Culturing of Melanocytes from the Equine Hair Follicle Outer Root Sheath

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Skin Sectioning for Histological Staining
2.2. Isolation of Forelock Hair Follicles
2.3. Processing of the Dissected Hair Follicles
2.4. Primary Air-Liquid-Interface Culture
2.5. Melanocyte Culture
2.6. L-DOPA Conversion Test
2.7. Melanin Content
2.8. Characterization of Melanocytes and Expression of Marker Proteins
2.9. Characterization of Melanocytes and Expression of Marker Genes
2.10. Statistical Analysis
3. Results
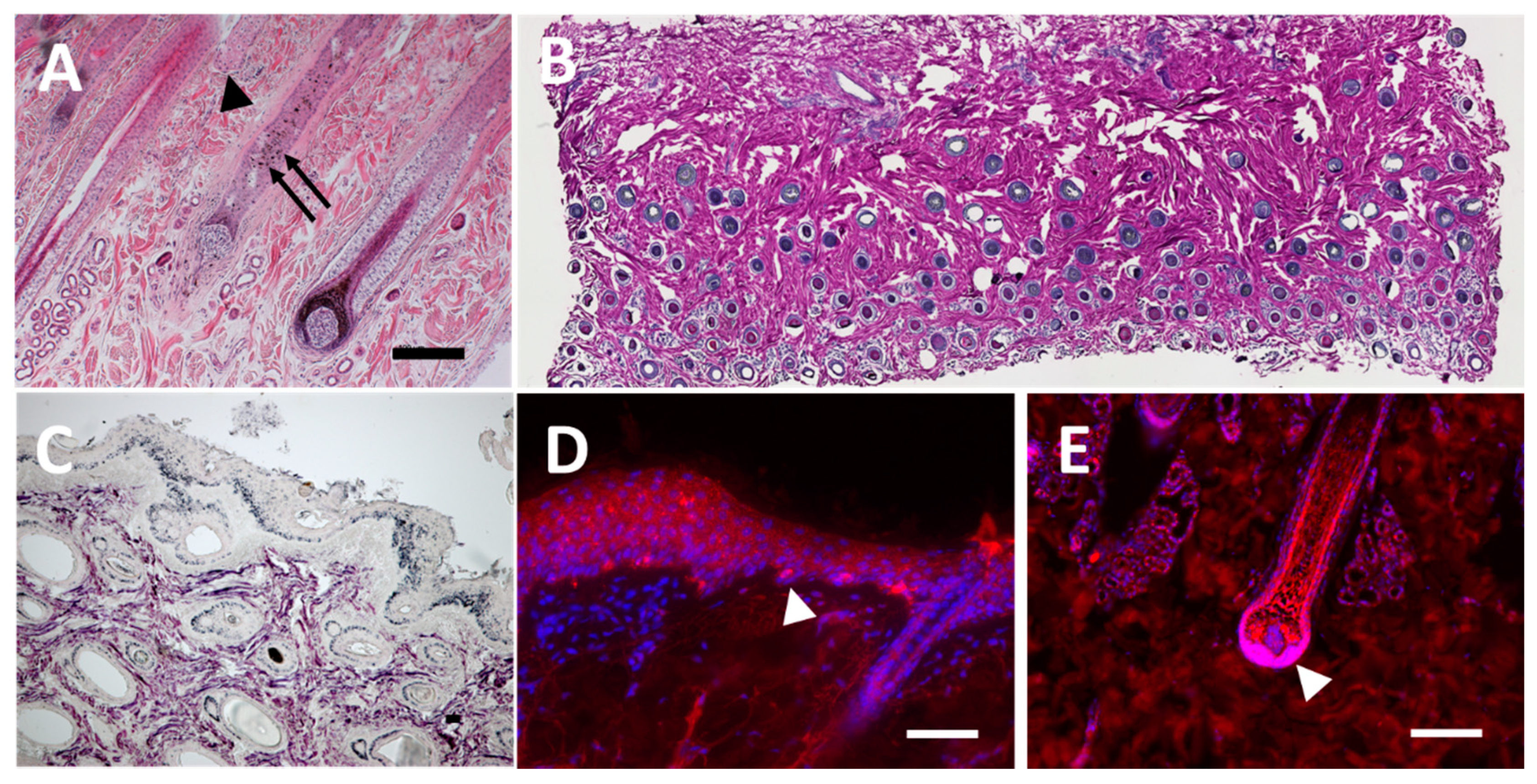
3.1. Staining of Histological Sections from Equine Skin
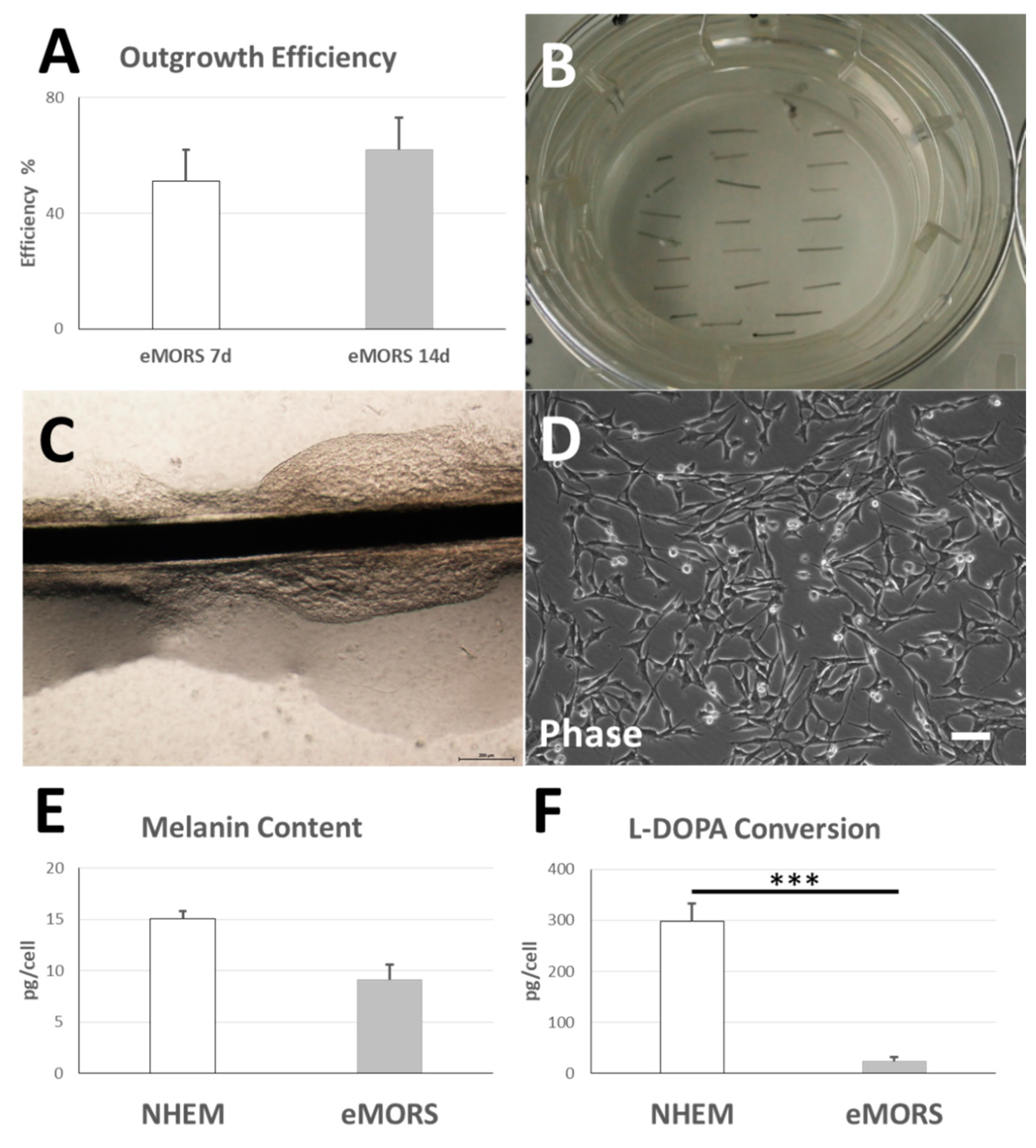
3.2. eMORS Culture Parameters
3.3. Melanin Content and Enzymatic L-DOPA Conversion Activity
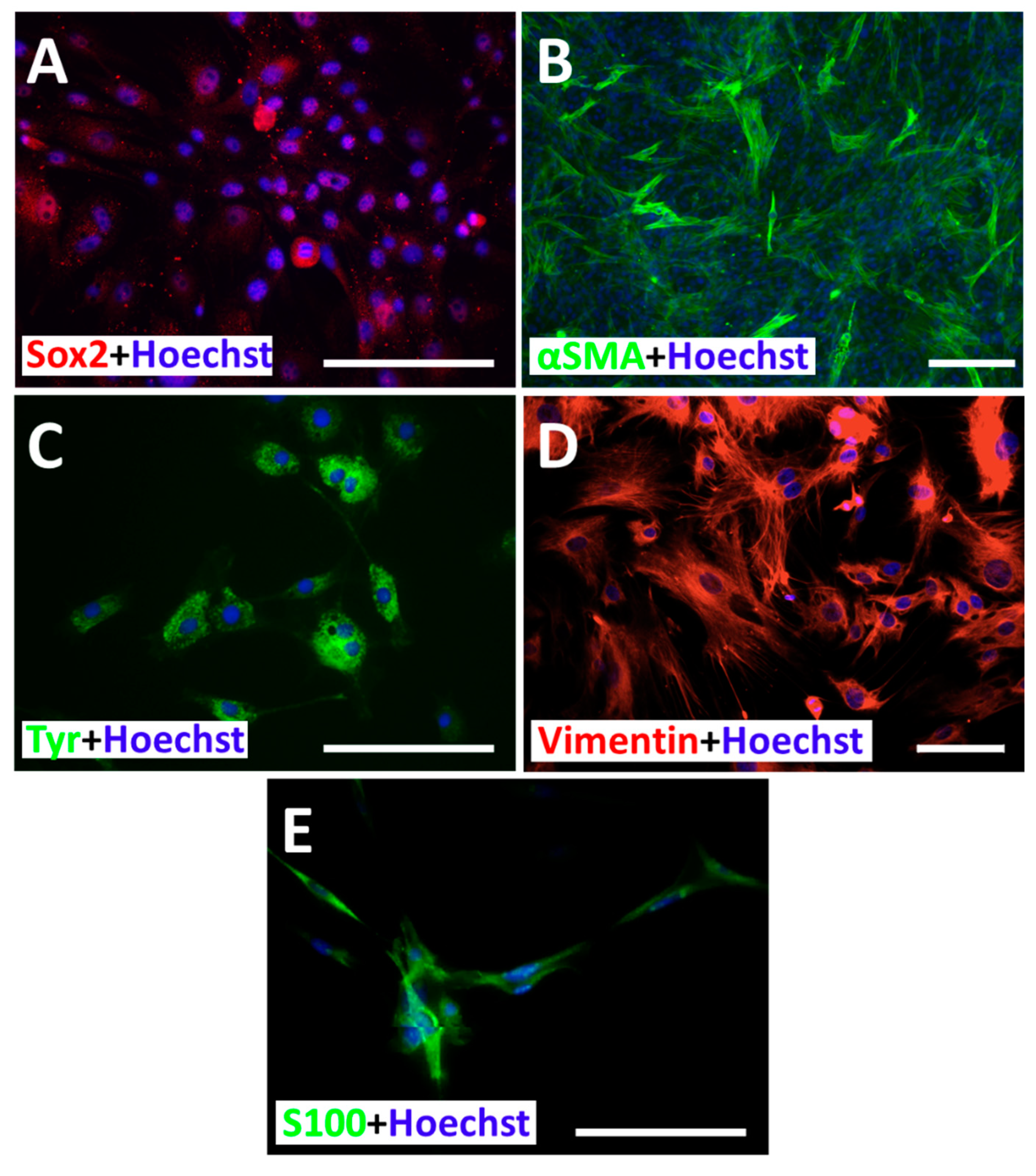
3.4. Subcellular Distribution of Sox2, α-SMA, Vimentin, Tyrosinase and S100ß
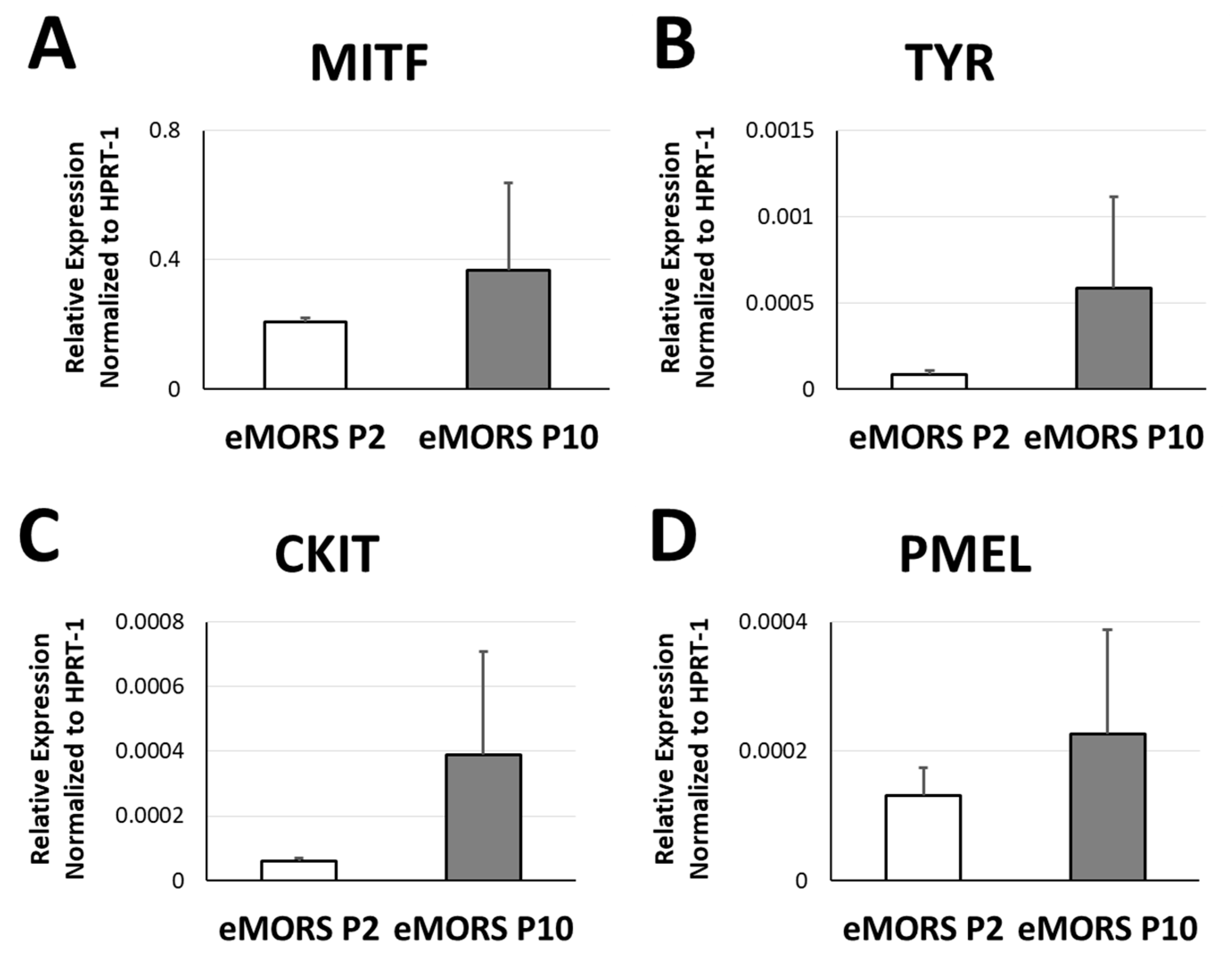
3.5. Gene Expressions of MITF, TYR, CKIT and PMEL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotsarelis, G.; Sun, T.T.; Lavker, R.M. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 1990, 61, 1329–1337. [Google Scholar] [CrossRef]
- Ma, H.-J.; Yue, X.-Z.; Wang, D.-G.; Li, C.-R.; Zhu, W.-Y. A modified method for purifying amelanotic melanocytes from human hair follicles. J. Dermatol. 2006, 33, 239–248. [Google Scholar] [CrossRef]
- Hoffman, R.M. The pluripotency of hair follicle stem cells. Cell Cycle 2006, 5, 232–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, D.J.; Swanson, N.; Pittelkow, M.; Peters, E.; Schallreuter, K. Melanocytes are not absent in lesional skin of long duration vitiligo. J. Pathol. 2000, 191, 407–416. [Google Scholar] [CrossRef]
- Na, G.Y.; Paek, S.H.; Park, B.C.; Kim, D.W.; Lee, W.J.; Lee, S.J.; Kim, M.K.; Kim, J.C. Isolation and characterization of outer root sheath melanocytes of human hair follicles. Br. J. Dermatol. 2006, 155, 902–909. [Google Scholar] [CrossRef]
- Zhu, W.Y.; Zhang, R.Z.; Ma, H.J.; Wang, D.G. Isolation and culture of amelanotic melanocytes from human hair follicles. Pigment. Cell Res. 2004, 17, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Kauser, S.B.; Thody, A.J.; Schallreuter, K.U.; Gummer, C.L.; Tobin, D.J. A Fully Functional Proopiomelanocortin/Melanocortin-1 Receptor System Regulates the Differentiation of Human Scalp Hair Follicle Melanocytes. Endocrinology 2005, 146, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Kauser, S.; Thody, A.J.; Schallreuter, K.U.; Gummer, C.L.; Tobin, D.J. β-Endorphin as a Regulator of Human Hair Follicle Melanocyte Biology. J. Investig. Dermatol. 2004, 123, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Limat, A.; French, L.E.; Blal, L.; Saurat, J.H.; Hunziker, T.; Salomon, D. Organotypic cultures of autologous hair follicle keratinocytes for the treatment of recurrent leg ulcers. J. Am. Acad. Dermatol. 2003, 48, 207–214. [Google Scholar] [CrossRef]
- Aasen, T.; Belmonte, J.C.I. Isolation and cultivation of human keratinocytes from skin or plucked hair for the generation of induced pluripotent stem cells. Nat. Protoc. 2010, 5, 371. [Google Scholar] [CrossRef]
- Seo, Y.-K.; Lee, D.-H.; Shin, Y.-H.; You, B.-Y.; Lee, K.-M.; Song, K.-Y.; Seo, S.-J.; Whang, S.-J.; Kim, Y.-J.; Yang, E.-K. Development of isolation and cultivation method for outer root sheath cells from human hair follicle and construction of bioartificial skin. J. Biotechnol. Bioprocess. Eng. 2003, 8, 151–157. [Google Scholar] [CrossRef]
- Nishimura, E.K. Melanocyte stem cells: A melanocyte reservoir in hair follicles for hair and skin pigmentation. Pigment. Cell Melanoma Res. 2011, 24, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xu, X.; Ma, H.; Yue, X.; Li, C.; Zhu, W. Optimization of the method for the culture of melanocyte precursors from hair follicles and their activation by 1, 25-dihydroxyvitamin D3. J. Exp. Therapeutic Med. 2013, 6, 967–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieckmann, C.; Milkova, L.; Hunziker, T.; Emmendörffer, A.; Simon, J.C. Human melanocytes can be isolated, propagated and expanded from plucked anagen hair follicles. J. Exp. Dermatol. 2010, 19, 543–545. [Google Scholar] [CrossRef] [PubMed]
- Savkovic, V.; Dieckmann, C.; Milkova, L.; Simon, J.C. Improved method of differentiation, selection and amplification of human melanocytes from the hair follicle cell pool. Exp. Dermatol. 2012, 21, 948–950. [Google Scholar] [CrossRef]
- Savkovic, V.; Dieckmann, C.; Simon, J.-C.; Schulz-Siegmund, M.; Hacker, M. Method for Deriving Melanocytes from the Hair Follicle Outer Root Sheath and Preparation for Grafting. World Patent WO2013060899A2, 2 May 2013. [Google Scholar]
- Marie, S.; Christina, D.; Katrin, R.; Simon, J.-C.; Vuk, S. Differentiating the Stem Cell Pool of Human Hair Follicle Outer Root Sheath into Functional Melanocytes. Stem Cells Tissue Repair 2014, 1210, 203–227. [Google Scholar]
- Korzynska, A.; Zychowicz, M. A Method of Estimation of the Cell Doubling Time on Basis of the Cell Culture Monitoring Data. Biocybern. Biomed. Eng. 2008, 28, 75–82. [Google Scholar]
- Cimadamore, F.; Shah, M.; Amador-Arjona, A.; Navarro-Peran, E.; Chen, C.; Huang, C.-T.; Terskikh, A.V. SOX2 modulates levels of MITF in normal human melanocytes, and melanoma lines in vitro. Pigment. Cell Melanoma Res. 2012, 25, 533–536. [Google Scholar] [CrossRef] [Green Version]
- El-Koraie, A.F.; Baddour, N.M.; Adam, A.G.; El-Kashef, E.H.; El Nahas, A.M. Cytoskeletal protein expression and regenerative markers in schistosomal nephropathy. Nephrol. Dial. Transplant. 2002, 17, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Shibahara, S.; Takeda, K.; Yasumoto, K.-I.; Udono, T.; Watanabe, K.-I.; Saito, H.; Takahashi, K. Microphthalmia-Associated Transcription Factor (MITF): Multiplicity in Structure, Function, and Regulation. J. Investig. Dermatol. Symp. Proc. 2001, 6, 99–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Jimenez, A.; Teruel-Puche, J.A.; Garcia-Ruiz, P.A.; Berna, J.; Rodríguez-López, J.N.; Tudela, J.; Garcia-Canovas, F. Action of 2,2’,4,4’-tetrahydroxybenzophenone in the biosynthesis pathway of melanin. Int. J. Biol. Macromol. 2017, 98, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Sülflow, K.; Schneider, M.; Loth, T.; Kascholke, C.; Schulz-Siegmund, M.; Hacker, M.C.; Simon, J.C.; Savkovic, V. Melanocytes from the outer root sheath of human hair and epidermal melanocytes display improved melanotic features in the niche provided by cGEL, oligomer-cross-linked gelatin-based hydrogel. J. Biomed. Mater. Res. Part A 2016, 104, 3115–3126. [Google Scholar] [CrossRef] [PubMed]
- Hornsby, P.J.; Sturek, M.; Harris, S.E.; Simonian, M.H. Serum and growth factor requirements for proliferation of human adrenocortical cells in culture: Comparison with bovine adrenocortical cells. Vitro 1983, 19, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Michler, J.K.; Hillmann, A.; Savkovic, V.; Mülling, C.K.W. Horse hair follicles: A novel dermal stem cell source for equine regenerative medicine. Cytom. Part A 2018, 93, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Wasmeier, C.; Hume, A.N.; Bolasco, G.; Seabra, M.C. Melanosomes at a glance. J. Cell Sci. 2008, 121, 3995–3999. [Google Scholar] [CrossRef] [Green Version]
- Chapman, S.W.; Metzger, N.; Grest, P.; Feige, K.; von Rechenberg, B.; Auer, J.A.; Hottiger, M.O. Isolation, establishment, and characterization of ex vivo equine melanoma cell cultures. In Vitro Cell. Dev. Biol. Anim. 2009, 45, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Seltenhammer, M.H.; Simhofer, H.; Scherzer, S.; Zechner, P.; Curik, I.; Sölkner, J.; Brandt, S.M.; Jansen, B.; Pehamberger, H.; Eisenmenger, E. Equine melanoma in a population of 296 grey Lipizzaner horses. Equine Vet. J. 2003, 35, 153–157. [Google Scholar] [CrossRef]
- Phillips, J.C.; Lembcke, L.M. Equine melanocytic tumors. Vet. Clin. N. Am. Equine Pract. 2013, 29, 673–687. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DLMS | Concentration |
|---|---|
| Basal Medium | 88% |
| L-Glutamine | 6 mM |
| Calcium Chloride | 0.2 mM |
| Epinephrine | 1 μM |
| Vitamin C | 50 μg/mL |
| Insulin | 5 μg/mL |
| StiMel8 Supplements | 1% Containing: FBS, rhEGF, rhFGF-b, Endothelin-1, Apo-Transferrin, Bovine Pituitary Extract Hydrocortisone Hemisuccinate |
| Penicillin/Streptomycin | 100 U/mL |
| Amphotericin | 2.5 µg/mL |
| Equine Serum | 10% |
| Protein | Primary Antibody | Secondary Antibody | Known Cross Reactivity in Horse |
|---|---|---|---|
| α-SMA | mouse anti-human αSMA, | goat anti mouse Dylight488, Biomol GmbH, Hamburg | - |
| mIgG1, Clone 1A4, Abcam Plc, ab5694 | cat.no: A90-516D2 | ||
| working dilution 1:100 | 1:500 in PBS | ||
| Vimentin | mouse monoclonal anti-human vimentin, C9080, Cy3-conjugated, Sigma Aldrich working dilution 1:500 | Does not apply | + |
| Sox2 | rabbit polyclonal anti-human Sox2, SAB2701974, Sigma Aldrich working dilution 1:100 | goat anti-rabbit PE, | - |
| Sigma-Aldrich, | |||
| cat.no: P9537 | |||
| 1:200 in PBS | |||
| Tyrosinase | mouse monoclonal anti-human tyrosinase, MFCD01322752, Sigma Aldrich working dilution 1:100 | goat anti mouse Dylight488, Biomol GmbH, Hamburg | - |
| cat.no: A90-516D2 | |||
| 1:500 in PBS | |||
| S100β | mouse monoclonal anti- mouse S100β, 612376, BD Transduction Laboratories working dilution 1:50 | goat anti mouse Alexa Fluor 488, Jackson ImmunoResearch via Dianova GmbH, Hamburg | - |
| cat.no: 115-545-062 | |||
| 1:500 in PBS |
| Gene | Primer Sequence |
|---|---|
| MITF for | CAAGGCAGGATCCATCAA |
| MITF rev | TGACATCATTAGCCTAGAATCAAGT |
| TYR for | CAGATCCCTGTACCTAGAAGAT |
| TYR rev | TTTCGAAGCAGTATGCACAA |
| CKIT for | ATCAGCGCATAACAGCCTAAT |
| CKIT rev | CCAGCAAAATCAGAGTTAATCG |
| PMEL for | ACTCTTTGACTCCTCACACAGC |
| PMEL rev | ATTTCAAATGGGGATCATAATGT |
| HPRT-1 for | CTTTTCCAAATCCTCAGCATAAT |
| HPRT-1 rev | AGGTCTTGTTCTGATCCTTCTGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Michler, J.K.; Bartella, A.; Sander, A.K.; Gaus, S.; Hahnel, S.; Zimmerer, R.; Simon, J.-C.; Savkovic, V.; Lethaus, B. Culturing of Melanocytes from the Equine Hair Follicle Outer Root Sheath. Processes 2021, 9, 177. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9010177
Li H, Michler JK, Bartella A, Sander AK, Gaus S, Hahnel S, Zimmerer R, Simon J-C, Savkovic V, Lethaus B. Culturing of Melanocytes from the Equine Hair Follicle Outer Root Sheath. Processes. 2021; 9(1):177. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9010177
Chicago/Turabian StyleLi, Hanluo, Jule Kristin Michler, Alexander Bartella, Anna Katharina Sander, Sebastian Gaus, Sebastian Hahnel, Rüdiger Zimmerer, Jan-Christoph Simon, Vuk Savkovic, and Bernd Lethaus. 2021. "Culturing of Melanocytes from the Equine Hair Follicle Outer Root Sheath" Processes 9, no. 1: 177. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9010177