2.2.1. Reactor
The reactor model is taken from [
32,
33]. The model describes a hydroformylation reaction in a continuous stirred tank reactor. CO and H
are provided to the homogeneous liquid reaction phase from a second, gaseous phase in the reactor. Other reactants and additives are provided in liquid form. Deviating from the aforementioned publications, we only consider the stationary case, and the equilibrium between the liquid and the gas phase is described by an artificial neural network (ANN) trained on the PC-SAFT equation of state [
34]. The activation function of the ANN, a hyperbolic tangent, is replaced by a piecewise linear approximation [
34]. Furthermore, the chemical species are different. The chain length of the olefin compared to the previous work is reduced by 2 and the new solvents are introduced. A list of all species involved in the reaction can be found in
Table 1. However, the original reaction kinetics were used because the deviations due to the different chain lengths are small and the influence of the polar solvent on the reaction is negligible [
49].
The mass balance is defined as
where
is the active catalyst concentration inside the reactor,
is the molar mass of the catalyst,
and
are the stoichiometric coefficients and reaction rates, respectively, for the eight reactions, RCT, taking place. The general form of the reaction rates
of all eight reactions in the reactor is given by
The reaction rate constants
are calculated using Arrhenius law and the parameters
are taken from the literature and can be found in [
33]. To calculate the concentration of the active catalyst
the following equation is used
where
is the total catalyst concentration.
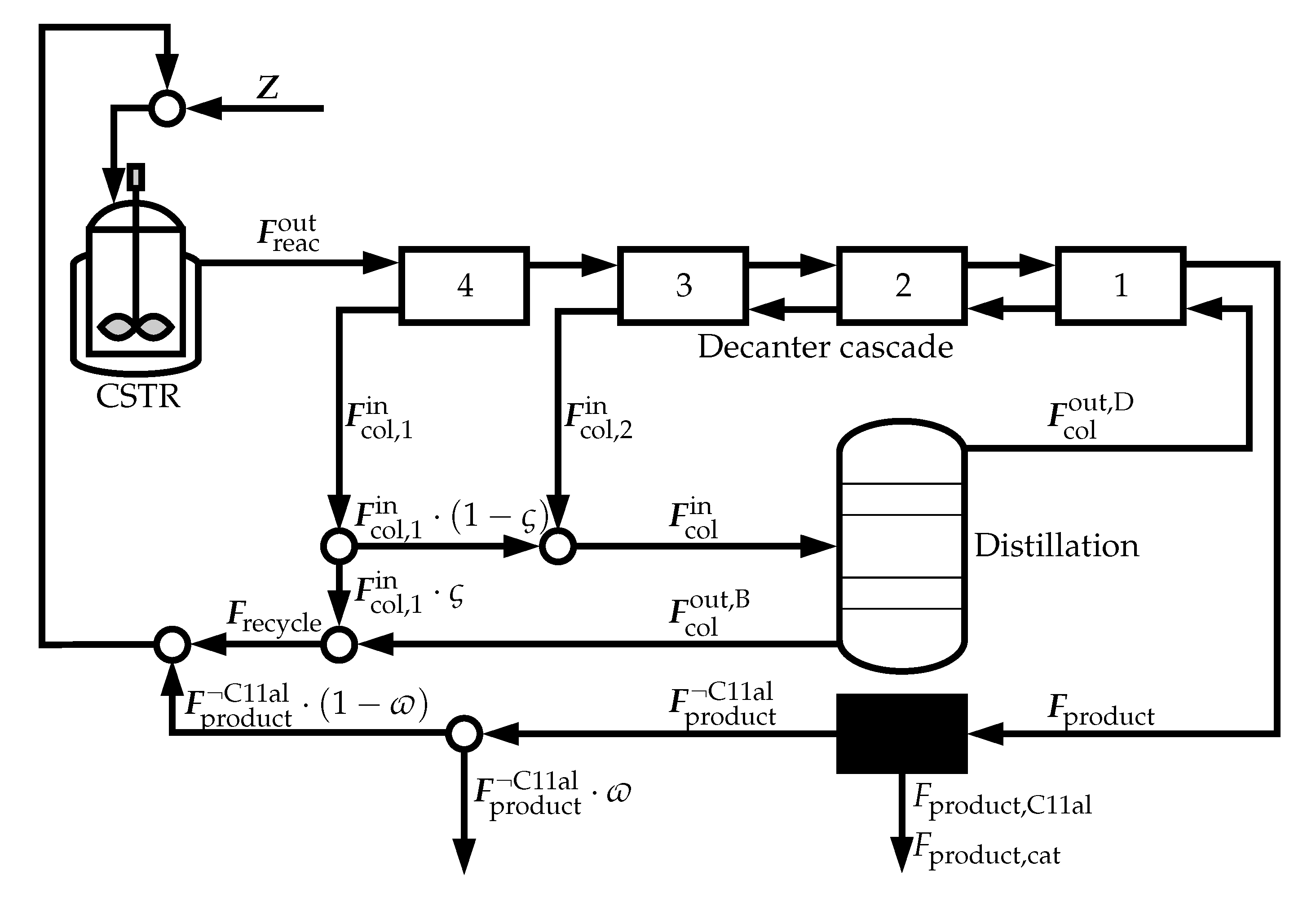
The reactor inlet is composed of three elements. The first of these is the feed stream, , which contains the make-up streams for the catalyst, the solvents, and the reactants. The second is the recycle stream, , coming from the decanter cascade, which recycles the polar solvent (thereby the catalyst), unused reactants, and some of the products. The final stream is the recycle coming from the black box, , minus the purged stream.
2.2.2. Downstream Processing
The reactor outlet, , is fed into the downstream section consisting of four decanters and a distillation column.
The distillation column is required to recover the extraction solvent. It is assumed to be operated at a pressure of 60 mbar, with a linear pressure drop of 30 mbar along the column. The requirement of the column is that it recovers all of the circulated extraction solvent at the top. Instead of a rigorous distillation column model, a separately calculated surrogate function is employed for each investigated polar solvent . The procedure to obtain the surrogate function can be split into two parts.
First, the relative volatilities
are calculated. An ensemble of 810 distillation columns with a different number of stages, reflux, and reboil ratios were simulated. Each distillation column is assumed to have constant molar flows, a constant molar overflow, and a total condenser. Furthermore, each is assumed to behave ideally, thus the gas phase is described by Dalton’s law
and the liquid phase is described by Raoult’s law
, where
and
are the liquid and vapor mole fractions, respectively. For the calculation of the vapor pressures the vapor pressure correlation
is used, with
T in K and
in mmHg. The parameters
can be found in
Table A1. Using these 810 simulations, mean temperatures for the top of the column,
, and the bottom of the column,
, can be calculated. These mean temperatures are used to calculate the relative volatilities using Equation (
5) and
where
denotes the highest boiling component, i.e., C11al. The obtained values for
can be found in
Table A3.
The optimal costs for the distillation are calculated using the constant
. Global optimizations using BARON [
53], with a Fenske–Underwood–Gilliland (FUG) shortcut model [
54], were conducted with 1000 varied feeds for each solvent. For these global optimizations the constraints are as follows:
DMF: 99% purity of DMF in the distillate
DSUC: 99% purity of DSUC + C10en in the distillate
THPO: 99% purity of THPO in the distillate
The varied purity requirements for the different polar solvent candidates are necessary because of their different separation properties. These properties will be explained later.
The total annualized costs (TAC) in
for the distillation column are calculated with a lumped cost function [
55]
using the parameters from
Table A2, where
is the vapor flow rate and
is the length of the column.
The values of
are used for a polynomial regression of the costs with respect to the circulated extraction solvent stream
and the recovery rate
,
with parameters
given in
Table A2. Equation (
8) is used as a surrogate for the distillation column.
The number of decanters is chosen because it was cost optimal in a previous study [
56]. Following the results of [
56] once again, the decanters are operated at a fixed temperature of
K. The decrease in temperature compared to the reaction conditions leads to a liquid–liquid phase split. The mass balances are defined as follows,
where the decanters are numbered from right to left, i.e.,
. The upper outlet of the decanters
is the less polar, product-rich phase with composition
and the lower outlet of the decanters
is the more polar, catalyst-rich phase with composition
.
The boiling points of the three solvents vary, with DMF being the easiest to separate and DSUC being the hardest to separate (see the relative volatilities
in
Table A3). Therefore, the separation properties of the three solvents regarding distillation vary. With DMF and THPO, it is possible to solely recover the solvent in the distillate stream. With DSUC, C10en becomes the component with the highest relative volatility. Therefore, we assume that all of the C10en contained in the column feed will leave through the distillate.
Because the components contained in the distillate flow differ depending on the separation properties of the solvents, the value of
needs to be calculated as
because the column itself is replaced by the aforementioned surrogate function in Equation (
8). Here,
is introduced as a vector of split factors for (
, C12an, C10en, C11al, cat) to capture the different separation behaviors of the mixture with the respective solvent:
Here, is used to make sure that only the circulated extraction solvent stream leaves the distillation column through the distillate.
Note that the fourth and the third decanter are only connected through the less polar flow of the fourth decanter. By doing this, the extraction solvent stream of the cascade bypasses the fourth decanter, allowing for an additional degree of freedom. The molar flow entering the column, , is composed of the more polar phase of the third decanter, , and a fraction of the more polar phase of the fourth decanter, ,
That fraction is determined by the split factor
. A large split factor leads to a smaller flow into the column. However, the composition of the mixture at the bottom of the column, and hence the temperature, also depends on the split factor. The literature suggests that the aldehyde will react with itself to the unwanted side product aldol at temperatures above
K [
57]. Therefore, the temperature at the bottom of the column is restricted to
K, the same temperature as inside the reactor, which should be safe. The split factor
can be used by the optimizer to achieve this temperature.
The catalyst inside the decanter cascade is described by the catalyst mass balance
where
is the catalyst in the more polar phase and
is the catalyst in the less polar phase. The partitioning of the catalyst between the two phases is calculated with
where
is the logarithmic partition coefficient for stage
n.
The activity coefficients
for the LLE are described by a modified UNIFAC (Dortmund) [
58] implementation. The corresponding LLE conditions,
are solved with a residuum less than 1 × 10
−9. For each of the LLE points, the partition coefficients
for the catalyst/ligand is calculated with COSMOtherm [
47], because the size of the catalyst/ligand complex prohibits the use of group contribution methods such as UNIFAC [
59]. To determine the composition of the two liquid phases and the catalyst distribution, a surrogate model
is employed,
where
is a parameterization of the binodal curve. To generate the input/output data for fitting the surrogate model, a parameter continuation [
60] in the orthogonal direction of the tie lines of the LLE is used.
The surrogate has a two-dimensional input space and a nine-dimensional output space. It consists of a second-degree polynomial and an ANN with a single hidden layer fit to the residuals of the polynomial. The ANN is generated with the “train” command of MATLABs deep learning toolbox. A more thorough description of the algorithm used to generate the surrogate is not the scope of this article and can be found in another publication of the authors in the present special issue of Processes [
61].
A previous optimization study yielded that the catalyst recovery plays an important role for the overall process cost [
56]. Therefore, the accuracy of the employed surrogate with regard to the partition coefficient
is of high importance. The maximum errors of the three surrogate models for
at the sample points are:

 ,
, 

{kind=link}
{kind=link}

