Investigation of Virulence Genes Detected in Antimicrobial-Resistance Pathogens Isolates for Five Countries across the World

Abstract
:1. Introduction
2. Materials and Methods
2.1. The Historical Data on Virulence Genes from the NCBI Pathogen Detection Isolates Browser
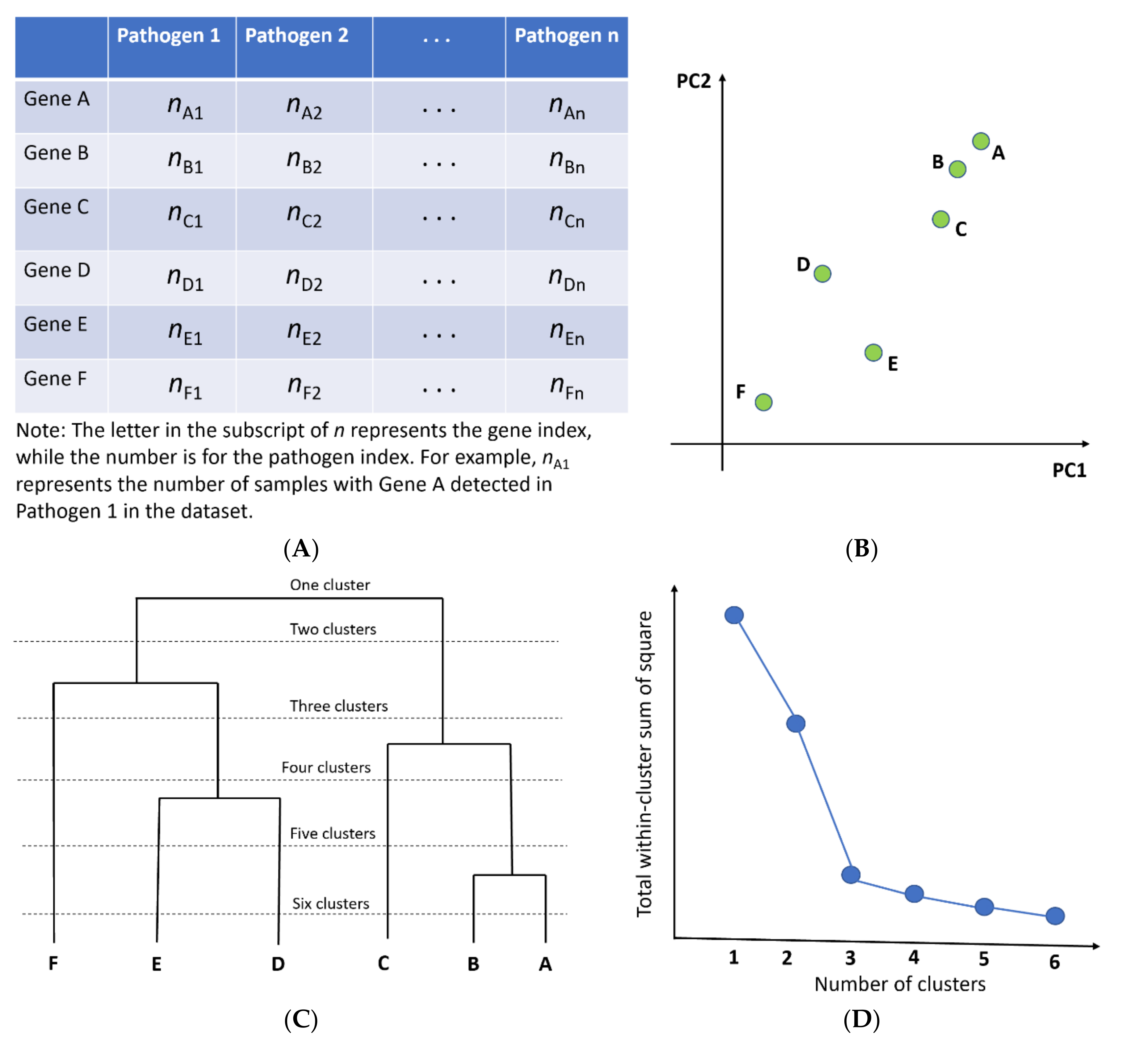
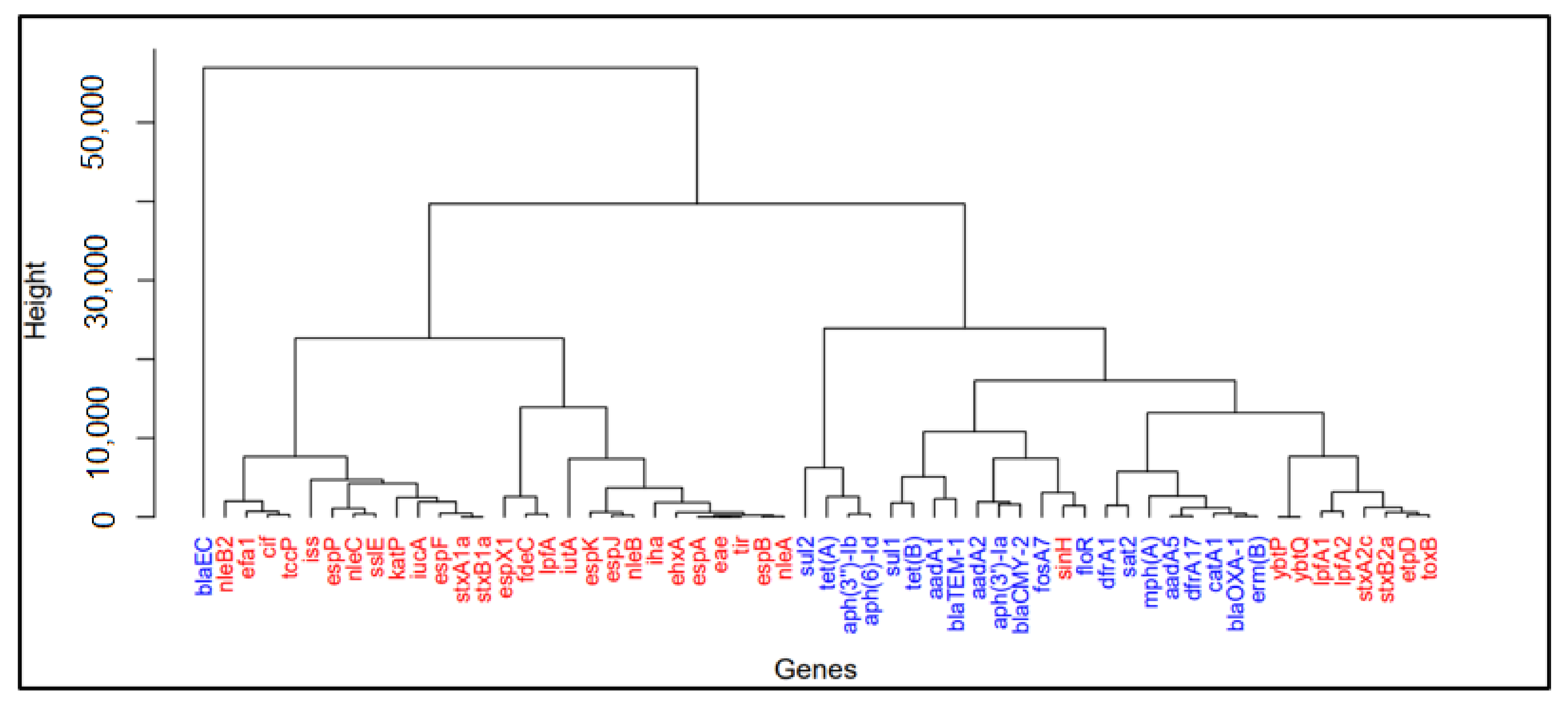
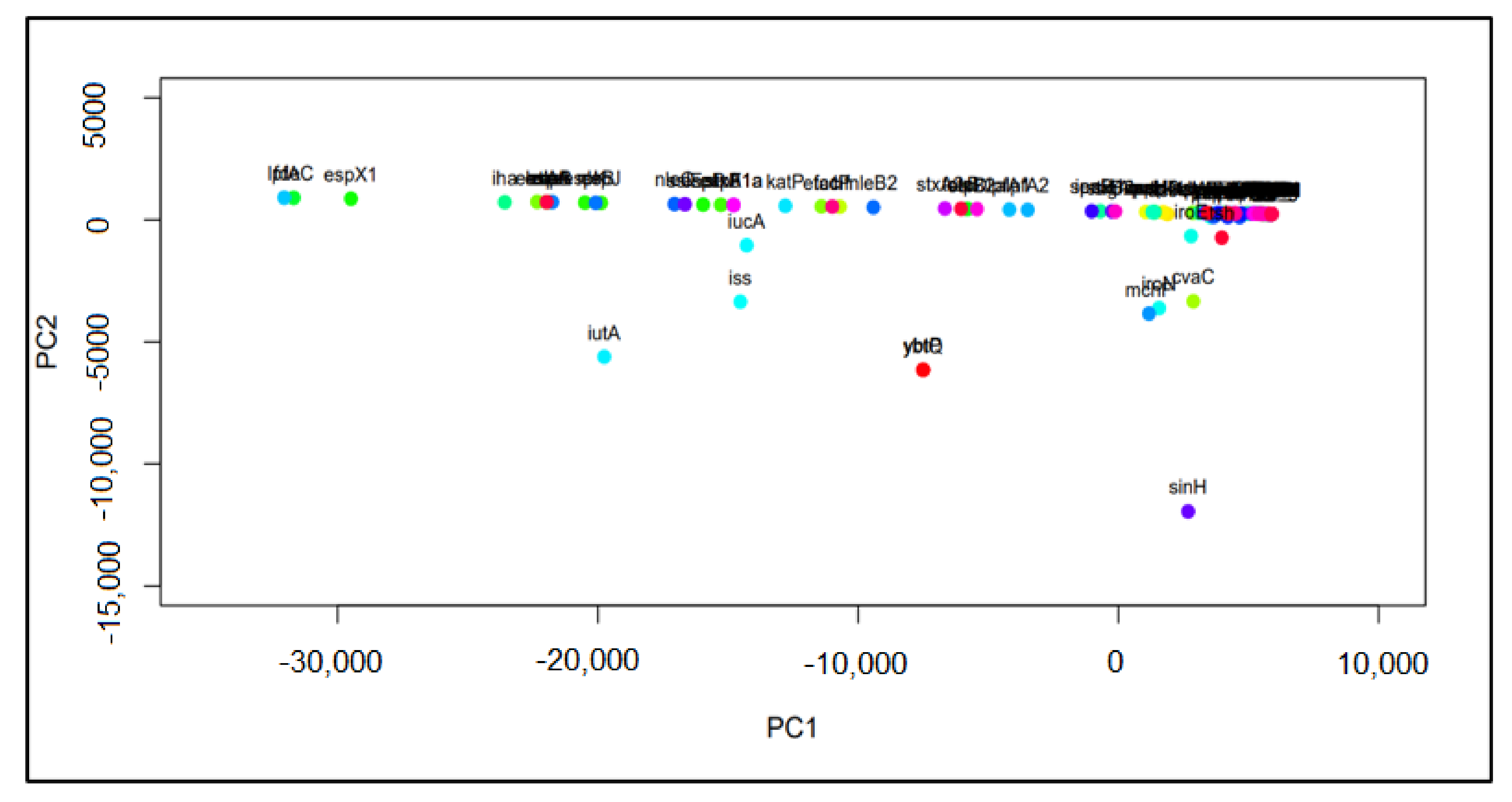
2.2. Principal Component Analysis (PCA) and Hierarchical Clustering
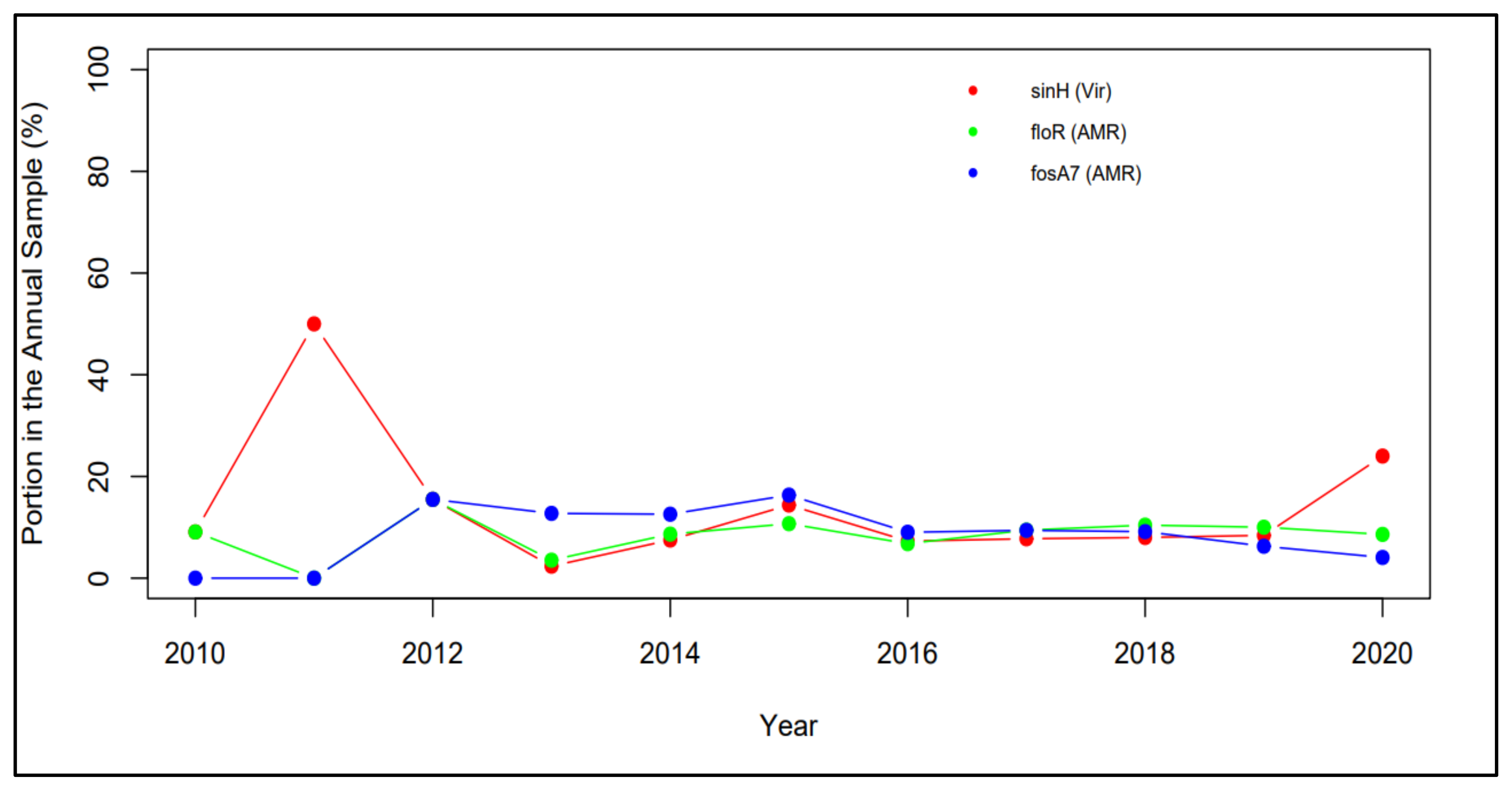
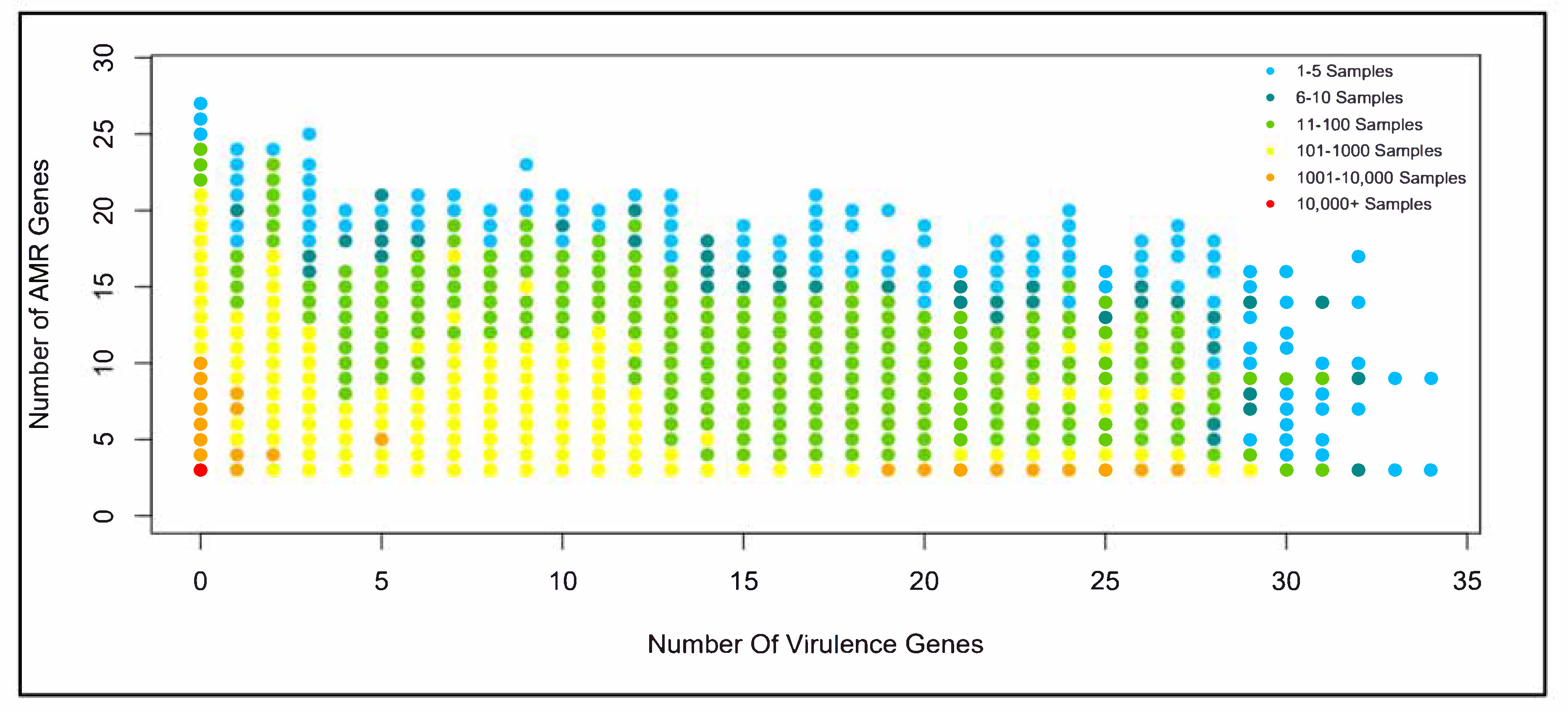
2.3. Time Profiles and Frequency Scatterplots
3. Results
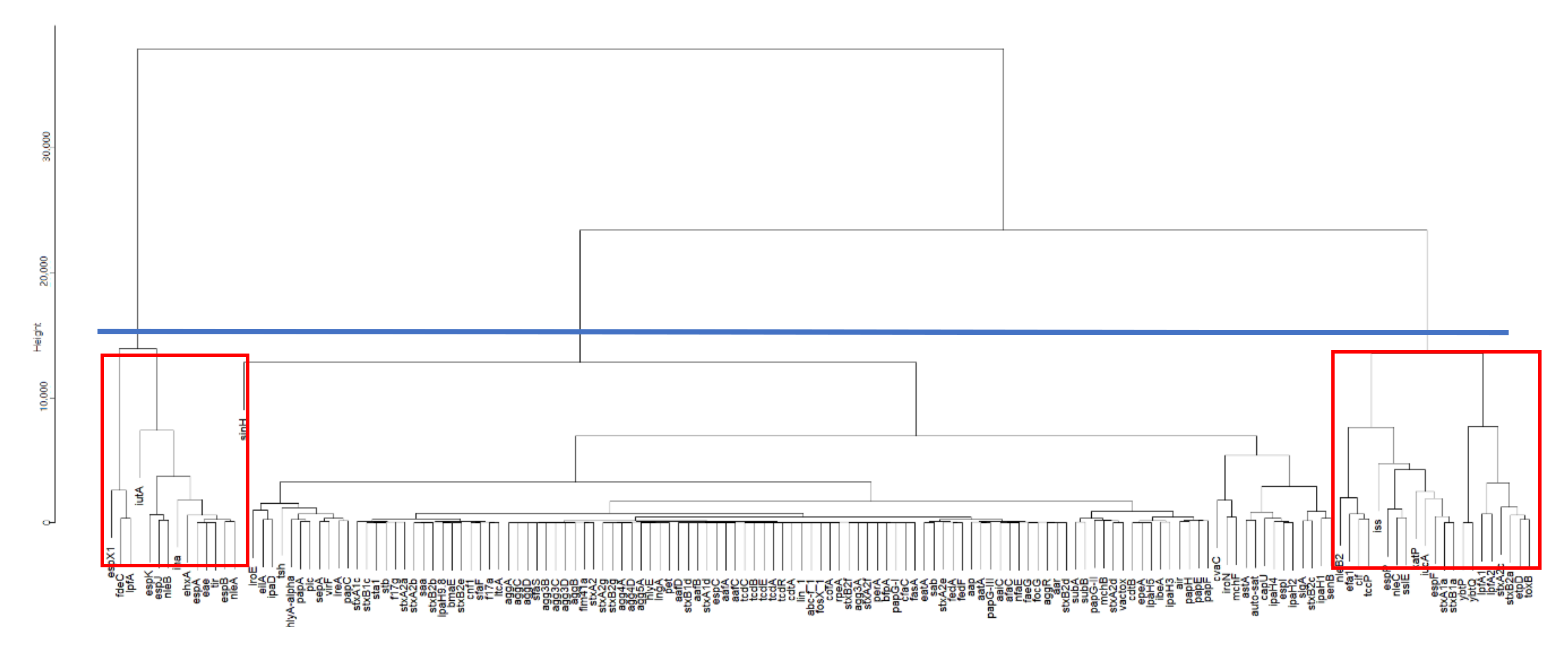
3.1. Important Virulence Genes in Samples from Australia, China, South Africa, the UK, and the US
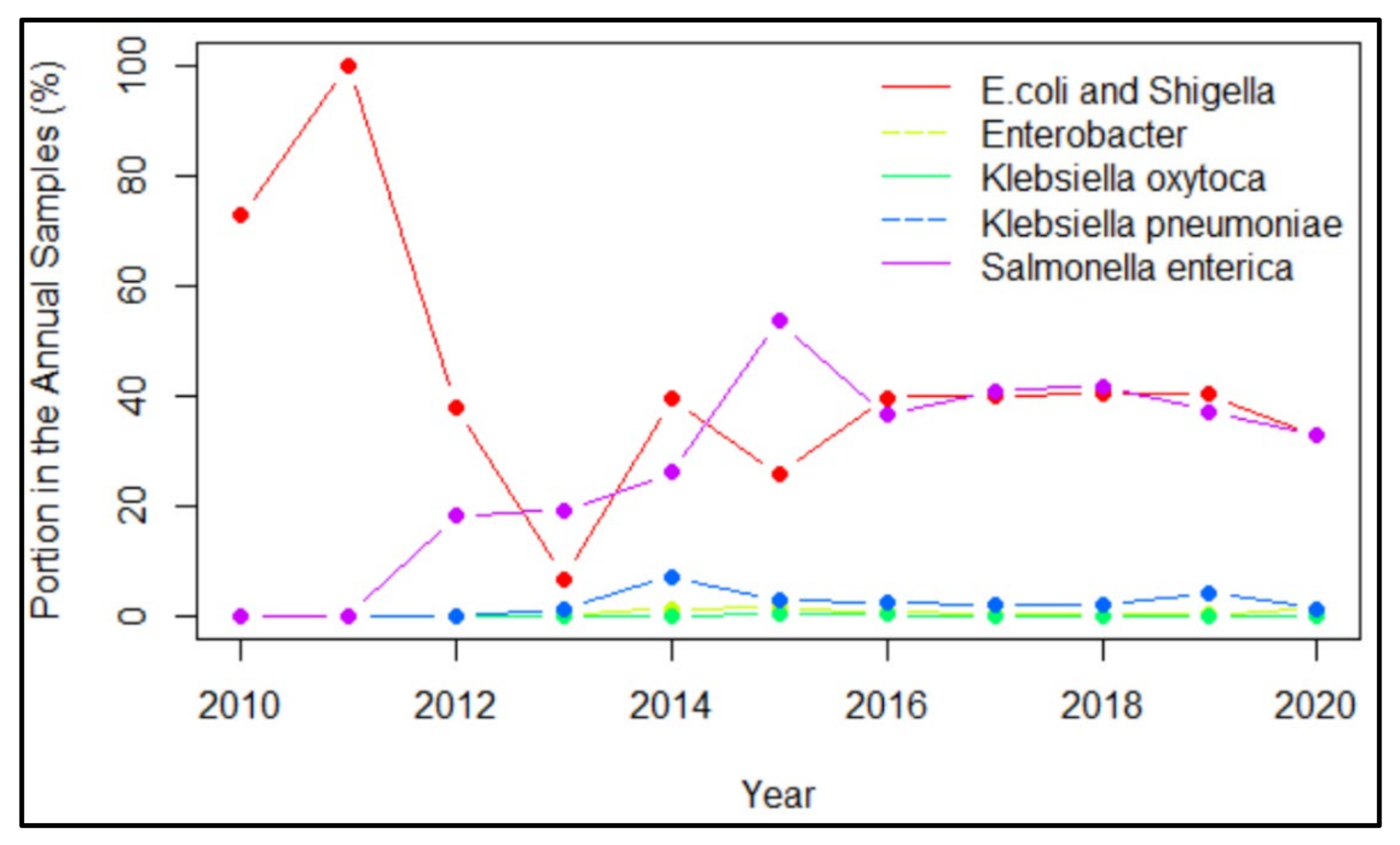
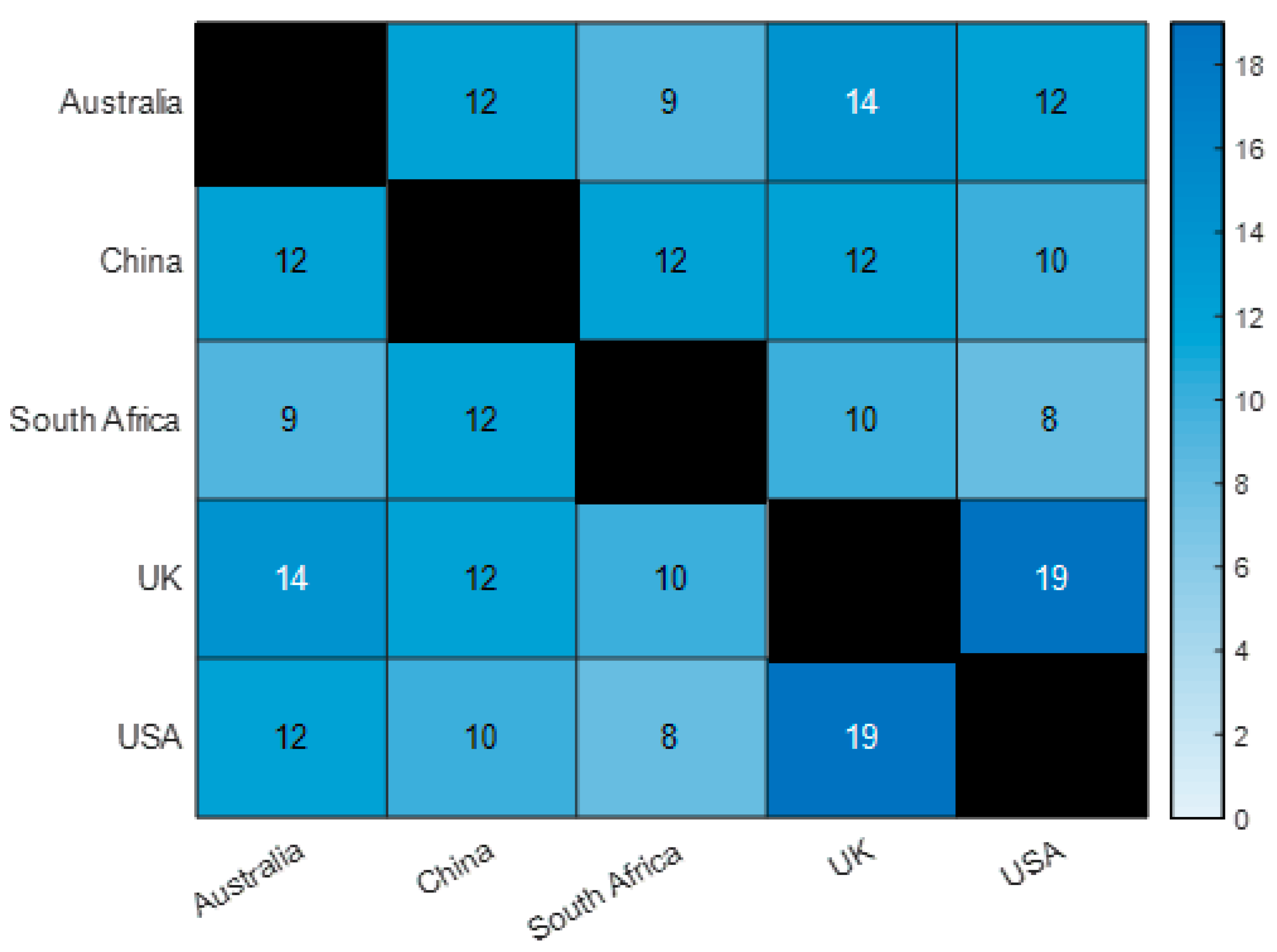
3.2. Important Pathogens Carrying Virulence Genes in Samples from Five Countries
3.3. Comparisons between AMR Genes and Virulence Genes
4. Discussion
4.1. Similarities of Virulence Genes across Five Selected Countries
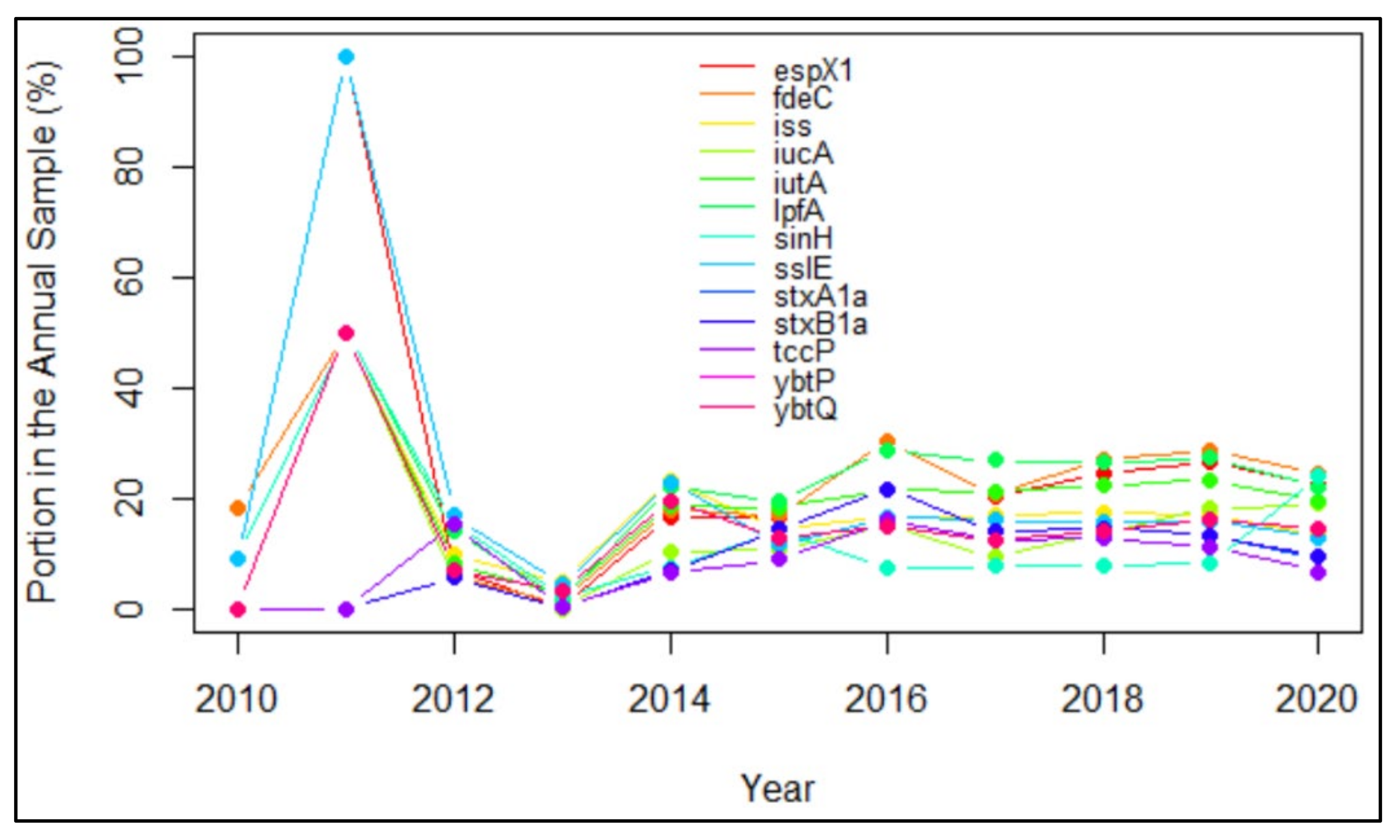
4.2. The Trend of Virulence Genes Indicated from Time Profiles
4.3. Correlation between Virulence Genes and AMR Genes
4.4. Limitations and Future Work
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Australia | China | South Africa | UK | US |
|---|---|---|---|---|
| air | astA | auto-sat | auto-sat | cif |
| astA | auto-sat | cvaC | espF | eae |
| espX1 | capU | fdeC | espX1 | efa1 |
| fdeC | cvaC | iha | etpD | ehxA |
| hlyA-alpha | eilA | iroN | fdeC | espA |
| ibeA | espX1 | iss | iha | espB |
| iha | fdeC | iutA | ireA | espF |
| ipaD | iha | lpfA | iss | espJ |
| ipaH1 | iroE | mchF | iutA | espK |
| iss | iroN | papA | lpfA | espP |
| iucA | iss | senB | lpfA1 | espX1 |
| iutA | iucA | sslE | lpfA2 | etpD |
| lpfA | iutA | vactox | mchF | fdeC |
| mchB | lpfA | ybtP | nleB2 | Iha |
| papE | mchF | ybtQ | pic | iss |
| papF | pic | aac(2′)-Ic | sepA | iucA |
| papH | sslE | blaA | sinH | iutA |
| pic | ybtP | erm(37) | sslE | katP |
| senB | ybtQ | fosX | stxA1a | lpfA |
| sepA | aadA2 | lin | stxA2c | lpfA1 |
| sigA | ant(3″)-IIa | aph(3″)-Ib | stxB1a | lpfA2 |
| sinH | aph(3″)-Ib | aph(6)-Id | stxB2a | nleA |
| sslE | aph(3′)-Ia | blaCTX-M-15 | stxB2c | nleB |
| stxB2c | aph(6)-Id | blaTEM-1 | virF | nleB2 |
| toxB | armA | sul1 | ybtP | nleC |
| tsh | blaEC | sul2 | ybtQ | sinH |
| virF | blaKPC-2 | aadA1 | sslE | |
| ybtP | blaOXA-23 | aph(3″)-Ib | stxA1a | |
| ybtQ | blaOXA-66 | aph(6)-Id | stxA2c | |
| aadA1 | blaSHV-11 | blaEC | stxB1a | |
| aadA5 | blaTEM-1 | blaOXA-1 | stxB2a | |
| ant(3″)-IIa | dfrA12 | blaTEM-1 | tccP | |
| aph(3″)-Ib | floR | catA1 | tir | |
| aph(3′)-Ia | fosA | dfrA1 | toxB | |
| aph(6)-Id | mcr-1.1 | mph(A) | ybtP | |
| blaADC-30 | mph(A) | sat2 | ybtQ | |
| blaEC | mph(E) | sul1 | aadA1 | |
| blaOXA-23 | msr(E) | sul2 | aadA2 | |
| blaOXA-66 | oqxA | tet(A) | aadA5 | |
| blaTEM-1 | oqxB | tet(B) | aph(3″)-Ib | |
| dfrA1 | aph(3′)-Ia | |||
| dfrA17 | aph(6)-Id | |||
| erm(B) | blaCMY-2 | |||
| mph(A) | blaEC | |||
| sat2 | blaOXA-1 | |||
| sul1 | blaTEM-1 | |||
| sul2 | catA1 | |||
| tet(A) | dfrA1 | |||
| tet(B) | dfrA17 | |||
| erm(B) | ||||
| floR | ||||
| fosA7 | ||||
| mph(A) | ||||
| sat2 | ||||
| sul1 | ||||
| sul2 | ||||
| tet(A) | ||||
| tet(B) |


References
- Welch, M.D. Why should cell biologists study microbial pathogens? Mol. Biol. Cell 2015, 26, 4295–4301. [Google Scholar] [CrossRef]
- Malik, B.; Bhattacharyya, S. Antibiotic Drug-Resistance as a Complex System Driven by Socio-Economic Growth and Antibiotic Misuse. Sci. Rep. 2019, 9, 9788. [Google Scholar] [CrossRef]
- Pechere, J.C. Patients’ Interviews and Misuse of Antibiotics. Clin. Infect. Dis. 2001, 33, S170–S173. [Google Scholar] [CrossRef] [Green Version]
- Anthouard, R.; DiRita, V. Chemical Biology Applied to the Study of Bacterial Pathogens. Infect. Immun. 2014, 83, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Totsika, M. Benefits and Challenges of Antivirulence Antimicrobials at the Dawn of the Post-Antibiotic Era. Drug Deliv. Lett. 2016, 6, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Liu, E.; Tang, A.; Ye, M.C.; Wang, K.; Jia, Q.; Huang, Z.J. Data-Driven Analysis of Antimicrobial Resistance in Foodborne Pathogens from Six States within the US. Int. J. Environ. Res. Public Health 2019, 16, 1811. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Wang, A.; Fu, M.; Wang, A.; Chen, K.; Jia, Q.; Huang, Z.J. Investigation of Incidents and Trends of Antimicrobial Resistance in Foodborne Pathogens in Eight Countries from Historical Sample Data. Int. J. Environ. Res. Public Health 2020, 17, 472. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Zheng, J.; Deng, T.; Peng, J.; Daniel, D.; Jia, Q.; Huang, Z.J. An Analysis of Antimicrobial Resistance of Clinical Pathogens from Historical Samples for Six Countries. Processes 2019, 7, 964. [Google Scholar] [CrossRef] [Green Version]
- Hua, M.; Huang, W.; Chen, A.; Rehmet, M.; Jin, C.; Huang, Z.J. Comparison of Antimicrobial Resistance Detected in Environmental and Clinical Isolates from Historical Data for the US. BioMed Res. Int. 2020, 2020, 1–11. [Google Scholar] [CrossRef]
- Lessa, F.; Mu, Y.; Bamberg, W.M.; Beldavs, Z.G.; Dumyati, G.; Dunn, J.R.; Farley, M.M.; Holzbauer, S.M.; Meek, J.I.; Phipps, E.C.; et al. Burden ofClostridium difficileInfection in the United States. N. Engl. J. Med. 2015, 372, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Buroni, S.; Chiarelli, L.R. Antivirulence compounds: A future direction to overcome antibiotic resistance? Future Microbiol. 2020, 15, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.J. Statistical process monitoring: Basics and beyond. J. Chemom. 2003, 17, 480–502. [Google Scholar] [CrossRef]
- Kourti, T. Application of latent variable methods to process control and multivariate statistical process control in industry. Int. J. Adapt. Control Signal Process. 2005, 19, 213–246. [Google Scholar] [CrossRef]
- Arnau, V.; Mars, S.; Marín, I. Iterative Cluster Analysis of Protein Interaction Data. Bioinformatics 2004, 21, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Smith, K.; Hyndman, R. Characteristic-Based Clustering for Time Series Data. Data Min. Knowl. Discov. 2006, 13, 335–364. [Google Scholar] [CrossRef]
- Bar-Joseph, Z.; Demaine, E.D.; Gifford, D.K.; Srebro, N.; Foley, A.M.; Jaakkola, T.S. K-ary clustering with optimal leaf ordering for gene expression data. Bioinformatics 2003, 19, 1070–1078. [Google Scholar] [CrossRef] [Green Version]
- Ihaka, R.; Gentleman, R. R: A Language for data analysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Thorndike, R.L. Who belongs in the family? Psychometrika 1953, 18, 267–276. [Google Scholar] [CrossRef]
- Easton, D.M.; Allsopp, L.P.; Phan, M.-D.; Moriel, D.G.; Goh, G.K.; Beatson, S.A.; Mahony, T.J.; Cobbold, R.N.; Schembri, M.A. The Intimin-Like Protein FdeC Is Regulated by H-NS and Temperature in Enterohemorrhagic Escherichia coli. Appl. Environ. Microbiol. 2014, 80, 7337–7347. [Google Scholar] [CrossRef] [Green Version]
- Léveillé, S.; Caza, M.; Johnson, J.R.; Clabots, C.; Sabri, M.; Dozois, C.M. Iha from an Escherichia coli Urinary Tract Infection Outbreak Clonal Group A Strain Is Expressed In Vivo in the Mouse Urinary Tract and Functions as a Catecholate Siderophore Receptor. Infect. Immun. 2006, 74, 3427–3436. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.G.; Blanco, M.; Valenzuela, P.; Slater, T.M.; Patel, S.D.; Dahbi, G.; López, C.; Barriga, X.F.; Blanco, J.E.; Gomes, T.A.T.; et al. Genes Related to Long Polar Fimbriae of Pathogenic Escherichia coli Strains as Reliable Markers To Identify Virulent Isolates. J. Clin. Microbiol. 2009, 47, 2442–2451. [Google Scholar] [CrossRef] [Green Version]
- Suez, J.; Porwollik, S.; Dagan, A.; Marzel, A.; Schorr, Y.I.; Desai, P.T.; Agmon, V.; McClelland, M.; Rahav, G.; Gal-Mor, O. Virulence Gene Profiling and Pathogenicity Characterization of Non-Typhoidal Salmonella Accounted for Invasive Disease in Humans. PLoS ONE 2013, 8, e58449. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, T.N.; Berlese, A.; Fernandes, F.F.; Milanezi, M.L.; Martinez, R.; Panunto-Castelo, A. The ferric aerobactin receptor IutA, a protein isolated on agarose column, is not essential for uropathogenic Escherichia coli infection. Revista Latino-Americana de Enfermagem 2012, 20, 340–345. [Google Scholar] [CrossRef]
- Koh, E.-I.; Hung, C.S.; Henderson, J.P. The Yersiniabactin-Associated ATP Binding Cassette Proteins YbtP and YbtQ Enhance Escherichia coli Fitness during High-Titer Cystitis. Infect. Immun. 2016, 84, 1312–1319. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.; Pan, H.; Gao, Q.; Xiong, L.; Zhou, Y.; Zhang, D.; Gao, S.; Liu, X. Aerobactin Synthesis Genes iucA and iucC Contribute to the Pathogenicity of Avian Pathogenic Escherichia coli O2 Strain E058. PLoS ONE 2013, 8, e57794. [Google Scholar] [CrossRef]
- Guyer, D.M.; Radulovic, S.; Jones, F.-E.; Mobley, H.L.T. Sat, the Secreted Autotransporter Toxin of Uropathogenic Escherichia coli, Is a Vacuolating Cytotoxin for Bladder and Kidney Epithelial Cells. Infect. Immun. 2002, 70, 4539–4546. [Google Scholar] [CrossRef] [Green Version]
- Tobe, T.; Beatson, S.A.; Taniguchi, H.; Abe, H.; Bailey, C.M.; Fivian, A.; Younis, R.; Matthews, S.; Marches, O.; Frankel, G.; et al. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. USA 2006, 103, 14941–14946. [Google Scholar] [CrossRef] [Green Version]
- Azpiroz, M.F.; Rodrı́guez, E.; Laviña, M. The Structure, Function, and Origin of the Microcin H47 ATP-Binding Cassette Exporter Indicate Its Relatedness to That of Colicin V. Antimicrob. Agents Chemother. 2001, 45, 969–972. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.J.; Wannemuehler, Y.M.; Nolan, L.K. Evolution of the iss Gene in Escherichia coli. Appl. Environ. Microbiol. 2008, 74, 2360–2369. [Google Scholar] [CrossRef] [Green Version]
- DeCanio, M.S.; Landick, R.; Haft, R.J.F. The non-pathogenic Escherichia coli strain W secretes SslE via the virulence-associated type II secretion system beta. BMC Microbiol. 2013, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Australia’s Economic Relationships with China. Available online: https://www.aph.gov.au/about_parliament/parliamentary_departments/parliamentary_library/pubs/briefingbook44p/china (accessed on 23 August 2020).
- Cheng, M. We Depend So Much More on Chinese Travellers Now. That Makes the Impact of This Coronavirus Novel. Available online: https://theconversation.com/we-depend-so-much-more-on-chinese-travellers-now-that-makes-the-impact-of-this-coronavirus-novel-130798 (accessed on 23 August 2020).
- Migration, Australia, 2018–2019. Available online: https://www.abs.gov.au/ausstats/[email protected]/Latestproducts/3412.0Main%20Features32018-19?opendocument&tabname=Summary&prodno=3412.0&issue=2018-19&num=&view= (accessed on 23 August 2020).
- South African Foreign Trade in Figures. Available online: https://santandertrade.com/en/portal/analyse-markets/south-africa/foreign-trade-in-figures#:~:text=South%20Africa%27s%20top%20trading%20partners,largest%20trading%20partner%20in%20Africa (accessed on 23 August 2020).
- The Uk’s Trade and Investment Relationship with Africa: 2016. Available online: https://www.ons.gov.uk/economy/nationalaccounts/balanceofpayments/articles/theukstradeandinvestmentrelationshipwithafrica/2016 (accessed on 23 August 2020).
- Weimer, M.; Vines, A. Uk-South Africa Relations and the Bilateral Forum. Available online: https://www.chathamhouse.org/publications/papers/view/175839/19481_sa-uk_links.pdf (accessed on 23 August 2020).
- How Many South Africans Have Left the Country. Available online: https://www.politicsweb.co.za/news-and-analysis/how-many-south-africans-have-left-the-country (accessed on 23 August 2020).
- Valeri, M.; Paccani, S.R.; Kasendra, M.; Nesta, B.; Serino, L.; Pizza, M.; Soriani, M. Pathogenic E. coli Exploits SslE Mucinase Activity to Translocate through the Mucosal Barrier and Get Access to Host Cells. PLoS ONE 2015, 10, e0117486. [Google Scholar] [CrossRef] [Green Version]
- Reiland, H.A.; Omolo, M.A.; Johnson, T.J.; Baumler, D.J. A Survey of Escherichia coli O157:H7 Virulence Factors: The First 25 Years and 13 Genomes. Adv. Microbiol. 2014, 4, 390–423. [Google Scholar] [CrossRef] [Green Version]
- Beceiro, A.; Tomás, M.; Bou, G. Antimicrobial Resistance and Virulence: A Successful or Deleterious Association in the Bacterial World? Clin. Microbiol. Rev. 2013, 26, 185–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisinger, E.; Isberg, R.R. Interplay between Antibiotic Resistance and Virulence During Disease Promoted by Multidrug-Resistant Bacteria. J. Infect. Dis. 2017, 215, S9–S17. [Google Scholar] [CrossRef] [Green Version]








| Australia | China | South Africa | UK | US |
|---|---|---|---|---|
| air | astA | auto-sat | auto-sat | cif |
| astA | auto-sat | cvaC | espF | eae |
| espX1 | capU | fdeC | espX1 | efa1 |
| fdeC | cvaC | iha | etpD | ehxA |
| hlyA-alpha | eilA | iroN | fdeC | espA |
| ibeA | espX1 | iss | iha | espB |
| iha | fdeC | iutA | ireA | espF |
| ipaD | iha | lpfA | iss | espJ |
| ipaH1 | iroE | mchF | iutA | espK |
| iss | iroN | papA | lpfA | espP |
| iucA | iss | senB | lpfA1 | espX1 |
| iutA | iucA | sslE | lpfA2 | etpD |
| lpfA | iutA | vactox | mchF | fdeC |
| mchB | lpfA | ybtP | nleB2 | iha |
| papE | mchF | ybtQ | pic | iss |
| papF | pic | sepA | iucA | |
| papH | sslE | sinH | iutA | |
| pic | ybtP | sslE | katP | |
| senB | ybtQ | stxA1a | lpfA | |
| sepA | stxA2c | lpfA1 | ||
| sigA | stxB1a | lpfA2 | ||
| sinH | stxB2a | nleA | ||
| sslE | stxB2c | nleB | ||
| stxB2c | virF | nleB2 | ||
| toxB | ybtP | nleC | ||
| tsh | ybtQ | sinH | ||
| virF | sslE | |||
| ybtP | stxA1a | |||
| ybtQ | stxA2c | |||
| stxB1a | ||||
| stxB2a | ||||
| tccP | ||||
| tir | ||||
| toxB | ||||
| ybtP | ||||
| ybtQ |
| Gene | Function | Species |
|---|---|---|
| fdeC | Intimin-like adhesin protein used to increase infectivity [19] | E. coli |
| iha | Adhesin protein used to increase infectivity [20] | E. coli |
| iss | Increased serum survival used for extraintestinal infection [29] | E. coli |
| iutA | Ferric (iron) aerobacter receptor used in colonization [23] | E. coli |
| lpfA | Long polar fimbriae adhesive protein used to increase infectivity [21] | E. coli, Salmonella enterica |
| sslE | Type II secretion system subunit used for biofilm promotion [30] | E. coli |
| ybtP | ATP binding cassette (ABC) proteins used in iron(III)-yersiniabactin import and for survival [24] | E. coli, Yersinia pestis |
| ybtQ | ATP binding cassette (ABC) proteins used in iron(III)-yersiniabactin import and for survival [24] | E. coli, Yersinia pestis |
| auto-sat | Autotransporter adenyltransferase cytotoxin in urinary tract infections [26] | E. coli |
| espX1 | Type III secretion system subunit used for effector protein injection [27] | E. coli |
| iucA | Aerobactin/siderophore synthesis used for iron uptake and colonization [25] | E. coli, Klebsiella pneumoniae |
| mchF | Microcin H47 ABC exporter used for inhibitory activity [28] | E. coli |
| sinH | Intimin-type protein used for colonization and infectivity [22] | Select Salmonella enterica serovars |
| Australia | China | South Africa | UK | US |
|---|---|---|---|---|
| Clostridium difficile | Citrobacter freundii | E. coli & Shigella | Clostridium difficile | E. coli & Shigella |
| Enterobacter | E. coli & Shigella | Enterobacter | E. coli & Shigella | Klebsiella pneumoniae |
| Mycobacterium tuberculosis | Enterobacter | Klebsiella pneumoniae | Klebsiella oxytoca | Salmonella enterica |
| Klebsiella oxytoca | Mycobacterium tuberculosis | Klebsiella pneumoniae | ||
| Klebsiella pneumoniae | Salmonella enterica | Salmonella enterica | ||
| Kluyvera intermedia | ||||
| Salmonella enterica |
| Australia | China | South Africa | UK | US |
|---|---|---|---|---|
| iha | espX1 | ybtP | auto-sat | sinH |
| iss | fdeC | ybtQ | espF | floR |
| iucA | iss | bla-TEM1 | ireA | fosA7 |
| senB | lpfA | mchF | ||
| sslE | sslE | pic | ||
| ybtP | aph(3″)-Ib | sepA | ||
| ybtQ | aph(6)-Id | sslE | ||
| aph(3″)-Ib | dfrA12 | stxA1a | ||
| aph(6)-Id | floR | stxB1a | ||
| blaTEM-1 | mcr-1.1 | stxB2a | ||
| sul1 | mph(A) | stxB2c | ||
| sul2 | ybtP | |||
| tet(A) | ybtQ | |||
| blaOXA-1 | ||||
| catA1 | ||||
| mph(A) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, K.; Gong, I.; Dong, A.; Yan, J.; Wang, M.; Huang, Z. Investigation of Virulence Genes Detected in Antimicrobial-Resistance Pathogens Isolates for Five Countries across the World. Processes 2020, 8, 1589. https://0-doi-org.brum.beds.ac.uk/10.3390/pr8121589
Cui K, Gong I, Dong A, Yan J, Wang M, Huang Z. Investigation of Virulence Genes Detected in Antimicrobial-Resistance Pathogens Isolates for Five Countries across the World. Processes. 2020; 8(12):1589. https://0-doi-org.brum.beds.ac.uk/10.3390/pr8121589
Chicago/Turabian StyleCui, Kevin, Iris Gong, Alvin Dong, Jacob Yan, Max Wang, and Zuyi Huang. 2020. "Investigation of Virulence Genes Detected in Antimicrobial-Resistance Pathogens Isolates for Five Countries across the World" Processes 8, no. 12: 1589. https://0-doi-org.brum.beds.ac.uk/10.3390/pr8121589