
One-Pot Synthesis of Ni0.05Ce0.95O2−δ Catalysts with Nanocubes and Nanorods Morphology for CO2 Methanation Reaction and in Operando DRIFT Analysis of Intermediate Species

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalysts Synthesis
2.2. Catalysts Characterization
2.3. Fixed-Bed Catalytic Test
3. Results
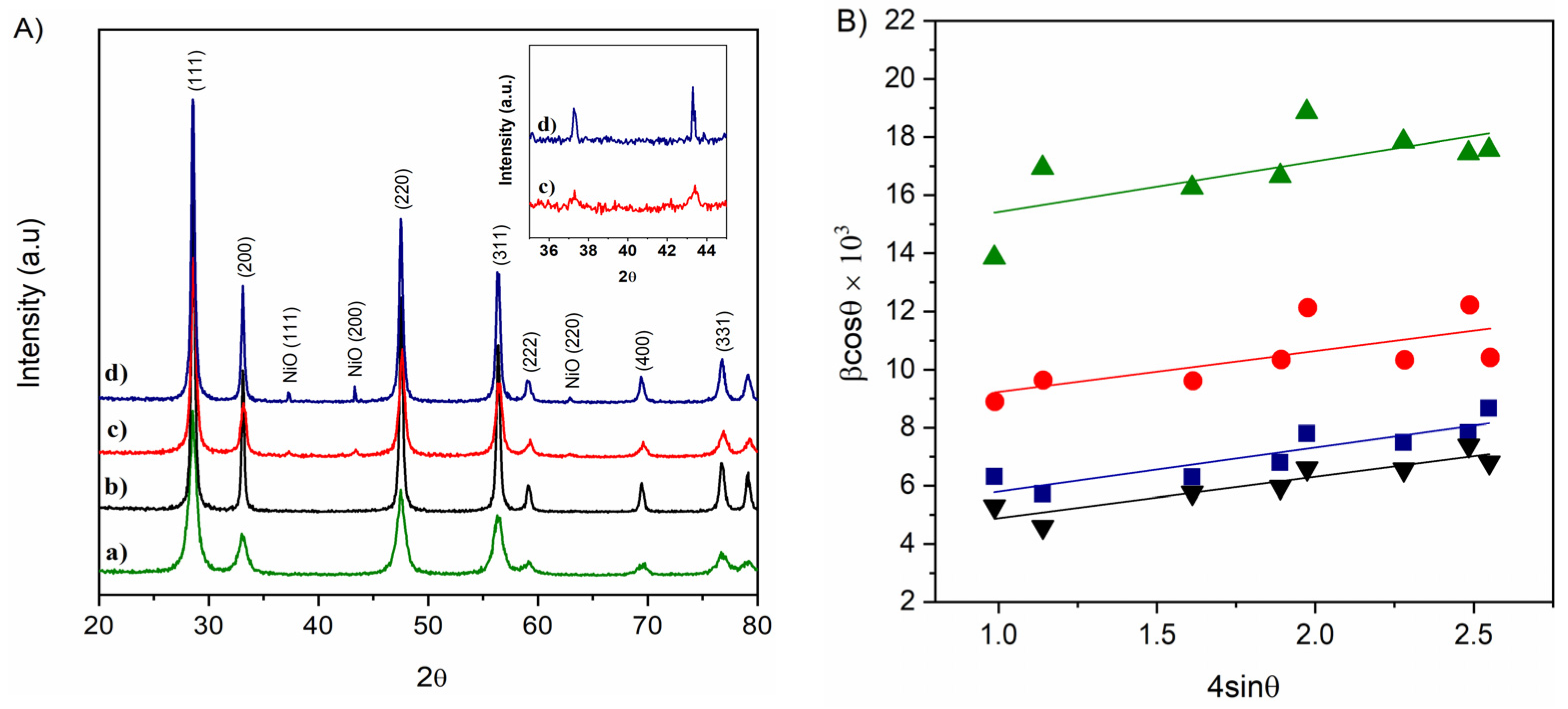
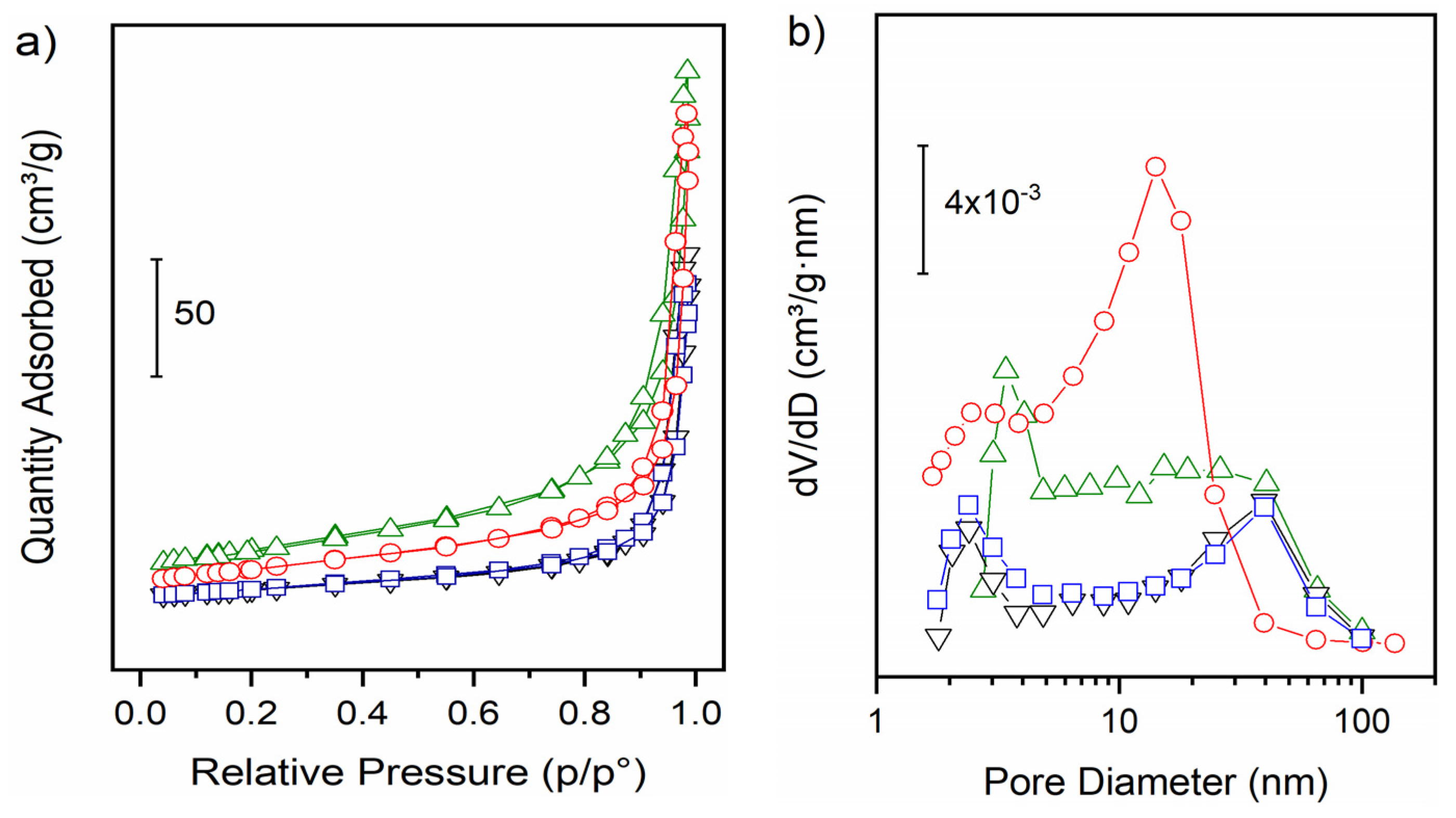
3.1. Structural and Morphological Characterization
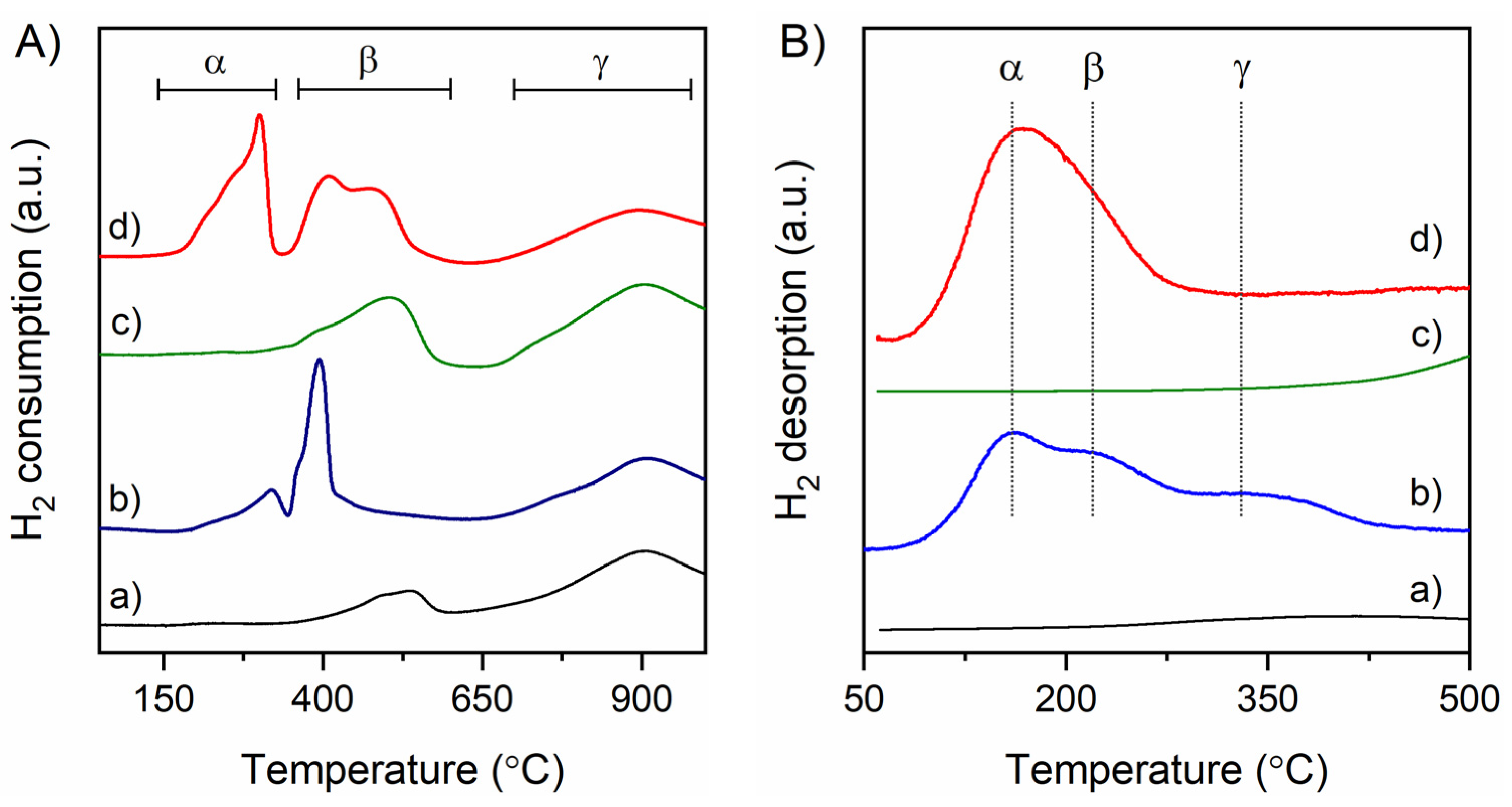
3.2. Temperature-Programmed Reduction (TPR) and Desorption (TPD) Characterization
3.3. TEM Characterization of the Synthesized Catalysts
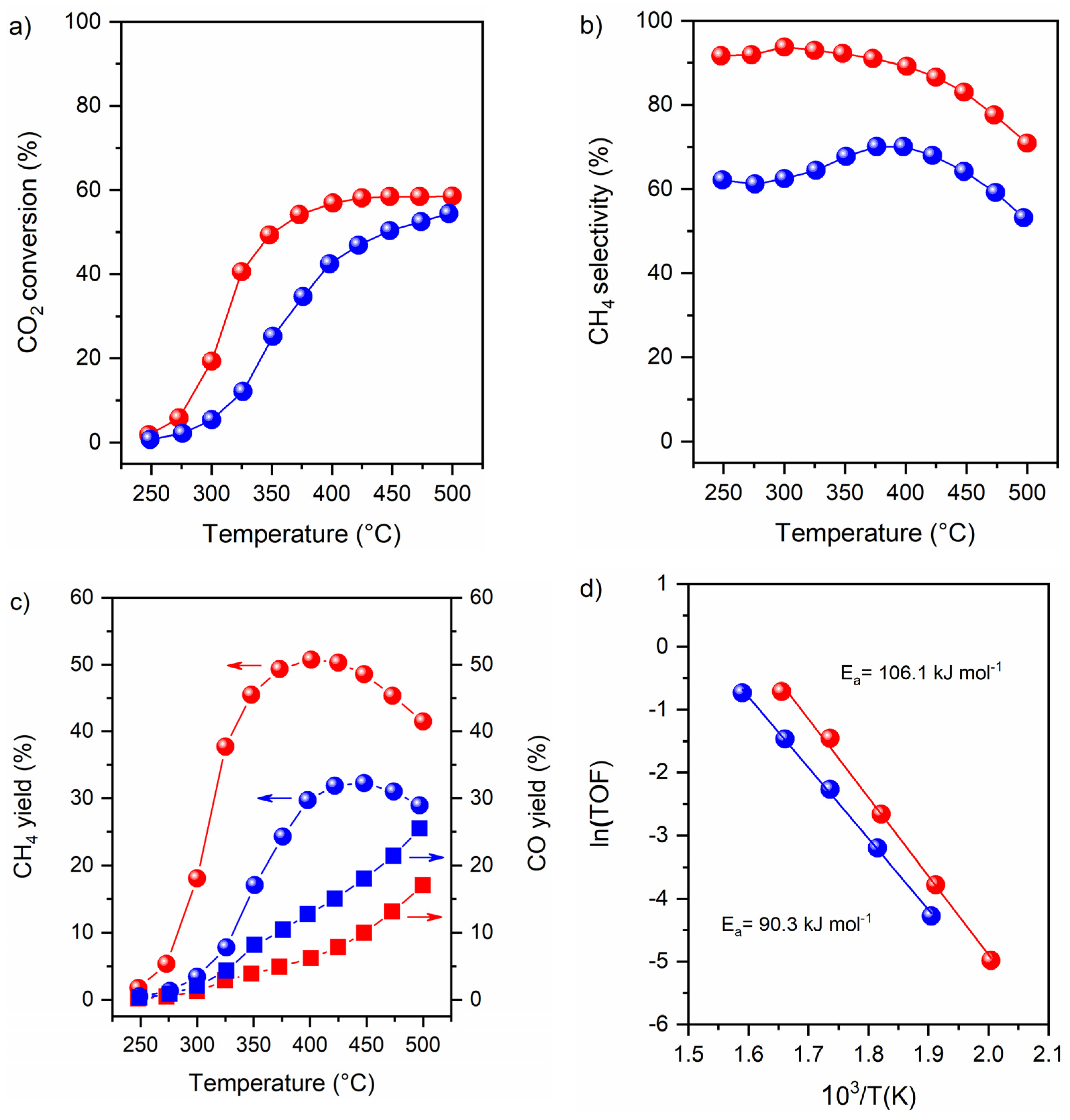
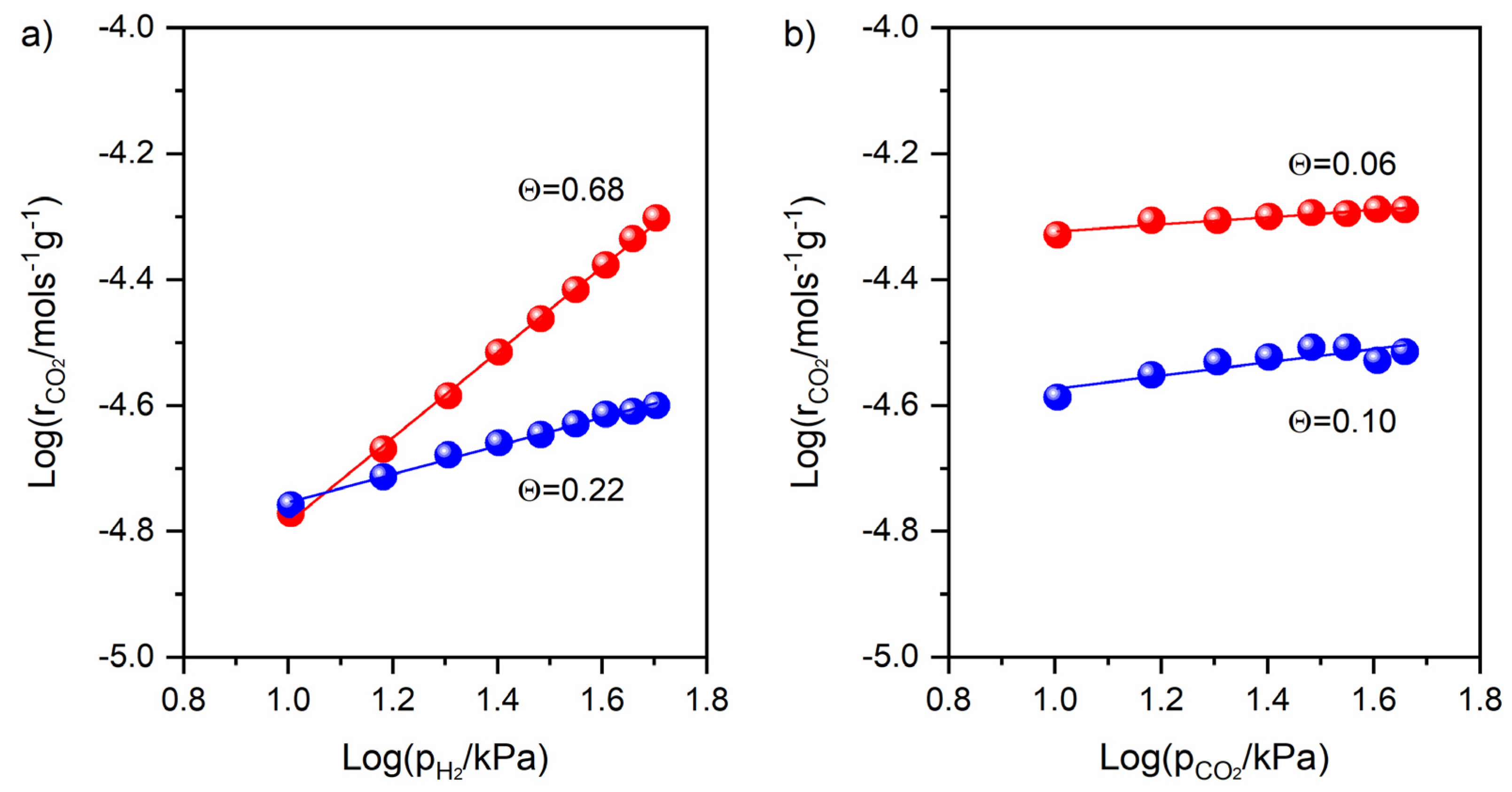
3.4. Catalytic Activity
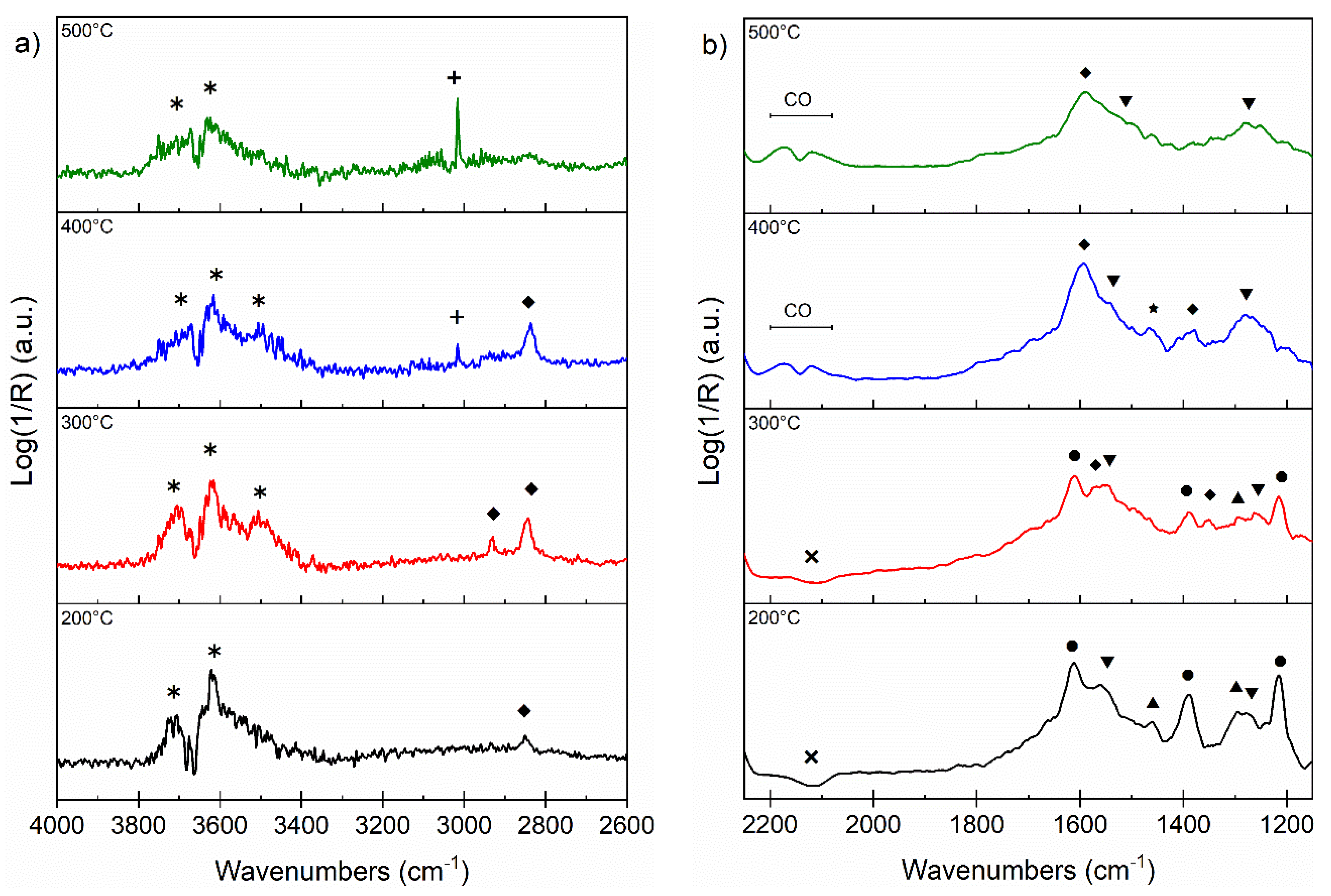
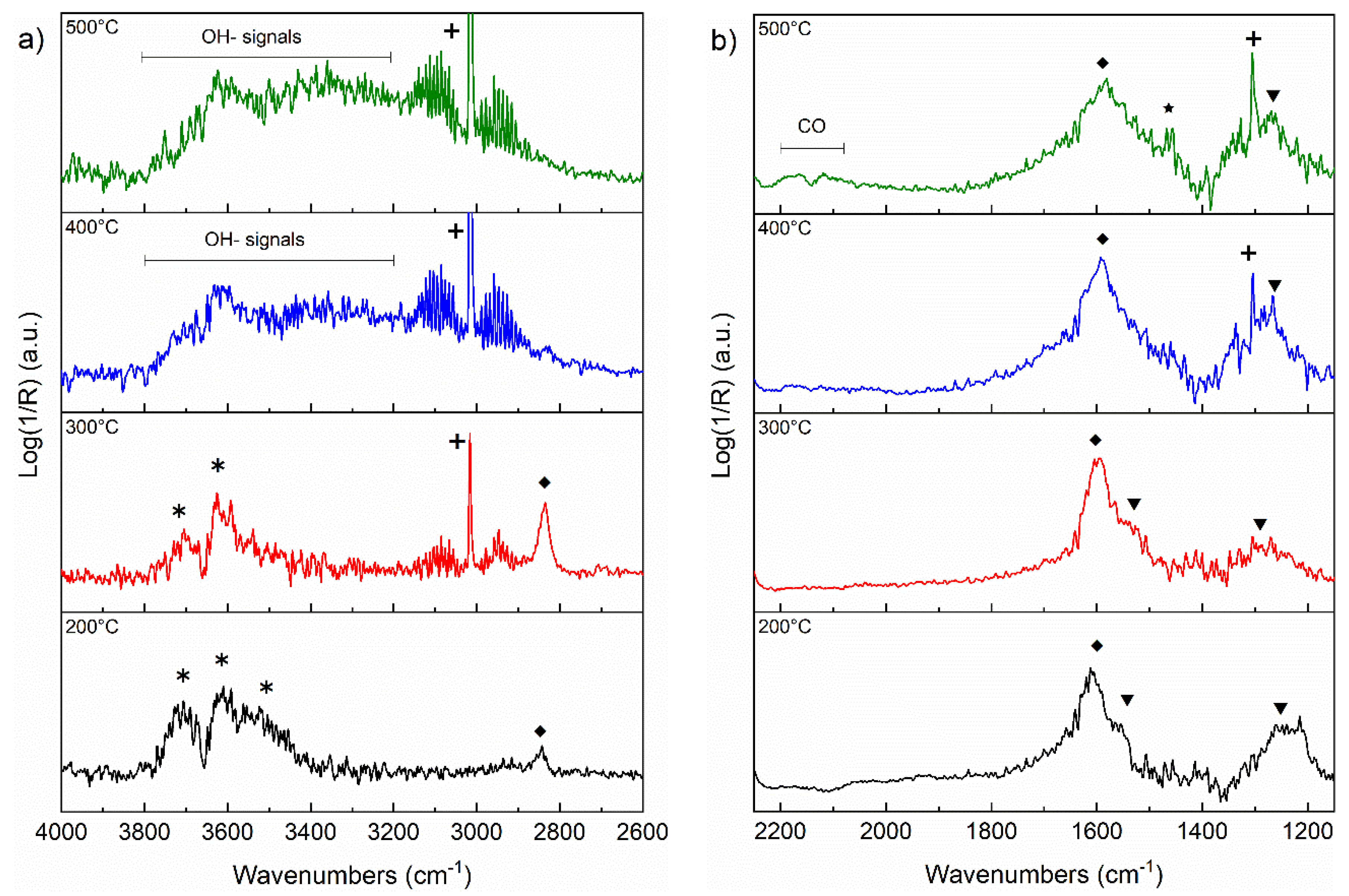
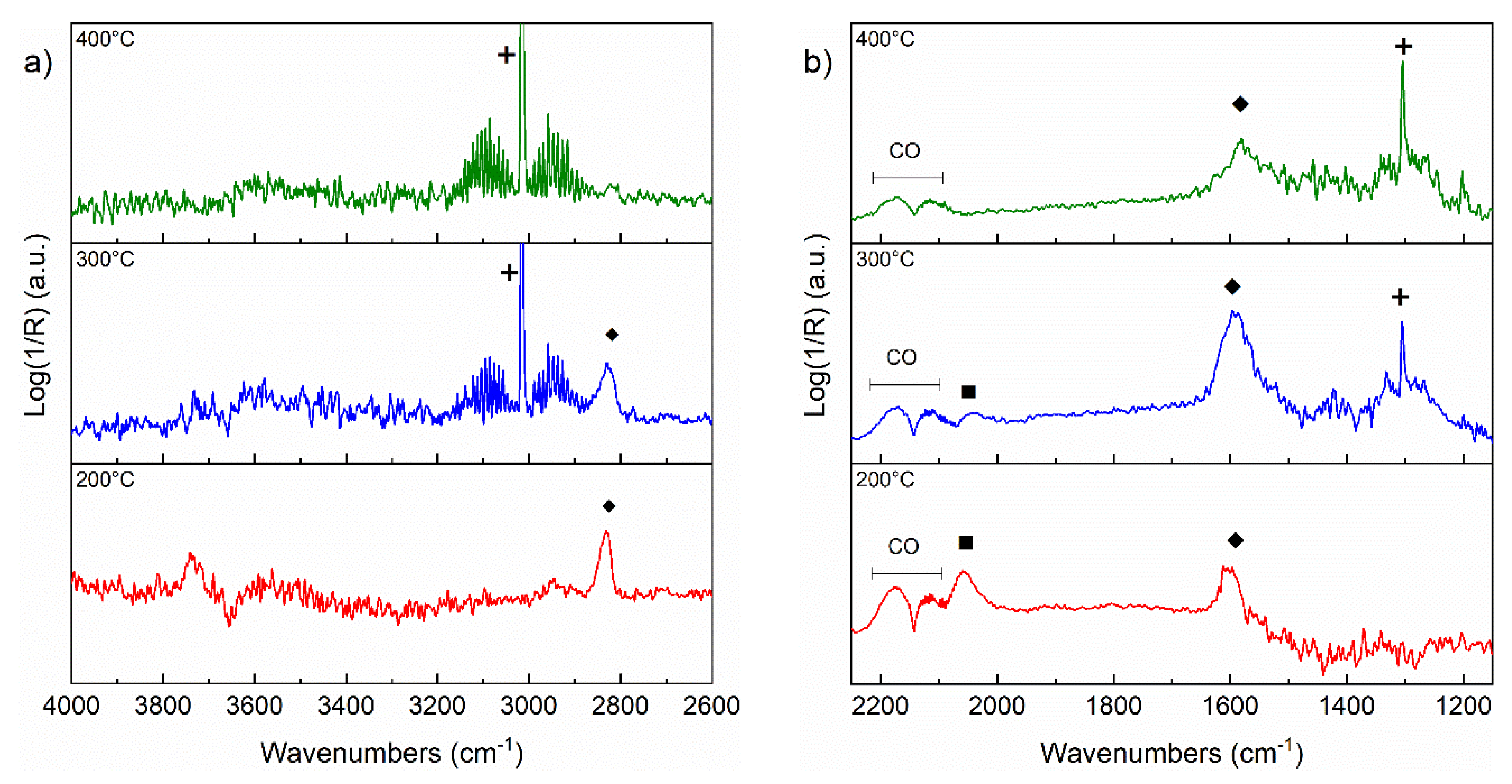
3.5. DRIFT
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catalysis Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [Green Version]
- Jomjaree, T.; Sintuya, P.; Srifa, A.; Koo-amornpattana, W.; Kiatphuengporn, S.; Assabumrungrat, S.; Sudoh, M.; Watanabe, R.; Fukuhara, C.; Ratchahat, S. Catalytic performance of Ni catalysts supported on CeO2 with different morphologies for low-temperature CO2 methanation. Catal. Today 2020. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Hidalgo, D.; Martín-Marroquín, J.M. Power-to-methane, coupling CO2 capture with fuel production: An overview. Renew. Sustain. Energy Rev. 2020, 132, 110057. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Fan, W.K.; Tahir, M. Recent trends in developments of active metals and heterogenous materials for catalytic CO2 hydrogenation to renewable methane: A review. J. Environ. Chem. Eng. 2021, 9, 105460. [Google Scholar] [CrossRef]
- Lee, W.J.; Li, C.; Prajitno, H.; Yoo, J.; Patel, J.; Yang, Y.; Lim, S. Recent trend in thermal catalytic low temperature CO2 methanation: A critical review. Catal. Today 2021, 368, 2–19. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Younas, M.; Loong Kong, L.; Bashir, M.J.K.; Nadeem, H.; Shehzad, A.; Sethupathi, S. Recent Advancements, Fundamental Challenges, and Opportunities in Catalytic Methanation of CO2. Energy Fuels 2016, 30, 8815–8831. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Jiang, X.; Zhu, J.; Liu, Z.; Guo, X.; Song, C. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RSC Adv. 2018, 8, 7651–7669. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Lee, Y.H.; Moon, D.H.; Ahn, J.Y.; Nguyen, D.D.; Chang, S.W.; Kim, S.S. Reaction Mechanism and Catalytic Impact of Ni/CeO2–x Catalyst for Low-Temperature CO2 Methanation. Ind. Eng. Chem. Res. 2019, 58, 8656–8662. [Google Scholar] [CrossRef]
- Shen, L.; Xu, J.; Zhu, M.; Han, Y.-F. Essential Role of the Support for Nickel-Based CO2 Methanation Catalysts. ACS Catal. 2020, 10, 14581–14591. [Google Scholar] [CrossRef]
- Lin, L.; Gerlak, C.A.; Liu, C.; Llorca, J.; Yao, S.; Rui, N.; Zhang, F.; Liu, Z.; Zhang, S.; Deng, K.; et al. Effect of Ni particle size on the production of renewable methane from CO2 over Ni/CeO2 catalyst. J. Energy Chem. 2021, 61, 602–611. [Google Scholar] [CrossRef]
- Wang, L.; Deo, S.; Dooley, K.; Janik, M.J.; Rioux, R.M. Influence of metal nuclearity and physicochemical properties of ceria on the oxidation of carbon monoxide. Chin. J. Catal. 2020, 41, 951–962. [Google Scholar] [CrossRef]
- Moeini, S.S.; Battocchio, C.; Casciardi, S.; Luisetto, I.; Lupattelli, P.; Tofani, D.; Tuti, S. Oxidized Palladium Supported on Ceria Nanorods for Catalytic Aerobic Oxidation of Benzyl Alcohol to Benzaldehyde in Protic Solvents. Catalysts 2019, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Li, W.; Liu, Q.; Lin, Q.; Zheng, X.; Huang, Q.; Guan, S.; Wang, X.; Wang, C.; Li, F. Typical crystal face effects of different morphology ceria on the activity of Pd/CeO2 catalysts for lean methane combustion. Fuel 2018, 233, 10–20. [Google Scholar] [CrossRef]
- Luisetto, I.; Tuti, S.; Di Bartolomeo, E. Co and Ni supported on CeO2 as selective bimetallic catalyst for dry reforming of methane. Int. J. Hydrogen Energy 2012, 37, 15992–15999. [Google Scholar] [CrossRef]
- Luisetto, I.; Tuti, S.; Battocchio, C.; Lo Mastro, S.; Sodo, A. Ni/CeO2–Al2O3 catalysts for the dry reforming of methane: The effect of CeAlO3 content and nickel crystallite size on catalytic activity and coke resistance. Appl. Catal. A Gen. 2015, 500, 12–22. [Google Scholar] [CrossRef]
- Luisetto, I.; Tuti, S.; Romano, C.; Boaro, M.; Di Bartolomeo, E.; Kesavan, J.K.; Kumar, S.S.; Selvakumar, K. Dry reforming of methane over Ni supported on doped CeO2: New insight on the role of dopants for CO2 activation. J. CO2 Util. 2019, 30, 63–78. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, S.; Dong, M.; Wang, J.; Fan, W. Ru/CeO2 catalyst with optimized CeO2 morphology and surface facet for efficient hydrogenation of ethyl levulinate to γ-valerolactone. J. Catal. 2020, 389, 60–70. [Google Scholar] [CrossRef]
- Marconi, E.; Tuti, S.; Luisetto, I. Structure-Sensitivity of CO2 Methanation over Nanostructured Ni Supported on CeO2 Nanorods. Catalysts 2019, 9, 375. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, I.; Quindimil, A.; Mariño, F.; De-La-Torre, U.; González-Velasco, J.R. Zr promotion effect in CO2 methanation over ceria supported nickel catalysts. Int. J. Hydrogen Energy 2019, 44, 1710–1719. [Google Scholar] [CrossRef]
- Ocampo, F.; Louis, B.; Roger, A.-C. Methanation of carbon dioxide over nickel-based Ce0.72Zr0.28O2 mixed oxide catalysts prepared by sol–gel method. Appl. Catal. A Gen. 2009, 369, 90–96. [Google Scholar] [CrossRef]
- Sakpal, T.; Lefferts, L. Structure-dependent activity of CeO2 supported Ru catalysts for CO2 methanation. J. Catal. 2018, 367, 171–180. [Google Scholar] [CrossRef]
- Ren, Z.; Peng, F.; Li, J.; Liang, X.; Chen, B. Morphology-Dependent Properties of Cu/CeO2 Catalysts for the Water-Gas Shift Reaction. Catalysts 2017, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Zhang, D.; Shi, L.; Gao, R.; Zhang, J. Morphology Dependence of Catalytic Properties of Ni/CeO2 Nanostructures for Carbon Dioxide Reforming of Methane. J. Phys. Chem. C 2012, 116, 10009–10016. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, L.; Wang, W.; Zhao, Y.; Zhang, G.; Ma, X.; Gong, J. Morphology control of ceria nanocrystals for catalytic conversion of CO2 with methanol. Nanoscale 2013, 5, 5582–5588. [Google Scholar] [CrossRef]
- Trovarelli, A.; Llorca, J. Ceria Catalysts at Nanoscale: How Do Crystal Shapes Shape Catalysis? ACS Catal. 2017, 7, 4716–4735. [Google Scholar] [CrossRef]
- Ye, R.-P.; Li, Q.; Gong, W.; Wang, T.; Razink, J.J.; Lin, L.; Qin, Y.-Y.; Zhou, Z.; Adidharma, H.; Tang, J.; et al. High-performance of nanostructured Ni/CeO2 catalyst on CO2 methanation. Appl. Catal. B Environ. 2020, 268, 118474. [Google Scholar] [CrossRef]
- Padi, S.P.; Shelly, L.; Komarala, E.P.; Schweke, D.; Hayun, S.; Rosen, B.A. Coke-free methane dry reforming over nano-sized NiO-CeO2 solid solution after exsolution. Catal. Commun. 2020, 138, 105951. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Perrichon, V.; Laachir, A.; Bergeret, G.; Fréty, R.; Tournayan, L.; Touret, O. Reduction of cerias with different textures by hydrogen and their reoxidation by oxygen. J. Chem. Soc. Faraday Trans. 1994, 90, 773–781. [Google Scholar] [CrossRef]
- Désaunay, T.; Bonura, G.; Chiodo, V.; Freni, S.; Couzinié, J.P.; Bourgon, J.; Ringuedé, A.; Labat, F.; Adamo, C.; Cassir, M. Surface-dependent oxidation of H2 on CeO2 surfaces. J. Catal. 2013, 297, 193–201. [Google Scholar] [CrossRef]
- Giordano, F.; Trovarelli, A.; de Leitenburg, C.; Giona, M. A Model for the Temperature-Programmed Reduction of Low and High Surface Area Ceria. J. Catal. 2000, 193, 273–282. [Google Scholar] [CrossRef]
- Xiao, Z.; Li, Y.; Hou, F.; Wu, C.; Pan, L.; Zou, J.; Wang, L.; Zhang, X.; Liu, G.; Li, G. Engineering oxygen vacancies and nickel dispersion on CeO2 by Pr doping for highly stable ethanol steam reforming. Appl. Catal. B Environ. 2019, 258, 117940. [Google Scholar] [CrossRef]
- Takeguchi, T.; Furukawa, S.-n.; Inoue, M. Hydrogen Spillover from NiO to the Large Surface Area CeO2–ZrO2 Solid Solutions and Activity of the NiO/CeO2–ZrO2 Catalysts for Partial Oxidation of Methane. J. Catal. 2001, 202, 14–24. [Google Scholar] [CrossRef]
- Varvoutis, G.; Lykaki, M.; Stefa, S.; Papista, E.; Carabineiro, S.A.C.; Marnellos, G.E.; Konsolakis, M. Remarkable efficiency of Ni supported on hydrothermally synthesized CeO2 nanorods for low-temperature CO2 hydrogenation to methane. Catal. Commun. 2020, 142, 106036. [Google Scholar] [CrossRef]
- Safa, S.; Hejazi, R.; Rabbani, M.; Azimirad, R. Hydrothermal synthesis of NiO nanostructures for photodegradation of 4-nitrophenol. Desalin. Water Treat. 2016, 57, 21982–21989. [Google Scholar] [CrossRef]
- Kumar, J.P.; Giri, S.D.; Sarkar, A. Mesoporous NiO with different morphology: Synthesis, characterization and their evaluation for oxygen evolution reaction. Int. J. Hydrogen Energy 2018, 43, 15639–15649. [Google Scholar] [CrossRef]
- Torrente-Murciano, L.; Gilbank, A.; Puertolas, B.; Garcia, T.; Solsona, B.; Chadwick, D. Shape-dependency activity of nanostructured CeO2 in the total oxidation of polycyclic aromatic hydrocarbons. Appl. Catal. B Environ. 2013, 132-133, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Qian, W.; Chu, W.; Wei, F. Crystal-plane effect of nanoscale CeO2 on the catalytic performance of Ni/CeO2 catalysts for methane dry reforming. Catal. Sci. Technol. 2016, 6, 3594–3605. [Google Scholar] [CrossRef]
- Wu, H.C.; Chang, Y.C.; Wu, J.H.; Lin, J.H.; Lin, I.K.; Chen, C.S. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: The influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Kesavan, J.K.; Luisetto, I.; Tuti, S.; Meneghini, C.; Iucci, G.; Battocchio, C.; Mobilio, S.; Casciardi, S.; Sisto, R. Nickel supported on YSZ: The effect of Ni particle size on the catalytic activity for CO2 methanation. J. CO2 Util. 2018, 23, 200–211. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Laachir, A.; Perrichon, V.; Badri, A.; Lamotte, J.; Catherine, E.; Lavalley, J.C.; El Fallah, J.; Hilaire, L.; Le Normand, F.; Quéméré, E.; et al. Reduction of CeO2 by hydrogen. Magnetic susceptibility and Fourier-transform infrared, ultraviolet and X-ray photoelectron spectroscopy measurements. J. Chem. Soc. Faraday Trans. 1991, 87, 1601–1609. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.-C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- López, J.M.; Gilbank, A.L.; García, T.; Solsona, B.; Agouram, S.; Torrente-Murciano, L. The prevalence of surface oxygen vacancies over the mobility of bulk oxygen in nanostructured ceria for the total toluene oxidation. Appl. Catal. B Environ. 2015, 174-175, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; LaCoste, J.D.; Khamidullina, N.G.; Fox, E.; Gang, D.D.; Hernandez, R.; Yan, H. Investigate interactions of water with mesoporous ceria using in situ VT-DRIFTS. Surf. Sci. 2020, 691, 121486. [Google Scholar] [CrossRef]
- Proaño, L.; Tello, E.; Arellano-Trevino, M.A.; Wang, S.; Farrauto, R.J.; Cobo, M. In-situ DRIFTS study of two-step CO2 capture and catalytic methanation over Ru,“Na2O”/Al2O3 Dual Functional Material. Appl. Surf. Sci. 2019, 479, 25–30. [Google Scholar] [CrossRef]
- Cárdenas-Arenas, A.; Quindimil, A.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Isotopic and in situ DRIFTS study of the CO2 methanation mechanism using Ni/CeO2 and Ni/Al2O3 catalysts. Appl. Catal. B Environ. 2020, 265, 118538. [Google Scholar] [CrossRef]
- Tada, S.; Nagase, H.; Fujiwara, N.; Kikuchi, R. What Are the Best Active Sites for CO2 Methanation over Ni/CeO2? Energy Fuels 2021, 35, 5241–5251. [Google Scholar] [CrossRef]
- Köck, E.-M.; Kogler, M.; Bielz, T.; Klötzer, B.; Penner, S. In Situ FT-IR Spectroscopic Study of CO2 and CO Adsorption on Y2O3, ZrO2, and Yttria-Stabilized ZrO2. J. Phys. Chem. C 2013, 117, 17666–17673. [Google Scholar] [CrossRef] [PubMed]
- Grünbacher, M.; Klötzer, B.; Penner, S. CO2 Reduction by Hydrogen Pre-Reduced Acceptor-Doped Ceria. ChemPhysChem 2019, 20, 1706–1718. [Google Scholar] [CrossRef]
- Jia, X.; Zhang, X.; Rui, N.; Hu, X.; Liu, C.-j. Structural effect of Ni/ZrO2 catalyst on CO2 methanation with enhanced activity. Appl. Catal. B Environ. 2019, 244, 159–169. [Google Scholar] [CrossRef]
- Solis-Garcia, A.; Louvier-Hernandez, J.F.; Almendarez-Camarillo, A.; Fierro-Gonzalez, J.C. Participation of surface bicarbonate, formate and methoxy species in the carbon dioxide methanation catalyzed by ZrO2-supported Ni. Appl. Catal. B Environ. 2017, 218, 611–620. [Google Scholar] [CrossRef]
- Tada, S.; Ochieng, O.J.; Kikuchi, R.; Haneda, T.; Kameyama, H. Promotion of CO2 methanation activity and CH4 selectivity at low temperatures over Ru/CeO2/Al2O3 catalysts. Int. J. Hydrogen Energy 2014, 39, 10090–10100. [Google Scholar] [CrossRef]
- Jin, T.; Zhou, Y.; Mains, G.J.; White, J.M. Infrared and x-ray photoelectron spectroscopy study of carbon monoxide and carbon dioxide on platinum/ceria. J. Phys. Chem. 1987, 91, 5931–5937. [Google Scholar] [CrossRef]
- Shido, T.; Iwasawa, Y. Regulation of reaction intermediate by reactant in the water-gas shift reaction on CeO2, in relation to reactant-promoted mechanism. J. Catal. 1992, 136, 493–503. [Google Scholar] [CrossRef]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.-I.; Onishi, T. Adsorption of carbon monoxide and carbon dioxide on cerium oxide studied by Fourier-transform infrared spectroscopy. Part 2.—Formation of formate species on partially reduced CeO2 at room temperature. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Ph. 1989, 85, 1451–1461. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | DW-H (nm) a | ε | a (Å) | DNiO (nm) b |
|---|---|---|---|---|
| nC | 40 | 1.4 × 10−3 | 5.41 | |
| Ni-nC | 32 | 1.5 × 10−3 | 5.41 | 71 |
| nR | 10 | 1.7 × 10−3 | 5.41 | |
| Ni-nR | 18 | 1.4 × 10−3 | 5.40 | 20 |
| Sample | SSA (m2g−1) | Pore Volume (cm3g−1) | Average Pore Diameter (nm) |
|---|---|---|---|
| nC | 32 | 2.4 × 10−1 | 25 |
| Ni-nC | 33 | 2.2 × 10−1 | 21 |
| nR | 92 | 3.6 × 10−1 | 19 |
| Ni-nR | 62 | 3.3 × 10−1 | 20 |
| Sample | Hydrogen Consumption/mmol g−1 | D (%) | d (nm) | |||
|---|---|---|---|---|---|---|
| α | β | γ | Total | |||
| nC | 0.16 | 1.05 | 1.20 | |||
| Ni-nC | 0.12 | 0.32 | 1.03 | 1.47 | 14 | 7 |
| nR | 0.38 | 0.79 | 1.17 | |||
| Ni-nR | 0.44 | 0.53 | 0.67 | 1.64 | 20 | 5 |
 |  |  |  |  |  |
| Bidentate bicarbonate | Bidentate carbonate | Monodentate carbonate | Carboxylate | Bidentate formate | Monodentate formate |
| 1612 νa(CO3) 1390 νs(CO3) 1216 δ(OH) | 1562 νa(CO3) 1272 νs(CO3) | 1462 νa(CO3) 1293 νs(CO3) | 1465 νa(CO2) | 2931 νa(CO2) + δ(CH) 2845 ν(CH) 1569 νa(CO2) 1352 νs(CO2) + δ(CH) | 1602 νa(CO2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luisetto, I.; Stendardo, S.; Senthil Kumar, S.M.; Selvakumar, K.; Kesavan, J.K.; Iucci, G.; Pasqual Laverdura, U.; Tuti, S. One-Pot Synthesis of Ni0.05Ce0.95O2−δ Catalysts with Nanocubes and Nanorods Morphology for CO2 Methanation Reaction and in Operando DRIFT Analysis of Intermediate Species. Processes 2021, 9, 1899. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9111899
Luisetto I, Stendardo S, Senthil Kumar SM, Selvakumar K, Kesavan JK, Iucci G, Pasqual Laverdura U, Tuti S. One-Pot Synthesis of Ni0.05Ce0.95O2−δ Catalysts with Nanocubes and Nanorods Morphology for CO2 Methanation Reaction and in Operando DRIFT Analysis of Intermediate Species. Processes. 2021; 9(11):1899. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9111899
Chicago/Turabian StyleLuisetto, Igor, Stefano Stendardo, Sakkarapalayam Murugesan Senthil Kumar, Karuppiah Selvakumar, Jagadesh Kopula Kesavan, Giovanna Iucci, Umberto Pasqual Laverdura, and Simonetta Tuti. 2021. "One-Pot Synthesis of Ni0.05Ce0.95O2−δ Catalysts with Nanocubes and Nanorods Morphology for CO2 Methanation Reaction and in Operando DRIFT Analysis of Intermediate Species" Processes 9, no. 11: 1899. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9111899