Predicting the Longitudinally and Radially Varying Gut Microbiota Composition using Multi-Scale Microbial Metabolic Modeling


Abstract
:1. Background
2. Results
2.1. A dynamic Framework for Simulating Spatially Differential Gut Microbiota Metabolism
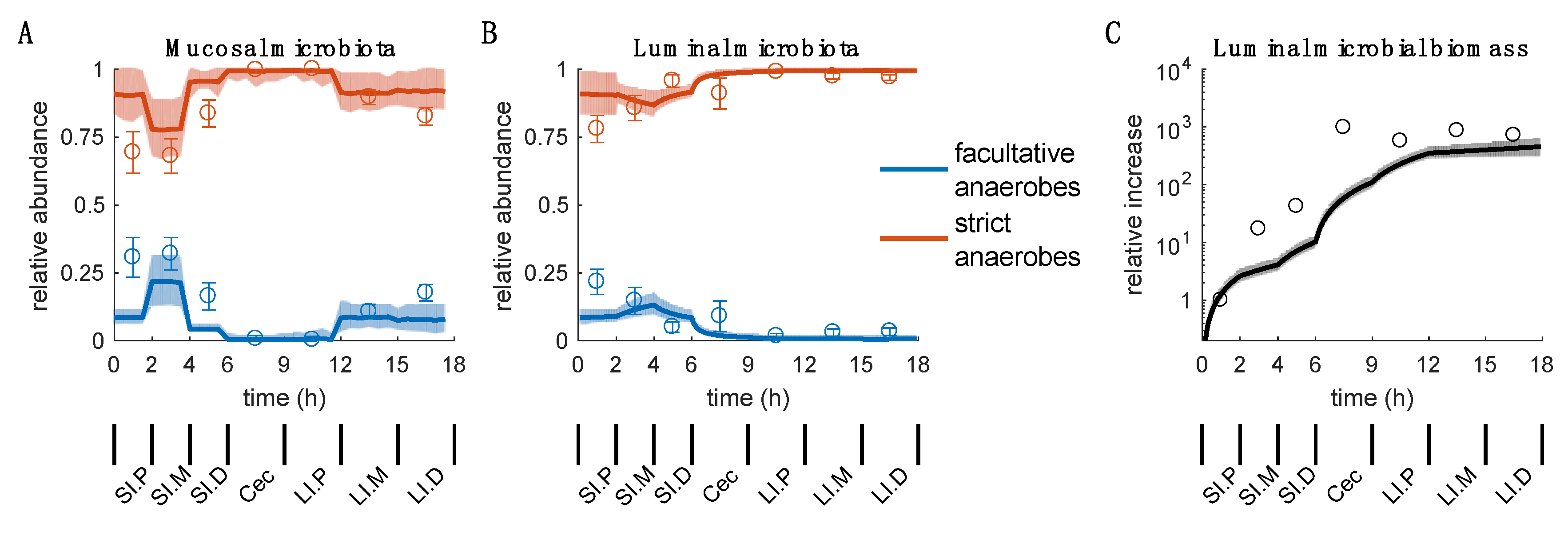
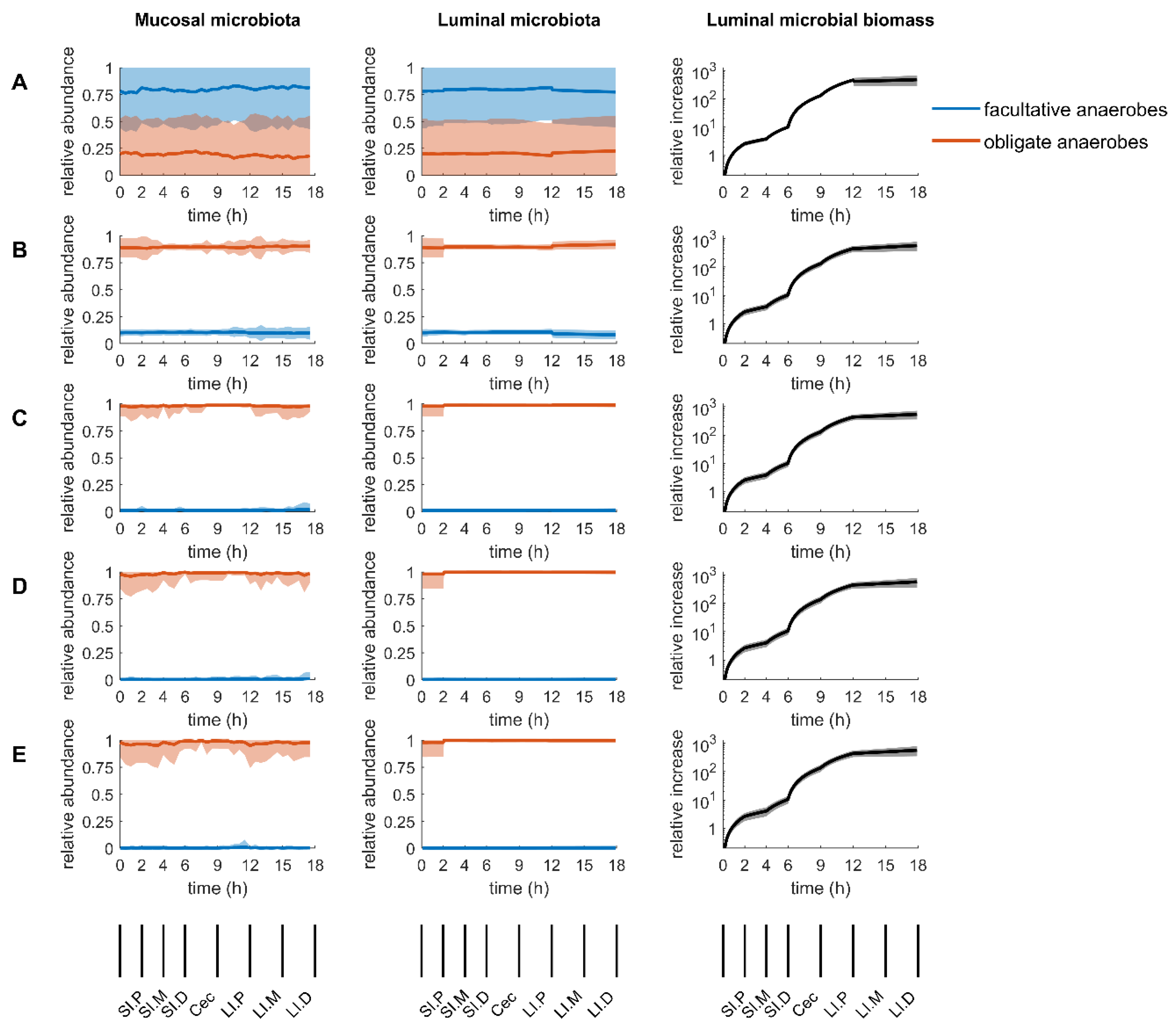
2.2. Overview of the Simulation Results
2.3. Mucosal Microbiota
2.4. Luminal Microbiota
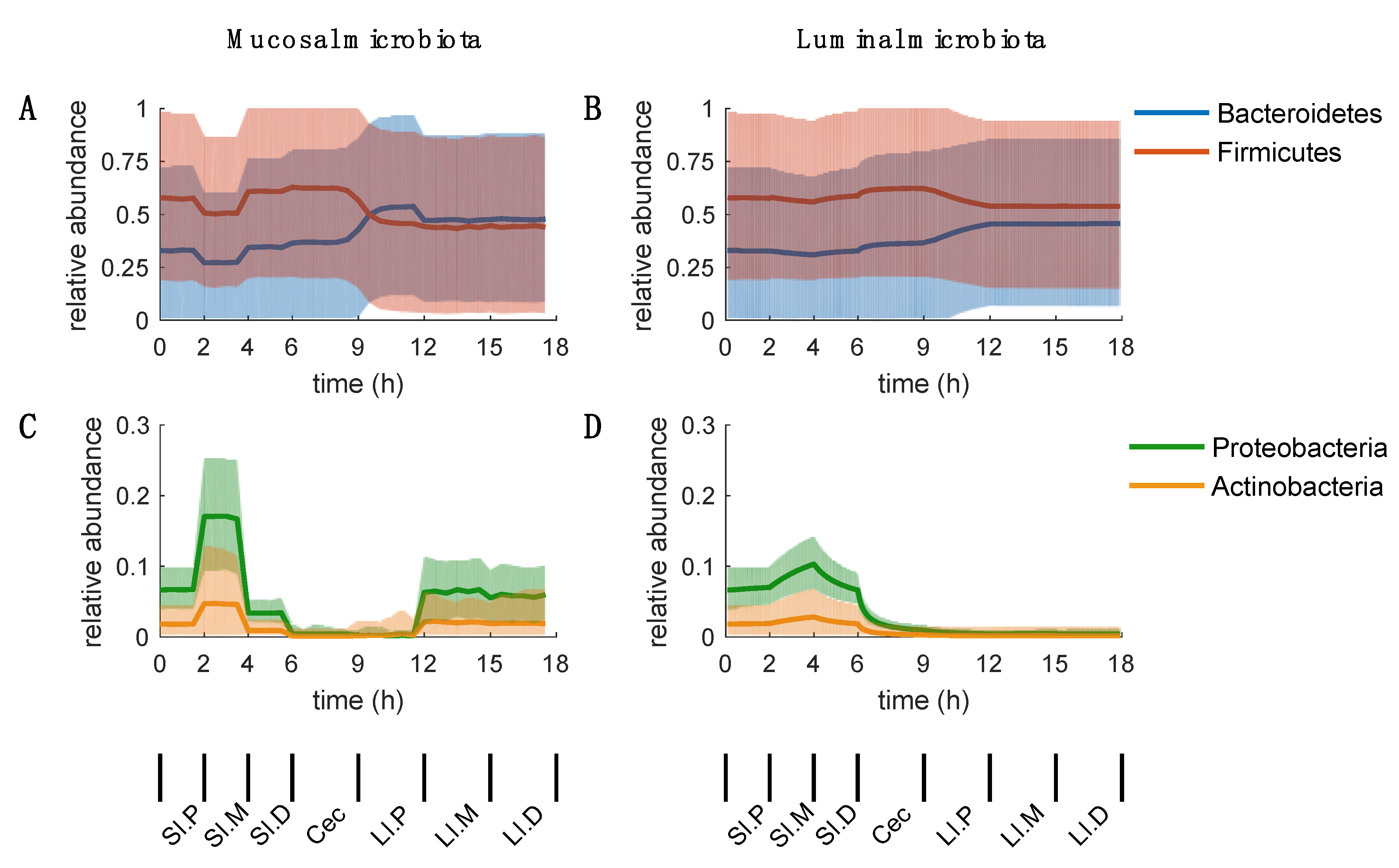
2.5. Inconclusive Firmicutes-to-Bacteroidetes Ratio
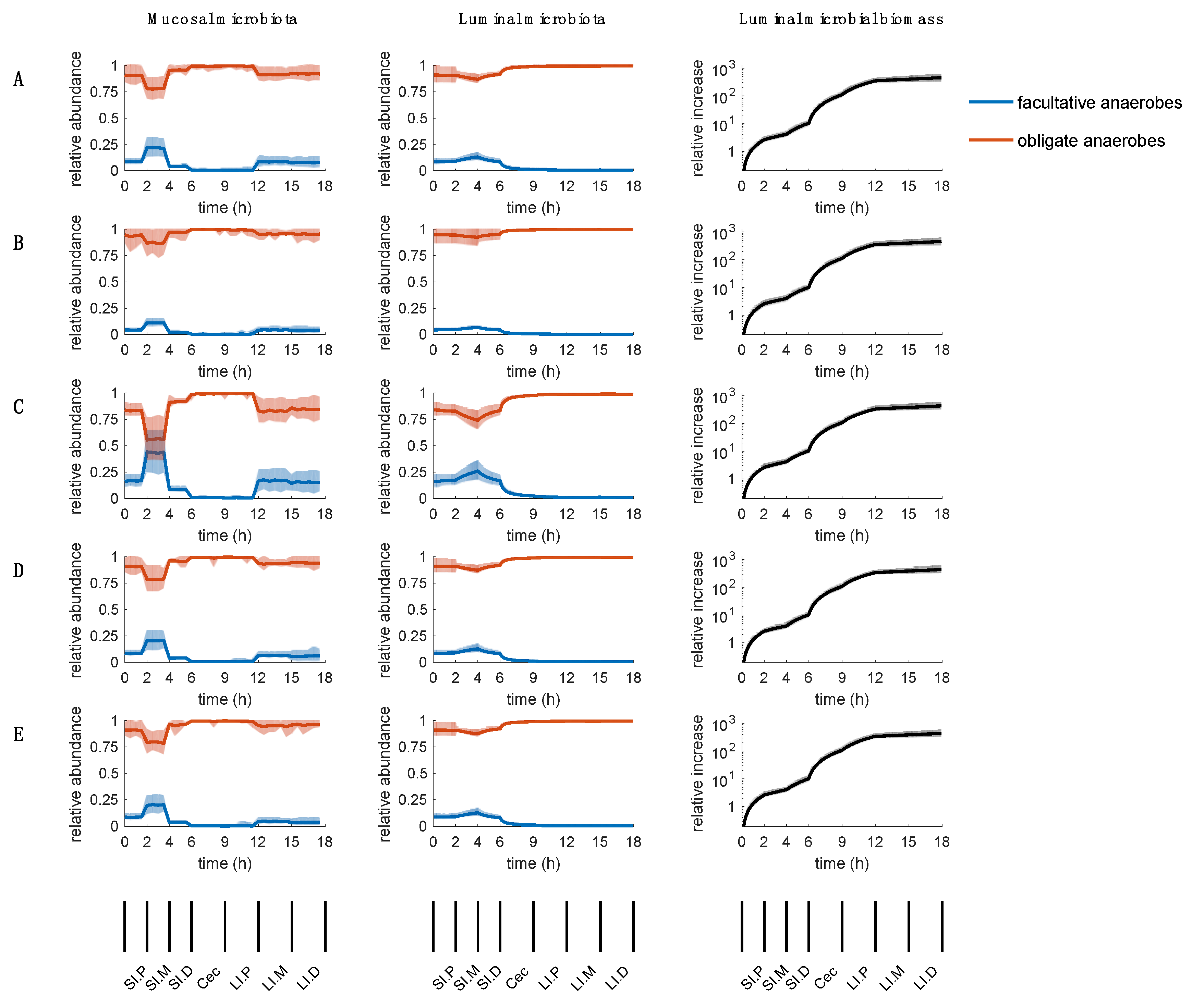
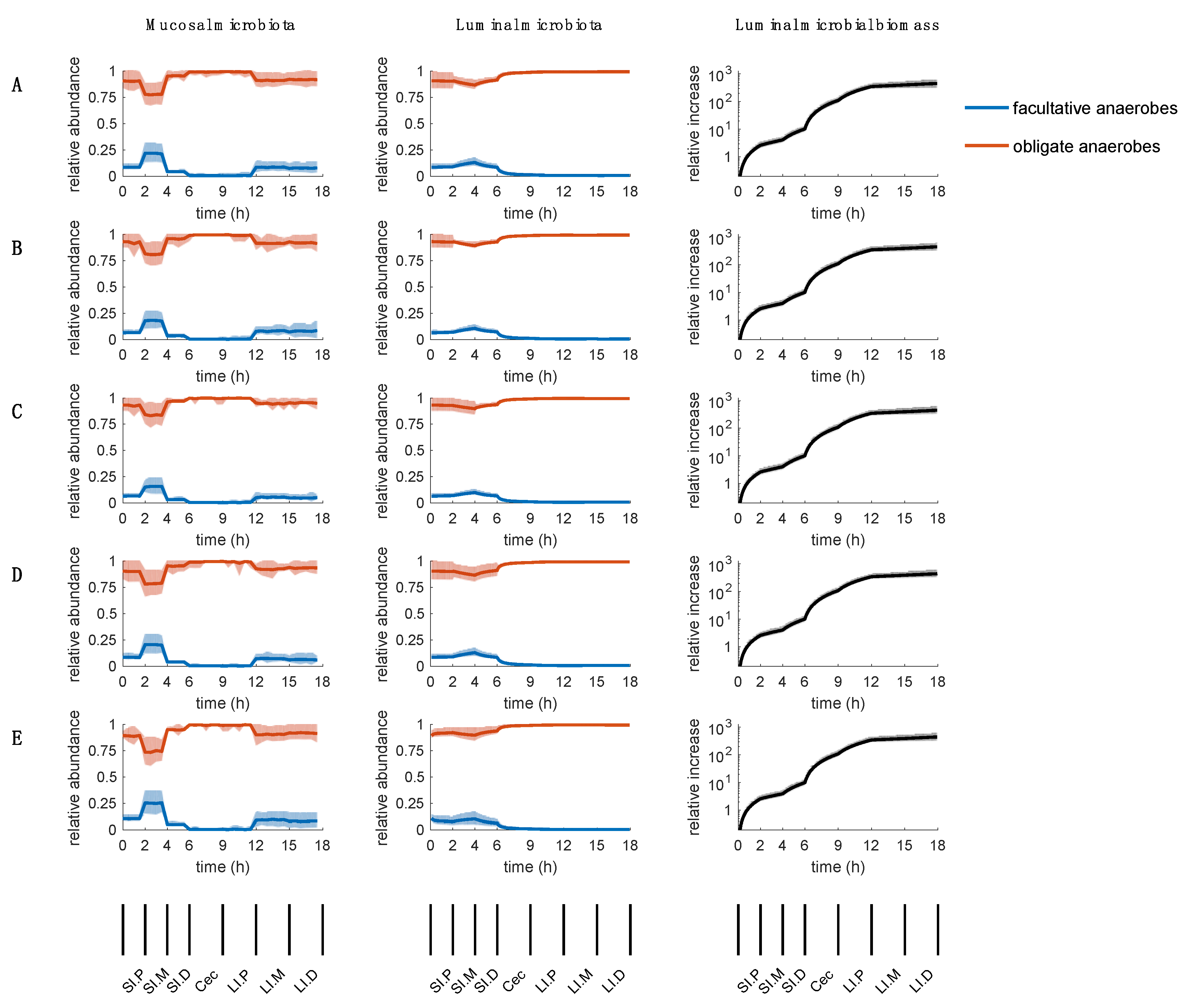
2.6. Sensitivity of Parameters
3. Discussion
3.1. A Dynamic Model Capturing the Spatial Distribution of Aerobes vs Anaerobes
3.2. Parameters Regarding Oxygen Availability and Uptake
3.3. Potential Future Extension of the Model
4. Materials and Methods
4.1. Metabolic Models and Nutrient Availability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Phylum | Ref. |
|---|---|---|
| Bacteroides thetaiotaomicron (B. thetaiotaomicron) | Bacteroidetes | [36,38] |
| Eubacterium rectale (E. rectale) | Firmicutes | [39] |
| Faecalibacterium prausnitzii (F. prausnitzii) | Firmicutes | [62] |
| Escherichia coli (E. coli) | Proteobacteria | [63] |
| Corynebacterium glutamicum (C. glutamicum) | Actinobacteria | [64] |
4.2. Mucosal Microbiota
| SI.P | SI.M | SI.D | Cecum | LI.P | LI.M | LI.D | |
|---|---|---|---|---|---|---|---|
| Default | 6.75 × 10−6 | 2.7 × 10−6 | 1.5 × 10−5 | 1.95 × 10−4 | 5.25 × 10−4 | 7.5 × 10−6 | 9 × 10−6 |
| Test 1 | 1 × 10−6 | 1 × 10−6 | 1 × 10−6 | 1 × 10−6 | 1 × 10−6 | 1 × 10−6 | 1 × 10−6 |
| Test 2 | 1 × 10−5 | 1 × 10−5 | 1 × 10−5 | 1 × 10−5 | 1 × 10−5 | 1 × 10−5 | 1 × 10−5 |
| Test 3 | 1 × 10−4 | 1 × 10−4 | 1 × 10−4 | 1 × 10−4 | 1 × 10−4 | 1 × 10−4 | 1 × 10−4 |
4.3. Luminal Microbiota
4.4. Connecting the Luminal and Mucosal Microbiota
4.5. Oxygen Availability
(mmol h−1g−1) | SI.P | SI.M | SI.D | Cecum | LI.P | LI.M | LI.D |
|---|---|---|---|---|---|---|---|
| Default | 1.6 × 10−6 | 1.6 × 10−6 | 1.6 × 10−6 | 1.6 × 10−6 | 1.6 × 10−6 | 1.6 × 10−6 | 1.6 × 10−6 |
| Test 1 | 0.8 × 10−6 | 0.8 × 10−6 | 0.8 × 10−6 | 0.8 × 10−6 | 0.8 × 10−6 | 0.8 × 10−6 | 0.8 × 10−6 |
| Test 2 | 3.2 × 10−6 | 3.2 × 10−6 | 3.2 × 10−6 | 3.2 × 10−6 | 3.2 × 10−6 | 3.2 × 10−6 | 3.2 × 10−6 |
| Test 3 | 1.6 × 10−6 | 1.52 × 10−6 | 1.44 × 10−6 | 1.36 × 10−6 | 1.28 × 10−6 | 1.2 × 10−6 | 1.12× 10−6 |
| Test 4 | 1.6 × 10−6 | 1.44 × 10−6 | 1.28 × 10−6 | 1.12 × 10−6 | 0.96 × 10−6 | 0.8 × 10−6 | 0.64× 10−6 |
| SI.P | SI.M | SI.D | Cecum | LI.P | LI.M | LI.D | |
|---|---|---|---|---|---|---|---|
| Default | 0.2 | 0.15 | 0.15 | 0.01 | 0.05 | 0.05 | 0 |
| Test 1 | 0.4 | 0.3 | 0.3 | 0.05 | 0.1 | 0.1 | 0 |
| Test 2 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 |
| Test 3 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 |
| Test 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
(mmol gdw−1h−1) | SI.P | SI.M | SI.D | Cecum | LI.P | LI.M | LI.D |
|---|---|---|---|---|---|---|---|
| Default | 0.24 | 0.59 | 0.11 | 0.008 | 0.003 | 0.21 | 0.18 |
| Test 1 | 2.4 | 2.4 | 2.4 | 2.4 | 2.4 | 2.4 | 2.4 |
| Test 2 | 0.24 | 0.24 | 0.24 | 0.24 | 0.24 | 0.24 | 0.24 |
| Test 3 | 0.024 | 0.024 | 0.024 | 0.024 | 0.024 | 0.024 | 0.024 |
| Test 4 | 0.0024 | 0.0024 | 0.0024 | 0.0024 | 0.0024 | 0.0024 | 0.0024 |
| Test 5 | 0.00024 | 0.00024 | 0.00024 | 0.00024 | 0.00024 | 0.00024 | 0.00024 |
4.6. Simulation Parameters
4.7. Availability of Data and Materials
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.A.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2015, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Lv, Y.; Li, J.; Krausz, K.W.; Shi, J.; Brocker, C.N.; Desai, D.; Amin, S.G.; Bisson, W.H.; et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 2015, 6, 10166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xie, C.; Nichols, R.G.; Chan, S.H.J.; Jiang, C.; Hao, R.; Smith, P.B.; Cai, J.; Simons, M.N.; Hatzakis, E.; et al. Farnesoid X receptor signaling shapes the gut microbiota and controls hepatic lipid metabolism. MSystems 2016, 1, e00070-16. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Cox, I.J.; Betrapally, N.S.; Heuman, D.M.; Schubert, M.L.; Ratneswaran, M.; Hylemon, P.B.; White, M.B.; Daita, K.; Noble, N.A.; et al. Systems biology analysis of omeprazole therapy in cirrhosis demonstrates significant shifts in gut microbiota composition and function. AJP Gastrointest. Liver Physiol. 2014, 307, G951–G957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, D.-H.; Kim, I.S.; Van Le, T.K.; Jung, I.-H.; Yoo, H.H.; Kim, D.-H. Gut microbiota-mediated drug interactions between lovastatin and antibiotics. Drug Metab. Dispos. 2014, 42, 1508–1513. [Google Scholar] [CrossRef]
- Wilson, I.D.; Nicholson, J.K. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl. Res. 2017, 179, 204–222. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.-Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, D.; Michael, M.; Rajput, H.; Patil, R.T. Dietary fibre in foods: A review. J. Food Sci. Technol. 2012, 49, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Compher, C.; Chen, E.Z.; Smith, S.A.; Shah, R.D.; Bittinger, K.; Chehoud, C.; Albenberg, L.G.; Nessel, L.; Gilroy, E.; et al. Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production. Gut 2016, 65, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–228. [Google Scholar] [CrossRef]
- Liang, X.; Bushman, F.D.; FitzGerald, G.A. Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc. Natl. Acad. Sci. USA 2015, 112, 10479–10484. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, H.E.; Jernberg, C.; Andersson, A.F.; Sjölund-Karlsson, M.; Jansson, J.K.; Engstrand, L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS ONE 2010, 5, e9836. [Google Scholar] [CrossRef]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.Z.; Li, J.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Gillevet, P.; Sikaroodi, M.; Keshavarzian, A.; Mutlu, E.A. Quantitative assessment of the human gut microbiome using multitag pyrosequencing. Chem. Biodivers. 2010, 7, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Albenberg, L.; Esipova, T.V.; Judge, C.P.; Bittinger, K.; Chen, J.; Laughlin, A.; Grunberg, S.; Baldassano, R.N.; Lewis, J.D.; Li, H.; et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 2014, 147, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Bittinger, K.; Li, X.; Abernethy, D.R.; Bushman, F.D.; FitzGerald, G.A. Bidirectional interactions between indomethacin and the murine intestinal microbiota. Elife 2015, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.S.; Bittinger, K.; Esipova, T.V.; Hou, L.; Chau, L.; Jiang, J.; Mesaros, C.; Lund, P.J.; Liang, X.; FitzGerald, G.A.; et al. Microbes vs. chemistry in the origin of the anaerobic gut lumen. Proc. Natl. Acad. Sci. USA 2018, 115, 4170–4175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swidsinski, A.; Sydora, B.C.; Doerffel, Y.; Loening-Baucke, V.; Vaneechoutte, M.; Lupicki, M.; Scholze, J.; Lochs, H.; Dieleman, L.A. Viscosity gradient within the mucus layer determines the mucosal barrier function and the spatial organization of the intestinal microbiota. Inflamm. Bowel Dis. 2007, 13, 963–970. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, D.; Palsson, B.O.; Feist, A.M. Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol. Syst. Biol. 2014, 9, 661. [Google Scholar] [CrossRef]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Varma, A.; Palsson, B.O. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl. Environ. Microbiol. 1994, 60, 3724–3731. [Google Scholar]
- Stolyar, S.; Van Dien, S.; Hillesland, K.L.; Pinel, N.; Lie, T.J.; Leigh, J.A.; Stahl, D.A. Metabolic modeling of a mutualistic microbial community. Mol. Syst. Biol. 2007, 3, 92. [Google Scholar] [CrossRef]
- Klitgord, N.; Segrè, D. Environments that induce synthetic microbial ecosystems. PLoS Comput. Biol. 2010, 6, e1001002. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Zarecki, R.; Eilam, O.; Segal, E.S.; Henry, C.S.; Kupiec, M.; Gophna, U.; Sharan, R.; Ruppin, E. Competitive and cooperative metabolic interactions in bacterial communities. Nat. Commun. 2011, 2, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomorrodi, A.R.; Maranas, C.D. OptCom: A multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput. Biol. 2012, 8, e1002363. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, R.A.; Olivier, B.G.; Röling, W.F.M.; Teusink, B.; Bruggeman, F.J. Community flux balance analysis for microbial consortia at balanced growth. PLoS ONE 2013, 8, e64567. [Google Scholar] [CrossRef] [PubMed]
- Zelezniak, A.; Andrejev, S.; Ponomarova, O.; Mende, D.R.; Bork, P.; Patil, K.R. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. USA 2015, 112, 6449–6454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinken, A.; Sahoo, S.; Fleming, R.M.T.; Thiele, I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 2013, 4, 28–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinken, A.; Thiele, I. Anoxic conditions promote species-specific mutualism between gut microbes in silico. Appl. Environ. Microbiol. 2015, 81, 4049–4061. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Thiele, I. Systematic prediction of health-relevant human-microbial co-metabolism through a computational framework. Gut Microbes 2015, 6, 120–130. [Google Scholar] [CrossRef]
- Shoaie, S.; Karlsson, F.; Mardinoglu, A.; Nookaew, I.; Bordel, S.; Nielsen, J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci. Rep. 2013, 3, 2532. [Google Scholar] [CrossRef]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; de Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Shoaie, S.; Bergentall, M.; Ghaffari, P.; Zhang, C.; Larsson, E.; Backhed, F.; Nielsen, J. The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 2015, 11, 834. [Google Scholar] [CrossRef] [PubMed]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2016, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.J.; Simons, M.N.; Maranas, C.D. SteadyCom: Predicting microbial abundances while ensuring community stability. PLoS Comput. Biol. 2017, 13, e1005539. [Google Scholar] [CrossRef] [PubMed]
- Henson, M.A.; Phalak, P. Suboptimal community growth mediated through metabolite crossfeeding promotes species diversity in the gut microbiota. PLoS Comput. Biol. 2018, 14, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, K.; Izallalen, M.; Mouser, P.; Richter, H.; Risso, C.; Mahadevan, R.; Lovley, D.R. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 2011, 5, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Hanly, T.J.; Henson, M.A. Dynamic flux balance modeling of microbial co-cultures for efficient batch fermentation of glucose and xylose mixtures. Biotechnol. Bioeng. 2011, 108, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.R.; Islam, M.M.; Maranas, C.D. d-OptCom: Dynamic multi-level and multi-objective metabolic modeling of microbial communities. ACS Synth. Biol. 2014, 3, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Wilken, S.; Saxena, M.; Petzold, L.; O’Malley, M. In silico identification of microbial partners to form consortia with anaerobic fungi. Processes 2018, 6, 7. [Google Scholar] [CrossRef]
- Harcombe, W.R.; Riehl, W.J.; Dukovski, I.; Granger, B.R.; Betts, A.; Lang, A.H.; Bonilla, G.; Kar, A.; Leiby, N.; Mehta, P.; et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014, 7, 1104–1115. [Google Scholar] [CrossRef]
- Van Hoek, M.J.A.; Merks, R.M.H. Emergence of microbial diversity due to cross-feeding interactions in a spatial model of gut microbial metabolism. BMC Syst. Biol. 2017, 11, 56. [Google Scholar] [CrossRef]
- Henson, M.; Phalak, P. Byproduct cross feeding and community stability in an in silico biofilm model of the gut microbiome. Processes 2017, 5, 13. [Google Scholar] [CrossRef]
- Bauer, E.; Zimmermann, J.; Baldini, F.; Thiele, I.; Kaleta, C. BacArena: Individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput. Biol. 2017, 13, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Phalak, P.; Chen, J.; Carlson, R.P.; Henson, M.A. Metabolic modeling of a chronic wound biofilm consortium predicts spatial partitioning of bacterial species. BMC Syst. Biol. 2016, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Song, H.S.; Cannon, W.; Beliaev, A.; Konopka, A. Mathematical modeling of microbial community dynamics: A methodological review. Processes 2014, 2, 711–752. [Google Scholar] [CrossRef]
- Van der Ark, K.C.H.; van Heck, R.G.A.; Martins Dos Santos, V.A.P.; Belzer, C.; de Vos, W.M. More than just a gut feeling: Constraint-based genome-scale metabolic models for predicting functions of human intestinal microbes. Microbiome 2017, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Magnúsdóttir, S.; Thiele, I. Modeling metabolism of the human gut microbiome. Curr. Opin. Biotechnol. 2018, 51, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef]
- Fong, S.S.; Marciniak, J.Y.; Palsson, B.O. Description and interpretation of adaptive evolution of Escherichia coli K-12 MG1655 by using a genome-scale in silico metabolic model. J. Bacteriol. 2003, 185, 6400–6408. [Google Scholar] [CrossRef]
- Ibarra, R.U.; Edwards, J.S.; Palsson, B.O. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 2002, 420, 186–189. [Google Scholar] [CrossRef]
- Chan, S.H.J.; Cai, J.; Wang, L.; Simons-Senftle, M.N.; Maranas, C.D. Standardizing biomass reactions and ensuring complete mass balance in genome-scale metabolic models. Bioinformatics 2017, 33, 3603–3609. [Google Scholar] [CrossRef]
- Rose, C.; Parker, A.; Jefferson, B.; Cartmell, E. The characterization of feces and urine: A review of the literature to inform advanced treatment technology. Crit. Rev. Environ. Sci. Technol. 2015, 45, 1827–1879. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Khan, M.T.; Paglia, G.; Rodionov, D.A.; Harmsen, H.J.M.; Thiele, I. Functional metabolic map of Faecalibacterium prausnitzii, a beneficial human gut microbe. J. Bacteriol. 2014, 196, 3289–3302. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Conrad, T.M.; Na, J.; Lerman, J.A.; Nam, H.; Feist, A.M.; Palsson, B.Ø. A comprehensive genome-scale reconstruction of Escherichia coli metabolism 2011. Mol. Syst. Biol. 2011, 7, 535. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, K.R.; Nielsen, J. In silico genome-scale reconstruction and validation of the Corynebacterium glutamicum metabolic network. Biotechnol. Bioeng. 2009, 102, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The long-term stability of the human gut microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef]
- Kiefer, P.; Heinzle, E.; Wittmann, C. Influence of glucose, fructose and sucrose as carbon sources on kinetics and stoichiometry of lysine production by Corynebacterium glutamicum. J. Ind. Microbiol. Biotechnol. 2002, 28, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Shinfuku, Y.; Sorpitiporn, N.; Sono, M.; Furusawa, C.; Hirasawa, T.; Shimizu, H. Development and experimental verification of a genome-scale metabolic model for Corynebacterium glutamicum. Microb. Cell Fact. 2009, 8, 43. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, S.H.J.; Friedman, E.S.; Wu, G.D.; Maranas, C.D. Predicting the Longitudinally and Radially Varying Gut Microbiota Composition using Multi-Scale Microbial Metabolic Modeling. Processes 2019, 7, 394. https://0-doi-org.brum.beds.ac.uk/10.3390/pr7070394
Chan SHJ, Friedman ES, Wu GD, Maranas CD. Predicting the Longitudinally and Radially Varying Gut Microbiota Composition using Multi-Scale Microbial Metabolic Modeling. Processes. 2019; 7(7):394. https://0-doi-org.brum.beds.ac.uk/10.3390/pr7070394
Chicago/Turabian StyleChan, Siu H. J., Elliot S. Friedman, Gary D. Wu, and Costas D. Maranas. 2019. "Predicting the Longitudinally and Radially Varying Gut Microbiota Composition using Multi-Scale Microbial Metabolic Modeling" Processes 7, no. 7: 394. https://0-doi-org.brum.beds.ac.uk/10.3390/pr7070394