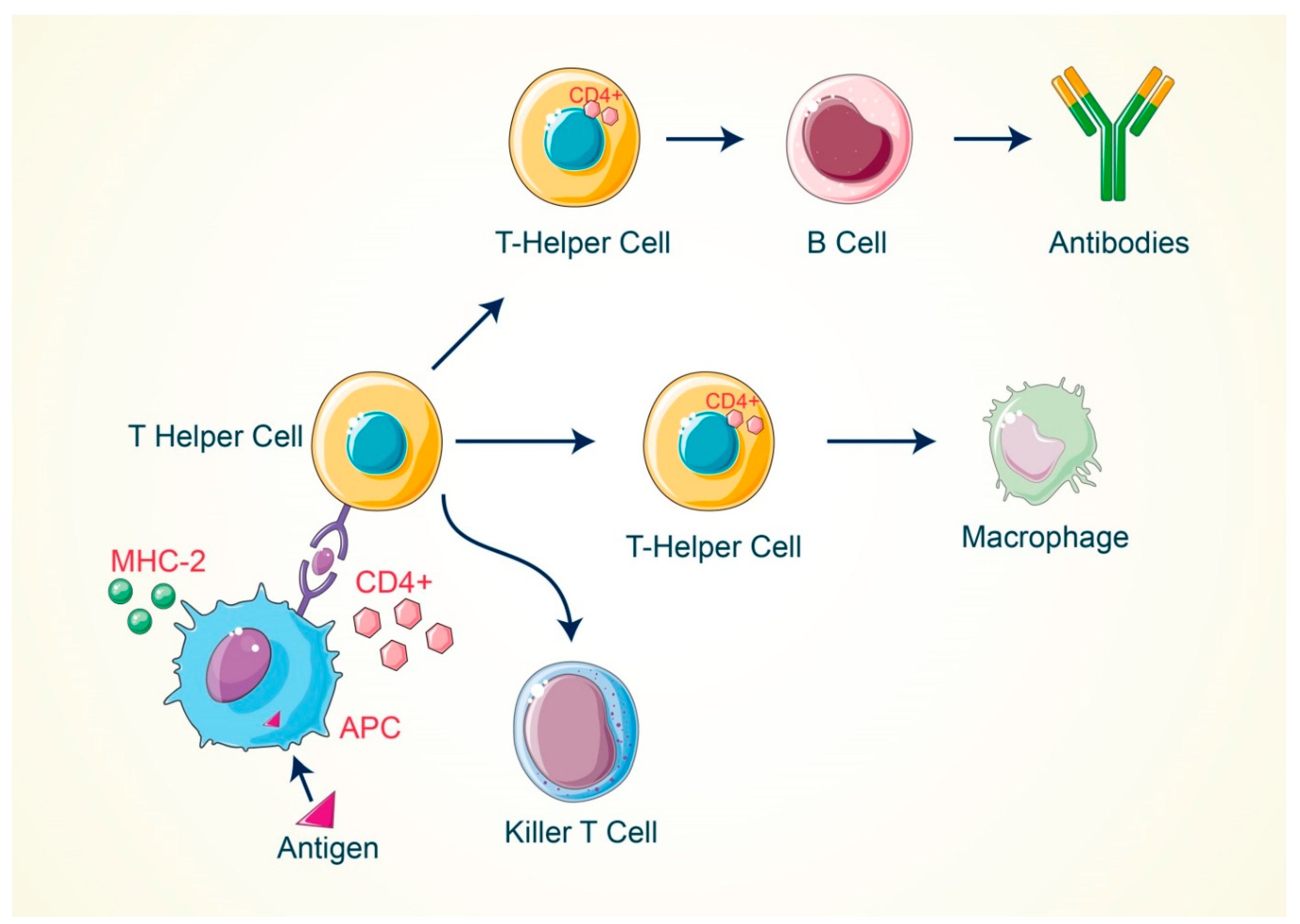
2. Activation of T Cell by Antigenic Response
3. Significance of T Cell in Cancer Treatment
4. The Causes of T Cell Failure for Recognizing Cancer Antigens
5. The Current Immunotherapies That Are Used in Cancer Treatment
5.1. Adoptive Cell Therapy
5.1.1. Tumor-Infiltrating Lymphocyte (TIL)
5.1.2. Engineered T-Cell Receptor (TCR) Therapy
5.1.3. Engineered Natural Killer (NK)-Cell Therapy
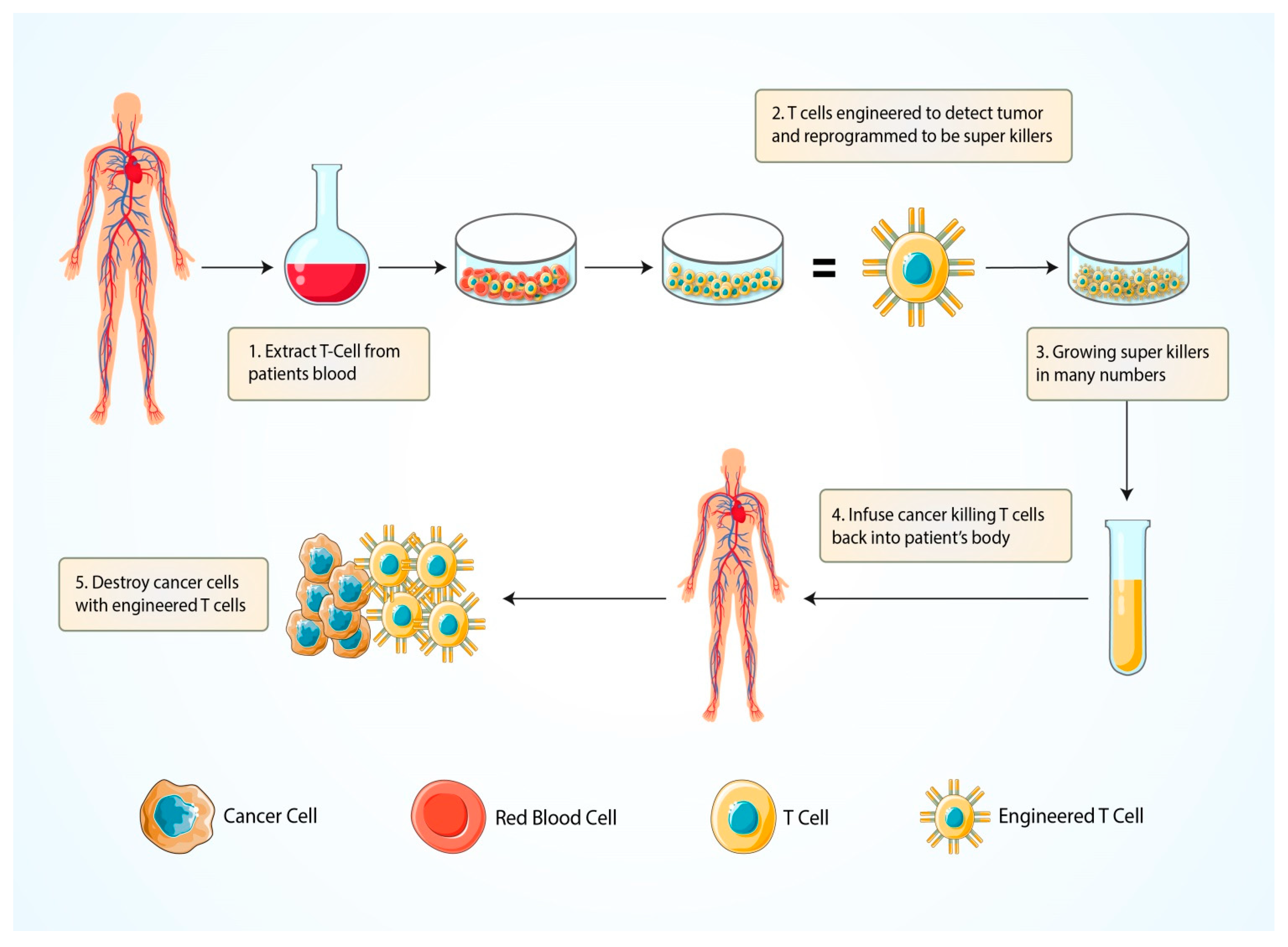
5.1.4. Chimeric Antigen Receptor (CAR) T-Cell Therapy
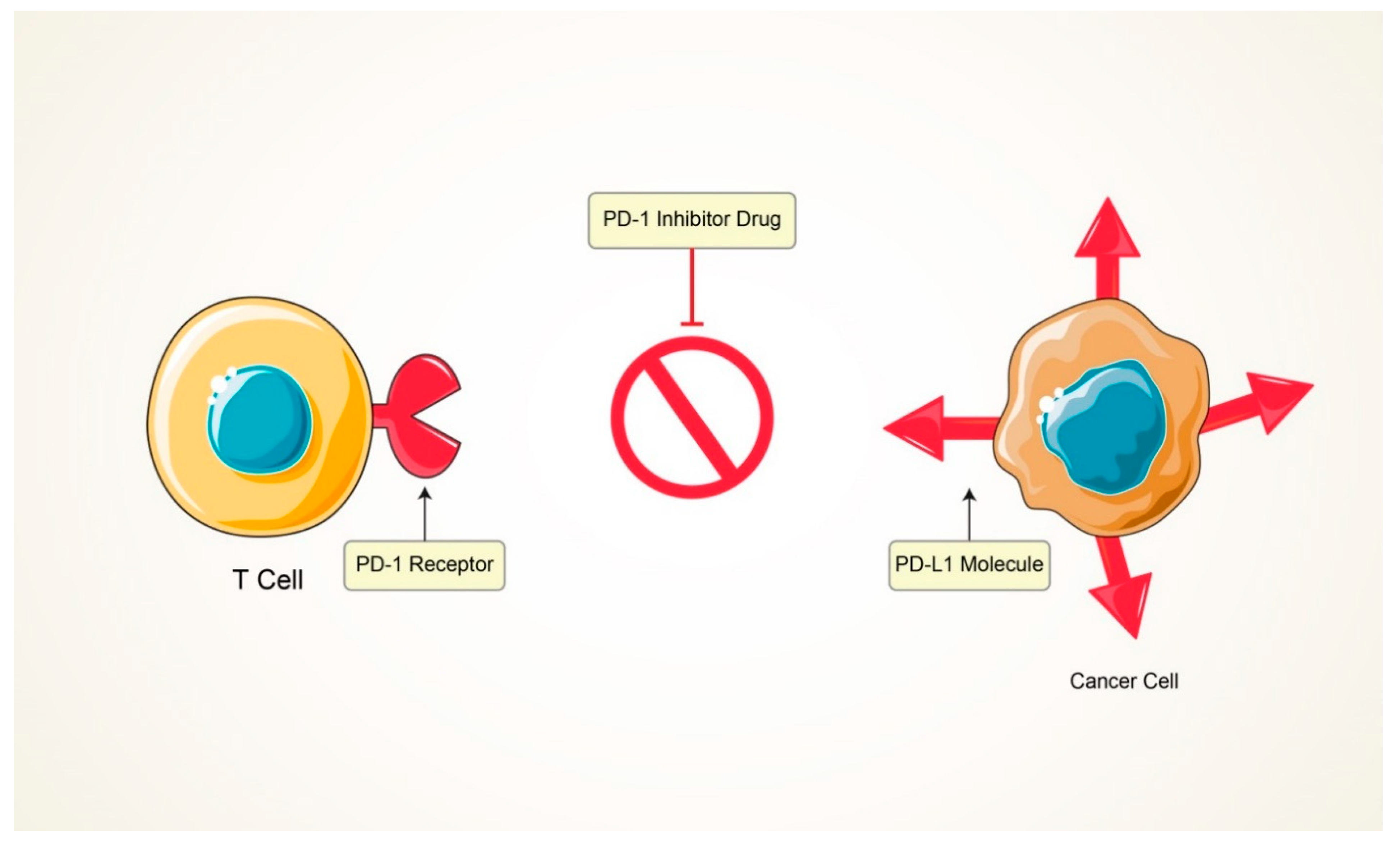
5.2. Immunomodulators
5.3. Antibody-Mediated Therapy
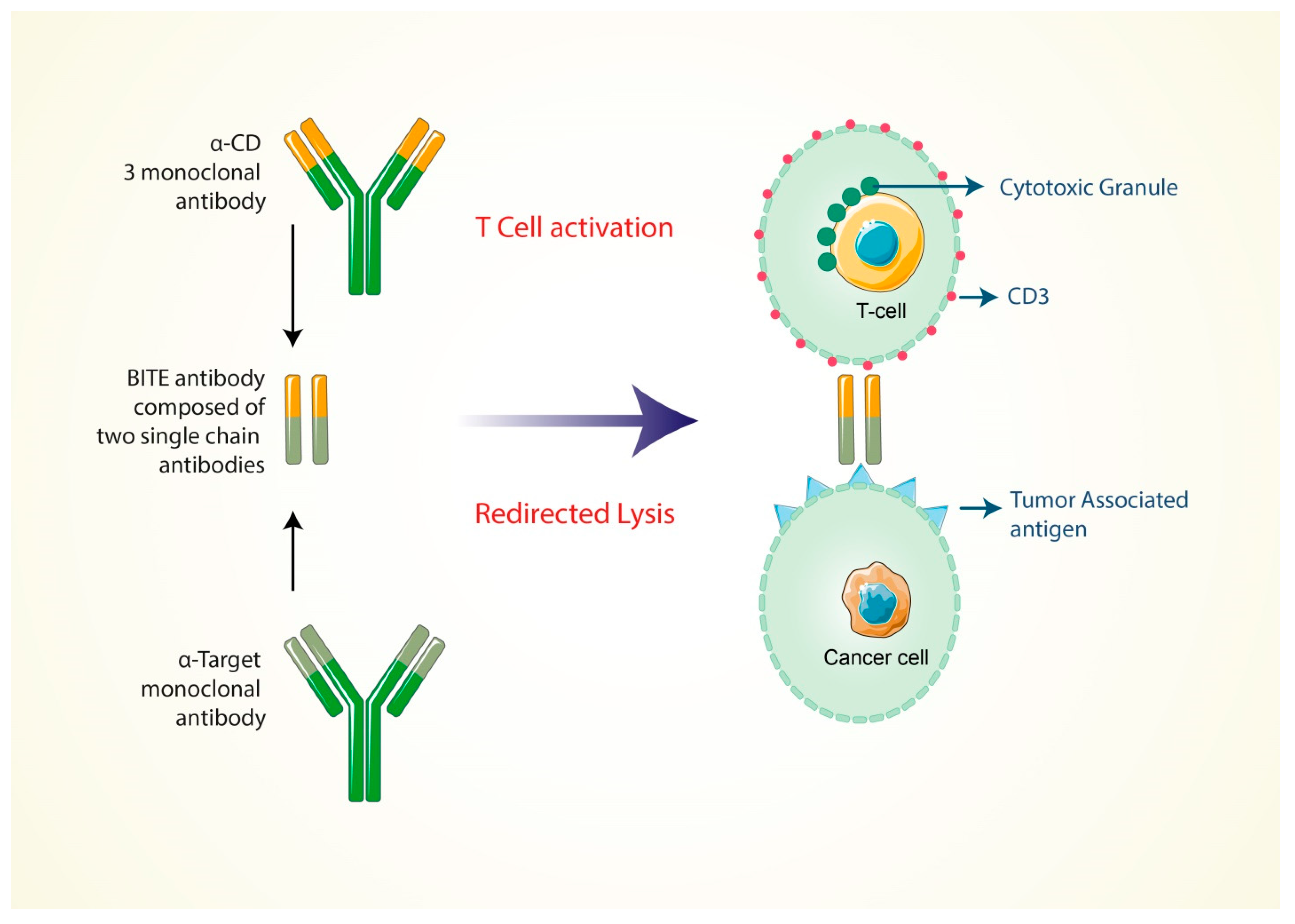
5.4. Formation of Bi-Specific T Cell-Engaging Antibodies for Cancer Therapy
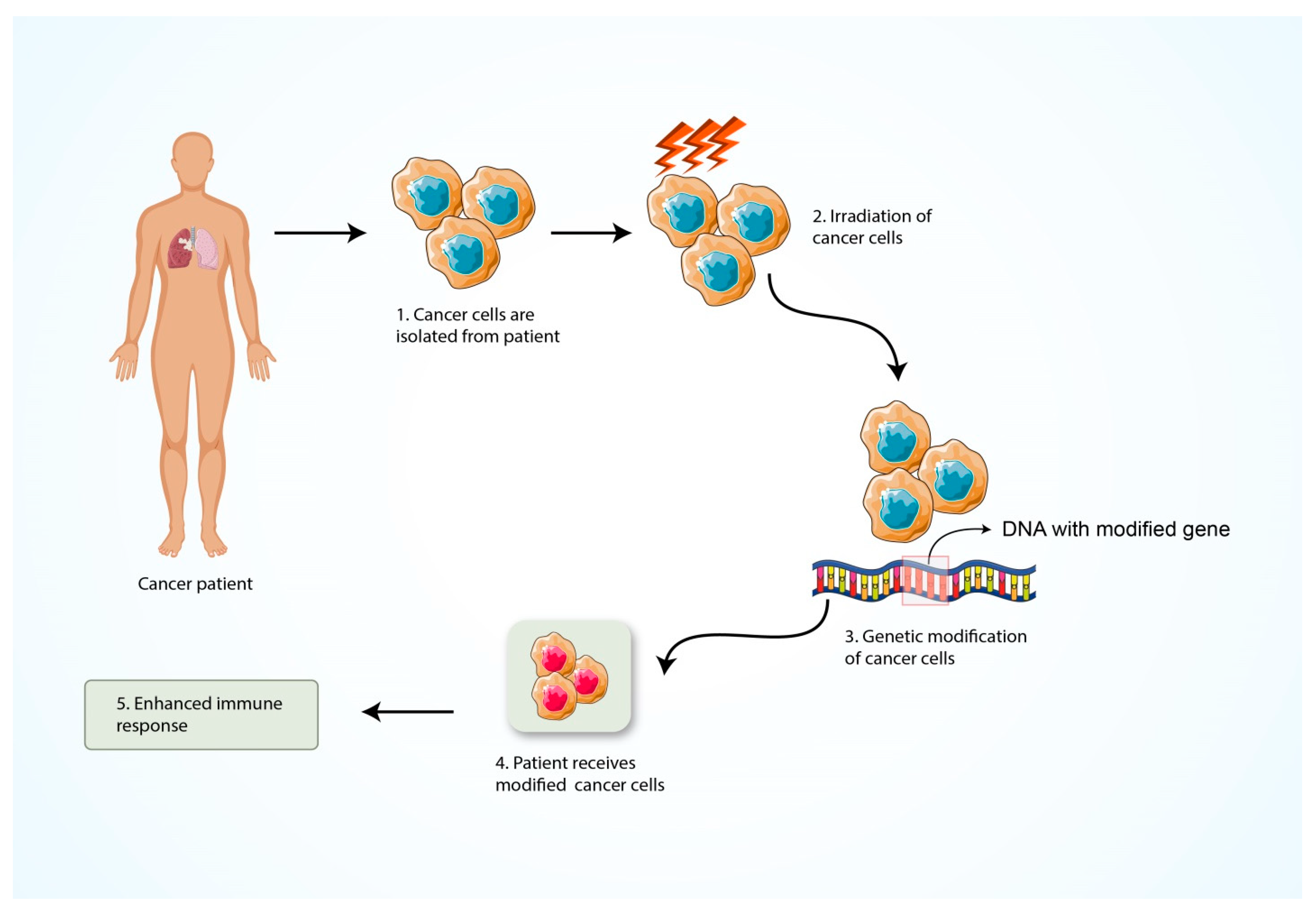
5.5. Formation of Cancer Vaccine
5.6. Nanoparticle-Based Cancer Immunotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigens | Type of Cancer They Cause | References |
|---|---|---|
| NY-ESO-1 | Esophageal Squamous Cell carcinoma | [138] |
| MAGEA-A3 | Melanoma | [139] |
| WT1 | Acute Myelocytic leukemia | [140] |
| hTERT | Viral mediated Cancer | [141] |
| Tyrosinase | Brain and Skin Cancer | [142] |
| gp 100 | Melanoma | [143] |
| MART-1 | Melanoma | [143] |
| Melan A | Melanoma | [143] |
| β catenin | Melanoma | [143] |
| MUC1 | Breast Cancer | [144] |
| CEA | Colon Cancer, Lung Cancer | [145] |
| Mam-A | Breast Cancer | [146] |
| Sialyl-Tn | Breast, Gastric, Lung, Colon, Esophageal, Prostate and Endometrial Cancer | [147] |
| α-fetoprotein | Hepatic Cancer | [148] |
| CA-125 | Ovarian Cancer | [149] |
| Ras, Src | Exhibited in Several Cancer Types | [150] |
| Mesothelin | Malignant Pleural Mesothelioma, Ovarian and Pancreatic Cancer | [151] |
| PSMA | Prostate Cancer | [151] |
| TPD52 | Prostate, Breast and Ovarian Cancer | [151] |
| PSA | Prostate Cancer | [151] |
| PAP | Prostate Cancer | [151] |
5.7. Inhibition of T Cell Exhaustion
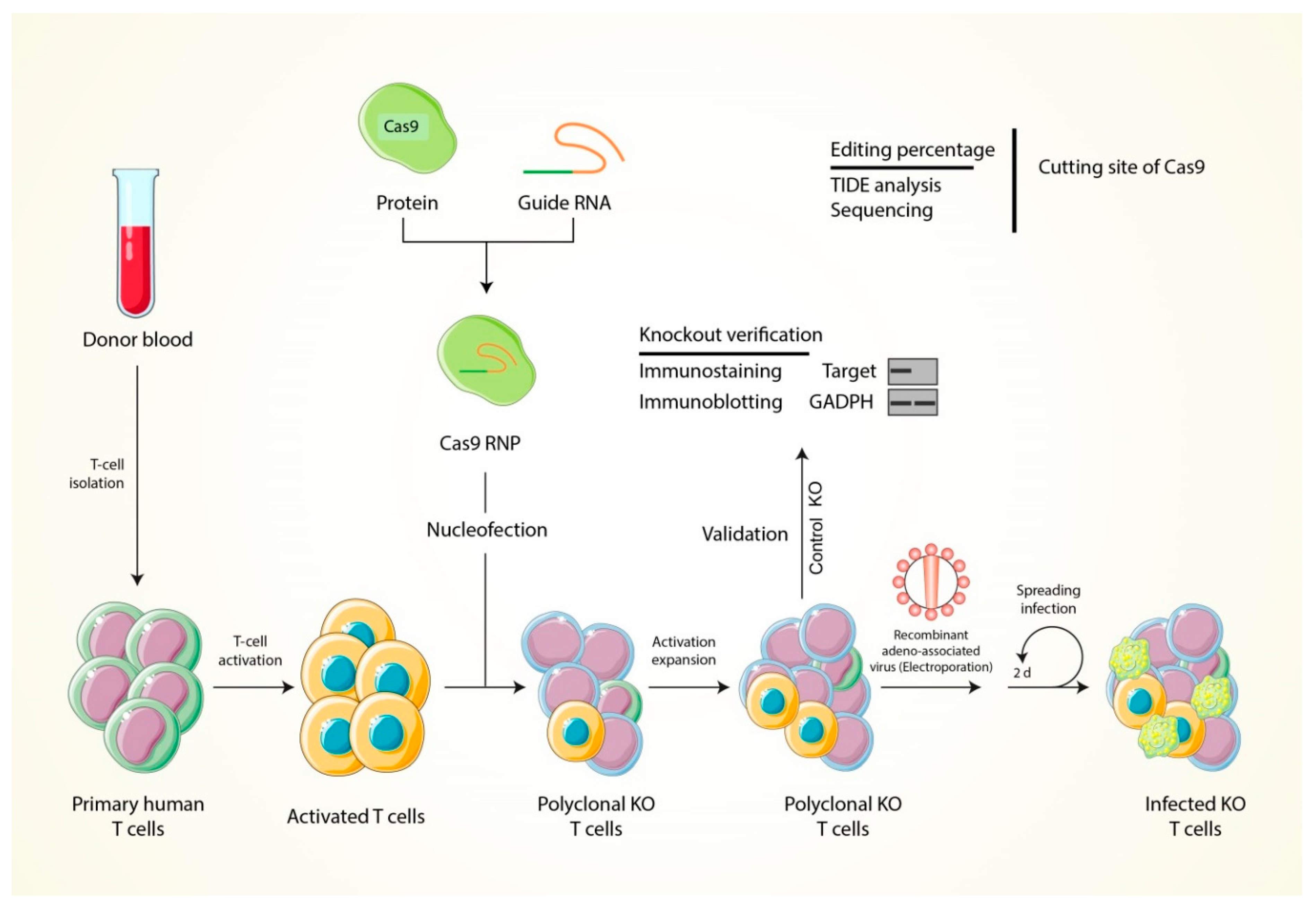
6. The Strategy of T Cell Modification through Using the CRISPR-Cas9 Genome Editing Tool
| Target Antigen | Malignancies | Endo Domains | References |
|---|---|---|---|
| Epidermal growth factor receptor | Gastric cancer | CD28+CD3ζ, 4-1BB | [169] |
| HER2 | Sarcoma, Glioblastoma, Osteosarcoma | CD28-CD3ζ | [170] |
| IL13Rα2 | Glioblastoma | CD3ζ | [171] |
| GD2 | Neuroblastoma | CD3ζ | [172,173] |
| FAP | Colon and ovarian cancer | CD8α, CD3ζ, 4-1BB | [174] |
| MSLN | Pancreatic cancer, Malignant pleural mesothelioma | CD3ζ and 4-1BB | [175] |
| CD171 | Refractory neuroblastoma | CD3ζ | [176] |
| EGFRvIII | Glioma | CD28+CD3ζ, 4-1BB | [177] |
| Carbonic anhydrase IX | Metastatic renal cell carcinoma | FcRγ | [169] |
| Alpha-folate receptor | Ovarian | FcRγ | [178] |
| Carcinoembryonic antigen | Liver metastasis | CD28+CD3ζ | [179] |
| ErbB2+MUC1 | Breast cancer | CD28, CD3ζ | [173] |
| Vascular endothelial growth factor receptor | Melanoma | CD28, CD3ζ | [180] |
| HER2+CD19 | Medulloblastoma | CD28, CD3ζ | [174] |
| NKG2D | Breast cancer | CD28+CD3ζ | [181] |
| S.N. | Immunotherapy Techniques | Their Mechanism of Action | Advantages | Limitations | References |
|---|---|---|---|---|---|
| 01 | Tumor-infiltrating lymphocyte (TIL) | Harvesting the Killer T cells in ex vivo expansion that have already been infiltrated from cancer patients, as well as activating and expanding the T cell population, is required for efficiently killing cancer cells. A large number of activated T cells are re-infused into patient’s body allowing them to destroy tumor cells. TILs have been widely used to treat solid tumors, metastatic melanoma, pancreatic cancer, colorectal cancer and various other cancers. | TILs are efficacious for melanoma patients. They are able to focus properly on tumor antigens. | Therapeutic delays owing to lengthy ex vivo expansion. MHC- I may be downregulated by tumor cells. | [169,182] |
| 02 | Engineered T-cell receptor (TCR) therapy | After isolating T cells from cancer patients, the engineered receptors are introduced into patient’s body for targeting specific cancer antigen and eradication of tumor cells. Modified T cells have exceptional activities and function in terms of having greater lifetime in tumor microenvironment. | Enhance functionality, polarization and working efficiency. | In most cases, it is monoclonal specificity. Inconceivable toxic effects may be found. | [170,182] |
| 03 | Engineered natural killer (NK) cell therapy | Augmenting the capability of NK cell antitumor responses via the introduction of antigen specificity by using genetic modification. CAR-mediated NK cell requires co-stimulatory molecule such as CD28, 4-1BB, CD134 in order to increase the proliferation and cytotoxicity effect against the solid tumor. | Less expensive, easy to be isolated, secreted safer cytokines (IFN-γ and GM-CSF). | Always have to be injected orally. | [82,83] |
| 04 | Chimeric antigen receptor (CAR) T-cell therapy | The CAR T-cell treatment is used to recognize unprocessed antigens that are represented on the surface of tumor cells. The Car T cells are attracted and guided to cancer target site without MHC molecule’s expression. CD4+ and CD8+ T cells carry out the primary killing methods as cytolysis by secretion of granzyme and perforin. | Large-scale production within short time, do not depend on MHC molecule, recognizes any surface antigens (Carbohydrate, protein and glycolipid). | CAR T cell can only target cell surface antigens. Lethal toxicity may be found owing to cytokine storm. | [175,182] |
| 05 | Immunomodulators | Enhancing cytokines’ secretion and act as an agonist for blocking the cancer progression and enhancing the potential activity of immune cells. | Can effectively suppress the cancer cells. | Inability to predict treatment efficacy and patient response. | [97] |
| 06 | Antibody-mediated therapy | Possesses cytotoxic effect against a tumor cell surface antigen and modifies the signal transduction cascade pathway within the tumor cells through the complement-dependent cytotoxicity (CDC) and/or antibody-dependent cellular cytotoxicity (ADCC). | Showed significant anticancer properties in the tumor microenvironment. | Inadequate pharmacokinetics and tissue accessibility as well as impaired interactions with the immune system. | [110] |
| 07 | Formation of bi-specific T-cell-engaging antibodies for cancer therapy | Enhance the specificity of the T-cell receptor, co-stimulation or presentation of peptide antigens independently. | Demonstrated promising efficacy in the cancer treatment. | When the bi-specific antibodies bind only the CD3-specific branch it shows low efficiency. | [120,121] |
| 08 | Formation of cancer vaccine | Enhance the immune system’s capability to signify and damage the cancer antigens more effectively. | Possesses an effective activity to provide the immunity towards the cancer antigens. | The efficacy of cancer vaccines has been limited. | [130] |
| 09 | Nanoparticle-based cancer immunotherapy | They can be employed as adjuvants or as carriers to carry molecules to a specified destination, equipped with certain ligands that encourage specific use and they can be applied in an immune-modulating activity. | Good pharmacokinetics, precise targeting of tumor cells, reduction of side effects and drug resistance. | Nanoparticles have no common feature other than their size. | [133] |
| 10 | Inhibition of T cell exhaustion | Inhibition of T cell exhaustion increase the efficacy of T cells in which it can effectively recognize and kill the cancer antigens. It shows an effective strategy to generate the CAR T cells that are resistant to exhaustion. | Can provide a hopeful result and it may become an important breakthrough in cancer immunotherapy. | More complex technique and also it is patient specific. | [63] |
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Crowther, M.D.; Svane, I.M.; Met, Ö. T-Cell Gene Therapy in Cancer Immunotherapy: Why It Is No Longer Just CARs on The Road. Cells 2020, 9, 1588. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.J.B. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood 2018, 132, 556. [Google Scholar] [CrossRef]
- Wei, G.; Ding, L.; Wang, J.; Hu, Y.; Huang, H.J.E.H. Advances of CD19-directed chimeric antigen receptor-modified T cells in refractory/relapsed acute lymphoblastic leukemia. Exp. Hematol. Oncol. 2017, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Mollanoori, H.; Shahraki, H.; Rahmati, Y.; Teimourian, S. CRISPR/Cas9 and CAR-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum. Immunol. 2018, 79, 876–882. [Google Scholar] [CrossRef]
- Havard, R.; Stephens, D.M. Anti-CD19 Chimeric Antigen Receptor T Cell Therapies: Harnessing the Power of the Immune System to Fight Diffuse Large B Cell Lymphoma. Curr. Hematol. Malig. Rep. 2018, 13, 534–542. [Google Scholar] [CrossRef]
- Kallam, A.; Vose, J.M. Recent Advances in CAR-T Cell Therapy for Non-Hodgkin Lymphoma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 751–757. [Google Scholar] [CrossRef]
- Ogasawara, K.; Dodds, M.; Mack, T.; Lymp, J.; Dell’Aringa, J.; Smith, J. Population Cellular Kinetics of Lisocabtagene Maraleucel, an Autologous CD19-Directed Chimeric Antigen Receptor T-Cell Product, in Patients with Relapsed/Refractory Large B-Cell Lymphoma. Clin. Pharmacokinet. 2021. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Ishizaki, Y.; Yukaya, N.; Kusuhara, K.; Kira, R.; Torisu, H.; Ihara, K.; Sakai, Y.; Sanefuji, M.; Pipo-Deveza, J.R.; Silao, C.L.; et al. PD1 as a common candidate susceptibility gene of subacute sclerosing panencephalitis. Hum. Genet. 2010, 127, 411–419. [Google Scholar] [CrossRef]
- Chevolet, I.; Speeckaert, R.; Schreuer, M.; Neyns, B.; Krysko, O.; Bachert, C.; Hennart, B.; Allorge, D.; van Geel, N.; Van Gele, M.; et al. Characterization of the in vivo immune network of IDO, tryptophan metabolism, PD-L1, and CTLA-4 in circulating immune cells in melanoma. Oncoimmunology 2015, 4, e982382. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Riddell, S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol. 2018, 200, 459–468. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sun, H.; Miao, K.; Deng, C.X. CRISPR-Cas9: From Genome Editing to Cancer Research. Int. J. Biol. Sci. 2016, 12, 1427–1436. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Frey, N.; Porter, D. Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2019, 25, e123–e127. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, M.K.; Chu, H.H.; McLachlan, J.B.; Moon, J.J. On the composition of the preimmune repertoire of T cells specific for Peptide-major histocompatibility complex ligands. Annu. Rev. Immunol. 2010, 28, 275–294. [Google Scholar] [CrossRef]
- Varma, R. TCR triggering by the pMHC complex: Valency, affinity, and dynamics. Sci. Signal. 2008, 1, pe21. [Google Scholar] [CrossRef]
- Kleiveland, C.R. Peripheral blood mononuclear cells. Impact Food Bioact. Health 2015, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Huppa, J.B.; Davis, M.M. T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 2003, 3, 973–983. [Google Scholar] [CrossRef]
- Mueller, D.L.; Jenkins, M.K.; Schwartz, R.H. Clonal expansion versus functional clonal inactivation: A costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu. Rev. Immunol. 1989, 7, 445–480. [Google Scholar] [CrossRef]
- Martin, P.J.; Ledbetter, J.A.; Morishita, Y.; June, C.H.; Beatty, P.G.; Hansen, J.A. A 44 kilodalton cell surface homodimer regulates interleukin 2 production by activated human T lymphocytes. J. Immunol. 1986, 136, 3282–3287. [Google Scholar]
- Gmünder, H.; Lesslauer, W. A 45-kDa human T-cell membrane glycoprotein functions in the regulation of cell proliferative responses. Eur. J. Biochem. 1984, 142, 153–160. [Google Scholar] [CrossRef]
- June, C.H.; Ledbetter, J.A.; Linsley, P.S.; Thompson, C.B. Role of the CD28 receptor in T-cell activation. Immunol. Today 1990, 11, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Lesslauer, W.; Koning, F.; Ottenhoff, T.; Giphart, M.; Goulmy, E.; van Rood, J.J. T90/44 (9.3 antigen). A cell surface molecule with a function in human T cell activation. Eur. J. Immunol. 1986, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, J.A.; Martin, P.J.; Nowinski, R.C.J.I. Monoclonal antibodies identifying a novel T-cell antigen and Ia antigens of human lymphocytes. Immunogenetics 1980, 10, 247–260. [Google Scholar] [CrossRef]
- Hara, T.; Fu, S.M.; Hansen, J.A. Human T cell activation. II. A new activation pathway used by a major T cell population via a disulfide-bonded dimer of a 44 kilodalton polypeptide (9.3 antigen). J. Exp. Med. 1985, 161, 1513–1524. [Google Scholar] [CrossRef]
- Jin, B.; Sun, T.; Yu, X.H.; Yang, Y.X.; Yeo, A.E. The effects of TLR activation on T-cell development and differentiation. Clin. Dev. Immunol. 2012, 2012, 836485. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantrell, D.A. T-cell antigen receptor signal transduction. Immunology 2002, 105, 369–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.R. The CD45 tyrosine phosphatase: A positive and negative regulator of immune cell function. Semin. Immunol. 2000, 12, 349–359. [Google Scholar] [CrossRef]
- Rossy, J.; Williamson, D.J.; Gaus, K. How does the kinase Lck phosphorylate the T cell receptor? Spatial organization as a regulatory mechanism. Front. Immunol. 2012, 3, 167. [Google Scholar] [CrossRef] [Green Version]
- Macián, F.; García-Cózar, F.; Im, S.H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 2002, 109, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Salsman, J.; Dellaire, G. Precision genome editing in the CRISPR era. Biochem. Cell Biol. Biochim. Et Biol. Cell. 2017, 95, 187–201. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.K. Costimulatory molecules and T-cell-B-cell interactions. Rheum. Dis. Clin. N. Am. 2004, 30, 175–191. [Google Scholar] [CrossRef]
- Fukunaga, A.; Miyamoto, M.; Cho, Y.; Murakami, S.; Kawarada, Y.; Oshikiri, T.; Kato, K.; Kurokawa, T.; Suzuoki, M.; Nakakubo, Y.; et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 2004, 28, e26–e31. [Google Scholar] [CrossRef] [PubMed]
- Oshikiri, T.; Miyamoto, M.; Shichinohe, T.; Suzuoki, M.; Hiraoka, K.; Nakakubo, Y.; Shinohara, T.; Itoh, T.; Kondo, S.; Katoh, H. Prognostic value of intratumoral CD8+ T lymphocyte in extrahepatic bile duct carcinoma as essential immune response. J. Surg. Oncol. 2003, 84, 224–228. [Google Scholar] [CrossRef]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Speiser, D.E.; Ho, P.C.; Verdeil, G. Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol. 2016, 16, 599–611. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
- Schultze, J.L.; Michalak, S.; Seamon, M.J.; Dranoff, G.; Jung, K.; Daley, J.; Delgado, J.C.; Gribben, J.G.; Nadler, L.M. CD40-activated human B cells: An alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J. Clin. Investig. 1997, 100, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Boon, T.; Cerottini, J.C.; Van den Eynde, B.; van der Bruggen, P.; Van Pel, A. Tumor antigens recognized by T lymphocytes. Annu. Rev. Immunol. 1994, 12, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Poccia, F.; Agrati, C.; Martini, F.; Capobianchi, M.R.; Wallace, M.; Malkovsky, M. Antiviral reactivities of gammadelta T cells. Microbes Infect. 2005, 7, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, X.; Zhou, W.L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Southam, C.M.; Brunschwig, A.; Levin, A.G.; Dizon, Q.S. Effect of leukocytes on transplantability of human cancer. Cancer 1966, 19, 1743–1753. [Google Scholar] [CrossRef]
- Cao, Y.; Trillo-Tinoco, J.; Sierra, R.A.; Anadon, C.; Dai, W.; Mohamed, E.; Cen, L.; Costich, T.L.; Magliocco, A.; Marchion, D.; et al. ER stress-induced mediator C/EBP homologous protein thwarts effector T cell activity in tumors through T-bet repression. Nat. Commun. 2019, 10, 1280. [Google Scholar] [CrossRef] [Green Version]
- Meydan, İ.; Kizil, G.; Demir, H.; Toptanci, B.C.; Kizil, M. In vitro DNA damage, protein oxidation protective activity and antioxidant potentials of almond fruit (Amygdalus trichamygdalus) parts (hull and drupe) using soxhlet ethanol extraction. Adv. Tradit. Med. 2020, 20, 571–579. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription Factor C/EBP Homologous Protein in Health and Diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Araki, K.; Morita, M.; Bederman, A.G.; Konieczny, B.T.; Kissick, H.T.; Sonenberg, N.; Ahmed, R. Translation is actively regulated during the differentiation of CD8(+) effector T cells. Nat. Immunol. 2017, 18, 1046–1057. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Hong, W.X.; Haebe, S.; Lee, A.S.; Westphalen, C.B.; Norton, J.A.; Jiang, W.; Levy, R. Intratumoral Immunotherapy for Early-stage Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3091–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Met, Ö.; Jensen, K.M.; Chamberlain, C.A.; Donia, M.; Svane, I.M. Principles of adoptive T cell therapy in cancer. Semin. Immunopathol. 2019, 41, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; June, C.H. Engineering T cells for cancer: Our synthetic future. Immunol. Rev. 2014, 257, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, R.; Donia, M.; Westergaard, M.C.; Pedersen, M.; Hansen, M.; Svane, I.M. Tumor infiltrating lymphocyte therapy for ovarian cancer and renal cell carcinoma. Hum. Vaccines Immunother. 2015, 11, 2790–2795. [Google Scholar] [CrossRef] [Green Version]
- Azimi, F.; Scolyer, R.A.; Rumcheva, P.; Moncrieff, M.; Murali, R.; McCarthy, S.W.; Saw, R.P.; Thompson, J.F. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2678–2683. [Google Scholar] [CrossRef]
- Sakellariou-Thompson, D.; Forget, M.A.; Hinchcliff, E.; Celestino, J.; Hwu, P.; Jazaeri, A.A.; Haymaker, C.; Bernatchez, C. Potential clinical application of tumor-infiltrating lymphocyte therapy for ovarian epithelial cancer prior or post-resistance to chemotherapy. Cancer Immunol. Immunother. CII 2019, 68, 1747–1757. [Google Scholar] [CrossRef]
- Buisseret, L.; Garaud, S.; de Wind, A.; Van den Eynden, G.; Boisson, A.; Solinas, C.; Gu-Trantien, C.; Naveaux, C.; Lodewyckx, J.N.; Duvillier, H.; et al. Tumor-infiltrating lymphocyte composition, organization and PD-1/ PD-L1 expression are linked in breast cancer. Oncoimmunology 2017, 6, e1257452. [Google Scholar] [CrossRef]
- Chung, Y.R.; Kim, H.J.; Jang, M.H.; Park, S.Y. Prognostic value of tumor infiltrating lymphocyte subsets in breast cancer depends on hormone receptor status. Breast Cancer Res. Treat. 2017, 161, 409–420. [Google Scholar] [CrossRef]
- Ping, Y.; Liu, C.; Zhang, Y. T-cell receptor-engineered T cells for cancer treatment: Current status and future directions. Protein Cell 2018, 9, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chodon, T.; Comin-Anduix, B.; Chmielowski, B.; Koya, R.C.; Wu, Z.; Auerbach, M.; Ng, C.; Avramis, E.; Seja, E.; Villanueva, A.; et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2457–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezvani, K. Adoptive cell therapy using engineered natural killer cells. Bone Marrow Transplant. 2019, 54, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell. Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310. [Google Scholar] [CrossRef]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Boissel, L.; Betancur-Boissel, M.; Lu, W.; Krause, D.S.; Van Etten, R.A.; Wels, W.S.; Klingemann, H. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology 2013, 2, e26527. [Google Scholar] [CrossRef] [Green Version]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.H.; Leung, W. NKAML: A pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef] [Green Version]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V. Immunotherapy: Tisagenlecleucel—The first approved CAR-T-cell therapy: Implications for payers and policy makers. Nat. Rev. Clin. Oncol. 2018, 15, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H. Adoptive immunotherapy for hematological malignancies using T cells gene-modified to express tumor antigen-specific receptors. Pharmaceuticals 2014, 7, 1049–1068. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Geyer, M.B.; Brentjens, R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: Interpreting clinical outcomes to date. Blood 2016, 127, 3312–3320. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Grupp, S.A.; Porter, D.L.; June, C.H. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Soyano, A.E.; Dholaria, B.; Knutson, K.L.; Lou, Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 8. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell Biol. 2018, 96, 21–33. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Subleski, J.J.; Wigginton, J.M.; Wiltrout, R.H. Immunotherapy of cancer by IL-12-based cytokine combinations. Expert Opin. Biol. Ther. 2007, 7, 1705–1721. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Parekh, D.J.; Bochner, B.H.; Dalbagni, G. Superficial and muscle-invasive bladder cancer: Principles of management for outcomes assessments. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 5519–5527. [Google Scholar] [CrossRef]
- Bettex-Galland, M.; Studer, U.E.; Walz, A.; Dewald, B.; Baggiolini, M. Neutrophil-activating peptide-1/interleukin-8 detection in human urine during acute bladder inflammation caused by transurethral resection of superficial cancer and bacillus Calmette-Guérin administration. Eur. Urol. 1991, 19, 171–175. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; White, D.E.; Steinberg, S.M. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: Identification of the antigens mediating response. Ann. Surg. 1998, 228, 307–319. [Google Scholar] [CrossRef]
- Thotathil, Z.; Jameson, M.B. Early experience with novel immunomodulators for cancer treatment. Expert Opin. Investig. Drugs 2007, 16, 1391–1403. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Hafeez, U.; Gan, H.K.; Scott, A.M. Monoclonal antibodies as immunomodulatory therapy against cancer and autoimmune diseases. Curr. Opin. Pharmacol. 2018, 41, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.P.; Weiner, L.M. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005, 23, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Pallasch, C.P.; Leskov, I.; Braun, C.J.; Vorholt, D.; Drake, A.; Soto-Feliciano, Y.M.; Bent, E.H.; Schwamb, J.; Iliopoulou, B.; Kutsch, N.; et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell 2014, 156, 590–602. [Google Scholar] [CrossRef] [Green Version]
- Trauth, B.C.; Klas, C.; Peters, A.M.; Matzku, S.; Möller, P.; Falk, W.; Debatin, K.M.; Krammer, P.H. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science 1989, 245, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mårlind, J.; Kaspar, M.; Trachsel, E.; Sommavilla, R.; Hindle, S.; Bacci, C.; Giovannoni, L.; Neri, D. Antibody-mediated delivery of interleukin-2 to the stroma of breast cancer strongly enhances the potency of chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 6515–6524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakhel, S.; Padilla, L.; Adan, J.; Masa, M.; Martinez, J.M.; Roque, L.; Coll, T.; Hervas, R.; Calvis, C.; Messeguer, R.; et al. S100P antibody-mediated therapy as a new promising strategy for the treatment of pancreatic cancer. Oncogenesis 2014, 3, e92. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Chung, H.; Banan, B.; Manning, P.T.; Ott, K.C.; Lin, S.; Capoccia, B.J.; Subramanian, V.; Hiebsch, R.R.; Upadhya, G.A.; et al. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett. 2015, 360, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, A.; Jimeno, A. Bispecific antibodies for cancer therapy: A review. Pharmacol. Ther. 2018, 185, 122–134. [Google Scholar] [CrossRef]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol. Med. 2018, 24, 50. [Google Scholar] [CrossRef]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef]
- Brischwein, K.; Parr, L.; Pflanz, S.; Volkland, J.; Lumsden, J.; Klinger, M.; Locher, M.; Hammond, S.A.; Kiener, P.; Kufer, P.; et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J. Immunother. 2007, 30, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeuerle, P.A.; Kufer, P.; Bargou, R. BiTE: Teaching antibodies to engage T-cells for cancer therapy. Curr. Opin. Mol. Ther. 2009, 11, 22–30. [Google Scholar]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Gore, L. Blinatumomab, a Bi-Specific Anti-CD19/CD3 BiTE(®) Antibody for the Treatment of Acute Lymphoblastic Leukemia: Perspectives and Current Pediatric Applications. Front. Oncol. 2014, 4, 63. [Google Scholar] [CrossRef] [Green Version]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef]
- Maetzel, D.; Denzel, S.; Mack, B.; Canis, M.; Went, P.; Benk, M.; Kieu, C.; Papior, P.; Baeuerle, P.A.; Munz, M.; et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat. Cell Biol. 2009, 11, 162–171. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.; Sur, M.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 102–106. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.-Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [PubMed] [Green Version]
- Jain, K.K. Personalized cancer vaccines. Expert Opin. Biol. Ther. 2010, 10, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Hu, X.L.; Saeed, M.; Chen, B.F.; Li, Y.P.; Yu, H.J. Overview of recent advances in liposomal nanoparticle-based cancer immunotherapy. Acta Pharmacol. Sin. 2019, 40, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Guan, C.; Chernyak, N.; Dominguez, D.; Cole, L.; Zhang, B.; Mirkin, C.A. RNA-Based Immunostimulatory Liposomal Spherical Nucleic Acids as Potent TLR7/8 Modulators. Small 2018, 14, e1803284. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Moynihan, K.D.; Zheng, Y.; Szeto, G.L.; Li, A.V.; Huang, B.; Van Egeren, D.S.; Park, C.; Irvine, D.J. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 2014, 507, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Xu, J.; Liang, C.; Chao, Y.; Jin, Q.; Wang, C.; Chen, M.; Liu, Z. Self-Supplied Tumor Oxygenation through Separated Liposomal Delivery of H(2)O(2) and Catalase for Enhanced Radio-Immunotherapy of Cancer. Nano Lett. 2018, 18, 6360–6368. [Google Scholar] [CrossRef]
- Pitchaimani, A.; Nguyen, T.D.T.; Aryal, S. Natural killer cell membrane infused biomimetic liposomes for targeted tumor therapy. Biomaterials 2018, 160, 124–137. [Google Scholar] [CrossRef]
- Auci, D.L.; Cecil, D.L.; Herendeen, D.; Broussard, E.K.; Liao, J.B.; Holt, G.E.; Disis, M.L. Clinical application of plasmid-based cancer vaccines. In Gene Therapy of Cancer; Elsevier: Amsterdam, The Netherlands, 2014; pp. 335–343. [Google Scholar]
- Oka, Y.; Tsuboi, A.; Elisseeva, O.A.; Udaka, K.; Sugiyama, H. WT1 as a novel target antigen for cancer immunotherapy. Curr. Cancer Drug Targets 2002, 2, 45–54. [Google Scholar] [CrossRef]
- Kita, K.; Nakase, K.; Miwa, H.; Masuya, M.; Nishii, K.; Morita, N.; Takakura, N.; Otsuji, A.; Shirakawa, S.; Ueda, T.; et al. Phenotypical characteristics of acute myelocytic leukemia associated with the t(8;21)(q22;q22) chromosomal abnormality: Frequent expression of immature B-cell antigen CD19 together with stem cell antigen CD34. Blood 1992, 80, 470–477. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kawakami, Y. Human tumor antigens recognized by T cells. Curr. Opin. Immunol. 1996, 8, 628–636. [Google Scholar] [CrossRef]
- Hegedűs, C.; Kovács, K.; Polgár, Z.; Regdon, Z.; Szabó, É.; Robaszkiewicz, A.; Forman, H.J.; Martner, A.; Virág, L. Redox control of cancer cell destruction. Redox Biol. 2018, 16, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Miyamoto, M.; Kato, K.; Fukunaga, A.; Shichinohe, T.; Kawarada, Y.; Hida, Y.; Oshikiri, T.; Kurokawa, T.; Suzuoki, M.; et al. CD4+ and CD8+ T cells cooperate to improve prognosis of patients with esophageal squamous cell carcinoma. Cancer Res. 2003, 63, 1555–1559. [Google Scholar] [PubMed]
- Snyder, L.A.; Goletz, T.J.; Gunn, G.R.; Shi, F.F.; Harris, M.C.; Cochlin, K.; McCauley, C.; McCarthy, S.G.; Branigan, P.J.; Knight, D.M. A MUC1/IL-18 DNA vaccine induces anti-tumor immunity and increased survival in MUC1 transgenic mice. Vaccine 2006, 24, 3340–3352. [Google Scholar] [CrossRef]
- Xiang, R.; Silletti, S.; Lode, H.N.; Dolman, C.S.; Ruehlmann, J.M.; Niethammer, A.G.; Pertl, U.; Gillies, S.D.; Primus, F.J.; Reisfeld, R.A. Protective immunity against human carcinoembryonic antigen (CEA) induced by an oral DNA vaccine in CEA-transgenic mice. Clin. Cancer Res. 2001, 7, 856s–864s. [Google Scholar] [PubMed]
- Tiriveedhi, V.; Tucker, N.; Herndon, J.; Li, L.; Sturmoski, M.; Ellis, M.; Ma, C.; Naughton, M.; Lockhart, A.C.; Gao, F.; et al. Safety and preliminary evidence of biologic efficacy of a mammaglobin-a DNA vaccine in patients with stable metastatic breast cancer. Clin. Cancer Res. 2014, 20, 5964–5975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, L.R.; Carrascal, M.A.; Barbas, A.; Ramalho, J.S.; Novo, C.; Delannoy, P.; Videira, P.A. Challenges in Antibody Development against Tn and Sialyl-Tn Antigens. Biomolecules 2015, 5, 1783–1809. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hong, Y.; Mizejewski, G.J. Engineering α-fetoprotein-based gene vaccines to prevent and treat hepatocellular carcinoma: Review and future prospects. Immunotherapy 2014, 6, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Yin, B.W.; Lloyd, K.O. Molecular cloning of the CA125 ovarian cancer antigen: Identification as a new mucin, MUC16. J. Biol. Chem. 2001, 276, 27371–27375. [Google Scholar] [CrossRef] [Green Version]
- Amara, S.; Tiriveedhi, V. The Five Immune Forces Impacting DNA-Based Cancer Immunotherapeutic Strategy. Int. J. Mol. Sci. 2017, 18, 650. [Google Scholar] [CrossRef] [Green Version]
- Aldrich, J.F.; Lowe, D.B.; Shearer, M.H.; Winn, R.E.; Jumper, C.A.; Kennedy, R.C. Vaccines and immunotherapeutics for the treatment of malignant disease. Clin. Dev. Immunol. 2010, 2010, 697158. [Google Scholar] [CrossRef]
- Wang, J.C.; Xu, Y.; Huang, Z.M.; Lu, X.J. T cell exhaustion in cancer: Mechanisms and clinical implications. J. Cell. Biochem. 2018, 119, 4279–4286. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363. [Google Scholar] [CrossRef]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Dong, X.; Xu, Q.; Zhu, L.; Wang, F.; Hou, Y.; Chao, C.C. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T-cell therapy. Cancer Med. 2019, 8, 4254–4264. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, R.A.; Debets, R.; Hart, E.; Hoogenboom, H.R.; Bolhuis, R.L.; Chames, P. A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther. 2001, 8, 1601–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.Y.; Cao, C.Y. The application of CRISPR-Cas9 genome editing tool in cancer immunotherapy. Brief. Funct. Genom. 2019, 18, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Piganeau, M.; Ghezraoui, H.; De Cian, A.; Guittat, L.; Tomishima, M.; Perrouault, L.; René, O.; Katibah, G.E.; Zhang, L.; Holmes, M.C.; et al. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res. 2013, 23, 1182–1193. [Google Scholar] [CrossRef] [Green Version]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- DiCarlo, J.E.; Norville, J.E.; Mali, P.; Rios, X.; Aach, J.; Church, G.M. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Lamers, C.H.; Klaver, Y.; Gratama, J.W.; Sleijfer, S.; Debets, R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem. Soc. Trans. 2016, 44, 951–959. [Google Scholar] [CrossRef]
- Kohn, D.B.; Dotti, G.; Brentjens, R.; Savoldo, B.; Jensen, M.; Cooper, L.J.; June, C.H.; Rosenberg, S.; Sadelain, M.; Heslop, H.E. CARs on track in the clinic. Mol. Ther. 2011, 19, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Wilkie, S.; van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.; Pereira, A.C.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Digiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R.; et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.T.; Song, H.; et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-cell Therapy for CEA+ Liver Metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef] [Green Version]
- Chinnasamy, D.; Tran, E.; Yu, Z.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013, 73, 3371–3380. [Google Scholar] [CrossRef] [Green Version]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaioannou, N.E.; Beniata, O.V.; Vitsos, P.; Tsitsilonis, O.; Samara, P. Harnessing the immune system to improve cancer therapy. Ann. Transl. Med. 2016, 4, 261. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ye, C.; Liu, J.; Zhang, D.; Kimata, J.T.; Zhou, P. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS ONE 2014, 9, e115987. [Google Scholar] [CrossRef] [Green Version]
- Li, J.F.; Norville, J.E.; Aach, J.; McCormack, M.; Zhang, D.; Bush, J.; Church, G.M.; Sheen, J. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 2013, 31, 688–691. [Google Scholar] [CrossRef]
- Chi, S.; Weiss, A.; Wang, H. A CRISPR-Based Toolbox for Studying T Cell Signal Transduction. BioMed Res. Int. 2016, 2016, 5052369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karthik, L.; Kumar, G.; Keswani, T.; Bhattacharyya, A.; Chandar, S.S.; Bhaskara Rao, K.V. Protease inhibitors from marine actinobacteria as a potential source for antimalarial compound. PLoS ONE 2014, 9, e90972. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Guan, X.; Du, T.; Jin, W.; Wu, B.; Liu, Y.; Wang, P.; Hu, B.; Griffin, G.E.; Shattock, R.J.; et al. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J. Gen. Virol. 2015, 96, 2381–2393. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.D.; Pozniak, C.D.; Walsh, G.S. Neuronal life and death: An essential role for the p53 family. Cell Death Differ. 2000, 7, 880–888. [Google Scholar] [CrossRef]
- Poletti, V.; Mavilio, F. Interactions between Retroviruses and the Host Cell Genome. Mol. Therapy. Methods Clin. Dev. 2018, 8, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Simhadri, V.L.; McGill, J.; McMahon, S.; Wang, J.; Jiang, H.; Sauna, Z.E. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther. Methods Clin. Dev. 2018, 10, 105–112. [Google Scholar] [CrossRef]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Freen-van Heeren, J.J.; Popović, B.; Guislain, A.; Wolkers, M.C. Human T cells employ conserved AU-rich elements to fine-tune IFN-γ production. Eur. J. Immunol. 2020, 50, 949–958. [Google Scholar] [CrossRef] [Green Version]
- Chicaybam, L.; Sodre, A.L.; Curzio, B.A.; Bonamino, M.H. An efficient low cost method for gene transfer to T lymphocytes. PLoS ONE 2013, 8, e60298. [Google Scholar] [CrossRef]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kurlander, R.J. Comparison of anti-CD3 and anti-CD28-coated beads with soluble anti-CD3 for expanding human T cells: Differing impact on CD8 T cell phenotype and responsiveness to restimulation. J. Transl. Med. 2010, 8, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.W.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017, 27, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017, 8, 17002–17011. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Saber, M.; Biswas, P.; Dey, D.; Kaium, M.A.; Islam, M.A.; Tripty, M.I.A.; Rahman, M.H.; Rahaman, T.I.; Biswas, M.Y.; Paul, P.; et al. A Comprehensive Review of Recent Advancements in Cancer Immunotherapy and Generation of CAR T Cell by CRISPR-Cas9. Processes 2022, 10, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/pr10010016
Al Saber M, Biswas P, Dey D, Kaium MA, Islam MA, Tripty MIA, Rahman MH, Rahaman TI, Biswas MY, Paul P, et al. A Comprehensive Review of Recent Advancements in Cancer Immunotherapy and Generation of CAR T Cell by CRISPR-Cas9. Processes. 2022; 10(1):16. https://0-doi-org.brum.beds.ac.uk/10.3390/pr10010016
Chicago/Turabian StyleAl Saber, Md., Partha Biswas, Dipta Dey, Md. Abu Kaium, Md. Aminul Islam, Miss Ismoth Ara Tripty, MD. Hasanur Rahman, Tanjim Ishraq Rahaman, Md. Yeaman Biswas, Priyanka Paul, and et al. 2022. "A Comprehensive Review of Recent Advancements in Cancer Immunotherapy and Generation of CAR T Cell by CRISPR-Cas9" Processes 10, no. 1: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/pr10010016