
PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment



1
OncoPharma Research Group, Center for Health Technology and Services Research (CINTESIS), Rua Doutor Plácido da Costa, 4200-450 Porto, Portugal
2
LAQV/REQUIMTE, Laboratory of Applied Chemistry, Department of Chemical Sciences, Faculty of Pharmacy, University of Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal
3
Department of Community Medicine, Health Information and Decision (MEDCIDS), Faculty of Medicine, University of Porto, 4200-319 Porto, Portugal
*
Author to whom correspondence should be addressed.
Processes 2021, 9(11), 2087; https://0-doi-org.brum.beds.ac.uk/10.3390/pr9112087
Submission received: 18 October 2021
/
Revised: 15 November 2021
/
Accepted: 18 November 2021
/
Published: 22 November 2021
(This article belongs to the Special Issue Studies of the Dosage Form and Stability of the Drug by Various Techniques)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Pharmacokinetics (PK) is a branch of pharmacology present and of vital importance for the research and development (R&D) of new drugs, post-market monitoring, and continued optimizations in clinical contexts. Ultimately, pharmacokinetics can contribute to improving patients’ clinical outcomes, helping enhance the efficacy of treatments, and reducing possible adverse side effects while also contributing to precision medicine. This article discusses the methods used to predict and study human pharmacokinetics and their evolution to the current physiologically based pharmacokinetic (PBPK) modeling and simulation methods. The importance of therapeutic drug monitoring (TDM) and PBPK as valuable tools for Model-Informed Precision Dosing (MIPD) are highlighted, with particular emphasis on antibiotic therapy since dosage adjustment of antibiotics can be vital to ensure successful clinical outcomes and to prevent the spread of resistant bacterial strains.
1. Introduction–Pharmacokinetics
Pharmacokinetics is the branch of pharmacology that studies the route and fate of substances administered to a living organism until their elimination (how the organism affects the drug), while pharmacodynamics studies the biochemical and physiologic effects of drugs (how a drug affects an organism) [1,2]. The International Union of Pure and Applied Chemistry (IUPAC) defines PK as the “Process of the uptake of drugs by the body, the biotransformation they undergo, the distribution of the drugs and their metabolites in the tissues, and the elimination of the drugs and their metabolites from the body over a period of time.” [3]. The acronym ADME encompasses the PK stages: absorption, distribution, metabolism, and excretion (Figure 1). The acronym LADME introduces considerations regarding the liberation of the active substance from the delivery system; ADMET or ADME-Tox add the toxicological aspect.
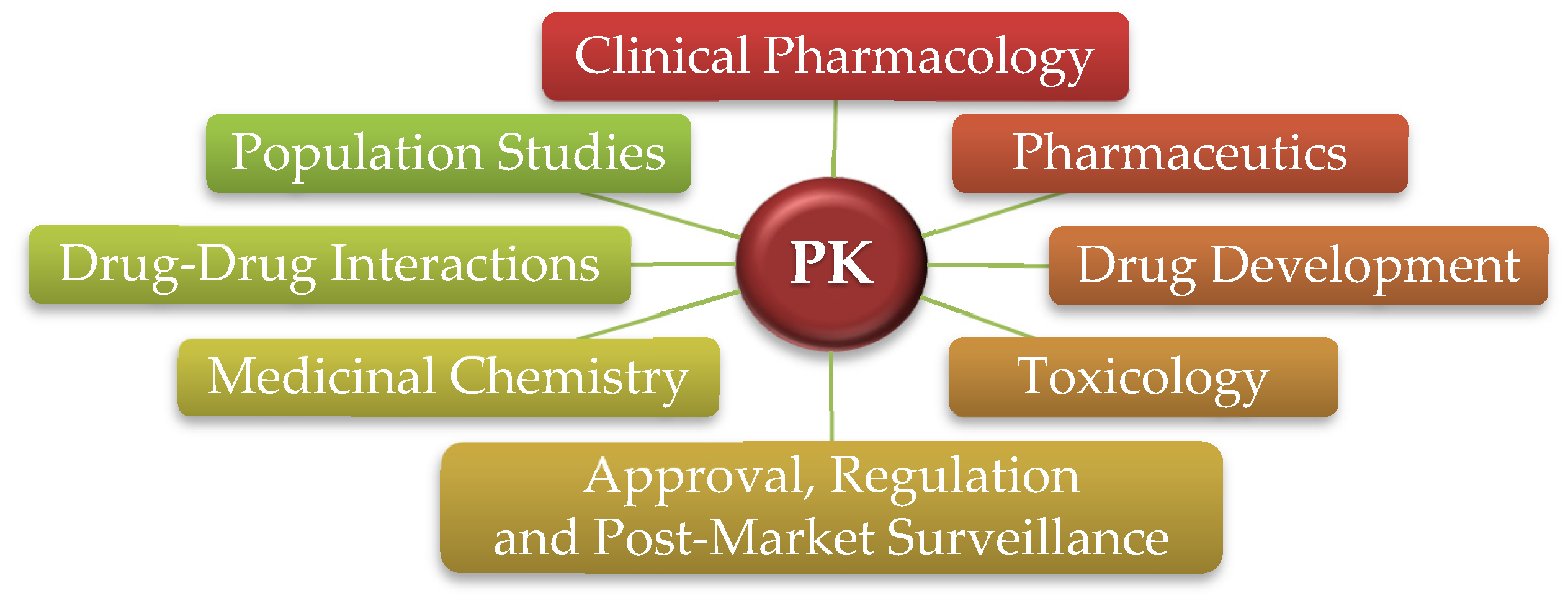
PK is a comprehensive area of pharmacology and an integral part of many fields, with countless applications and inestimable value. It has an important role throughout the research and development (R&D) process of new drugs, including during regulatory review and approval and post-market monitoring and surveillance, which extends to PK’s usefulness in helping design clinical trials and its vital significance in both clinical settings and improving patient care and outcomes (Figure 2).
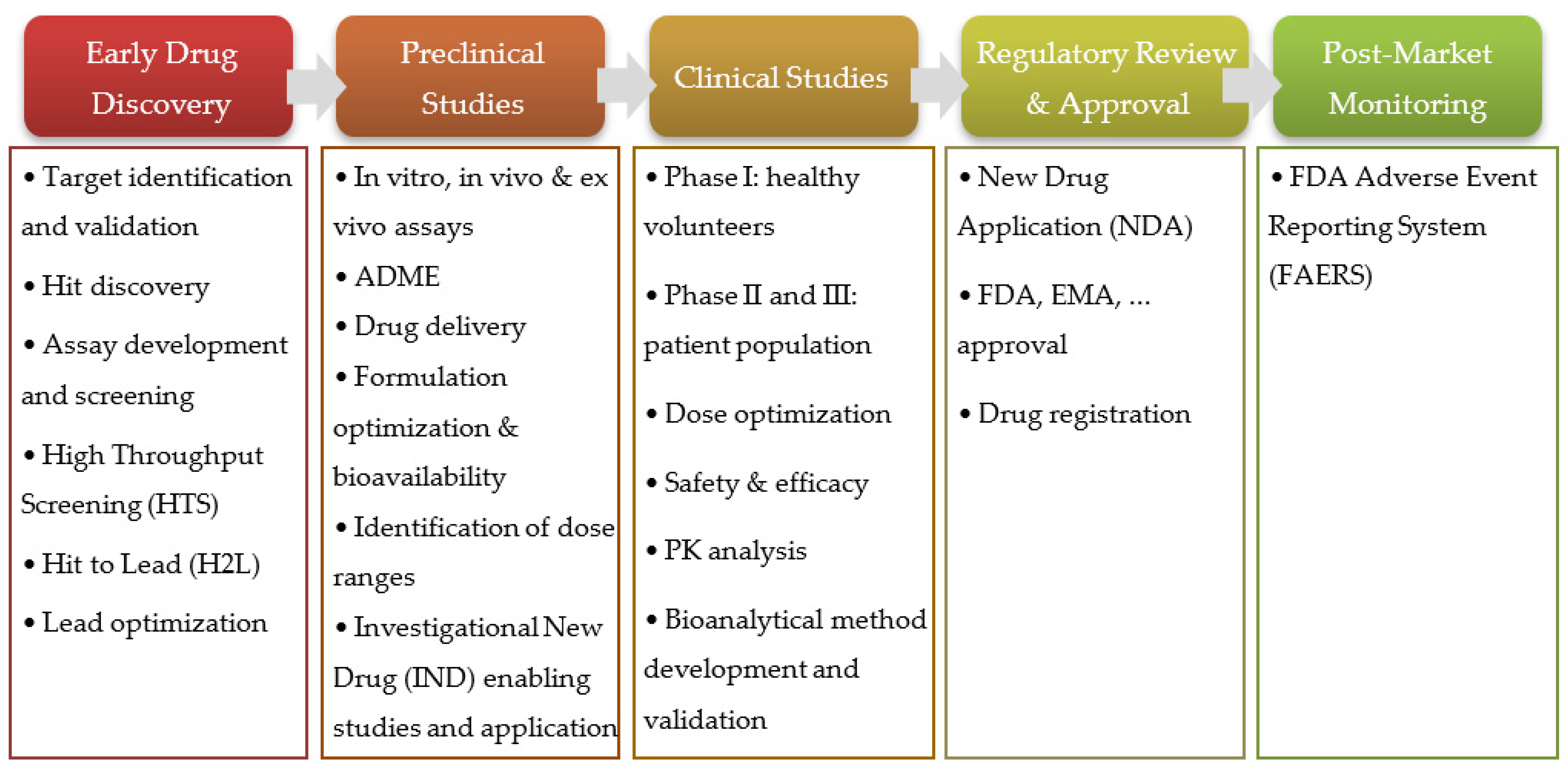
The R&D for new therapeutic agents is long, complex, difficult, and expensive, and a multitude of procedures are required for a new drug to be approved and commercialized. Only about 12 percent of drugs entering clinical trials are ultimately approved for introduction by the FDA, and recent studies estimate the development and approval of new drugs take, on average, seven to nine years. The cost of introducing a new drug can range from 1 billion USD to more than 2 billion USD [4,5,6]. This process can be divided into five stages, depicted in Figure 3 [7].
During R&D of new therapeutic agents, some of the most important factors to consider and evaluate are related to LADME properties, including solubility, lipophilicity, permeability, modification, or degradation due to chemical stability and metabolism, transport, specificity, and targeting. Furthermore, bioactivity and toxicology are crucial PD aspects to assess.
In fact, poor PK properties, such as low bioavailability, were responsible for the failure of about 40% of lead compounds 30 years ago and remain one of the main motives preventing the progression of new drug candidates to further stages [8]. Since then, with the acknowledgement of ADME properties’ major impacts on clinical outcomes, technological innovations, and in silico tools and software packages’ development, dramatic changes and reductions of the time, human resources, and financial investment necessary to achieve new advancements have been observed, both in the R&D process and clinical applications.
Predicting many of the relevant physicochemical, pharmacological, and pharmacokinetic properties and the disposition attributes of drugs using in silico methods can rapidly identify PK liabilities, such as poor bioavailability, high metabolism and clearance, the potential for drug–food and drug–drug interactions (DDI), the need for dose adjustments, and particular alterations in special populations. Identifying these PK liabilities has become essential from the early stages of new drugs’ R&D to the clinical setting, and ensuring the best outcome and minimal side effects for patients.
As such, there have been increased interest and investment in drug metabolism and pharmacokinetics (DMPK) and PK/PD relationship studies, including in the development of improved software packages. Most modern tools to model and simulate PK profiles can accelerate drug discovery and help design clinical trials, analyze clinical data in all stages of clinical evaluation, obtain regulatory approval, and conduct post-market monitoring and surveillance to quickly identify adequate therapeutic solutions [9,10,11,12,13]. The importance of TDM and PBPK as valuable tools for MIPD is evaluated, with particular emphasis on antibiotic therapy, since dosage adjustment of this class of drugs is crucial to ensure the treatment’s efficacy and the prevention of toxic effects.
2. Prediction of ADME Properties and PK Modeling and Simulation
Numerous methods have been explored to predict, study, and model human pharmacokinetics. To predict ADME and other physicochemical properties, a multitude of quantitative and mechanistic approaches can be applied. These include interspecies allometric scaling [14], in vitro-to-in vivo extrapolations (IVIVE) [15], quantitative statistical methods such as quantitative structure-activity relationships (QSAR) or quantitative structure-property relationships (QSPR) [16,17], principal component analysis (PCA), multivariate analysis (MVA) [18], and other in silico methods [19].
Early preclinical and clinical PK studies were merely descriptive, verifying concentrations in blood/plasma (Cp) over time, tracing the Cp profile, and determining the time necessary to reach the peak concentration (Tmax).
Currently, quantitative PK parameters requiring mathematical formulas can be evaluated, including volume of distribution, clearance (CL), elimination rate constants (Ke), and mean residence time (MRT), among many others. Many commercial platforms and software packages are available and user-friendly, making PK modeling and simulation more accessible without requiring extensive mathematical, modeling, or programming experience.
There are three main modeling and simulation approaches: (a) “top-down”, which refers to models built on observed data (from general to specific); (b) “bottom-up” models, which require an in-depth mechanistic description of human physiology (from specific to general); and (c) “middle-out”, which combines both strategies [20,21].
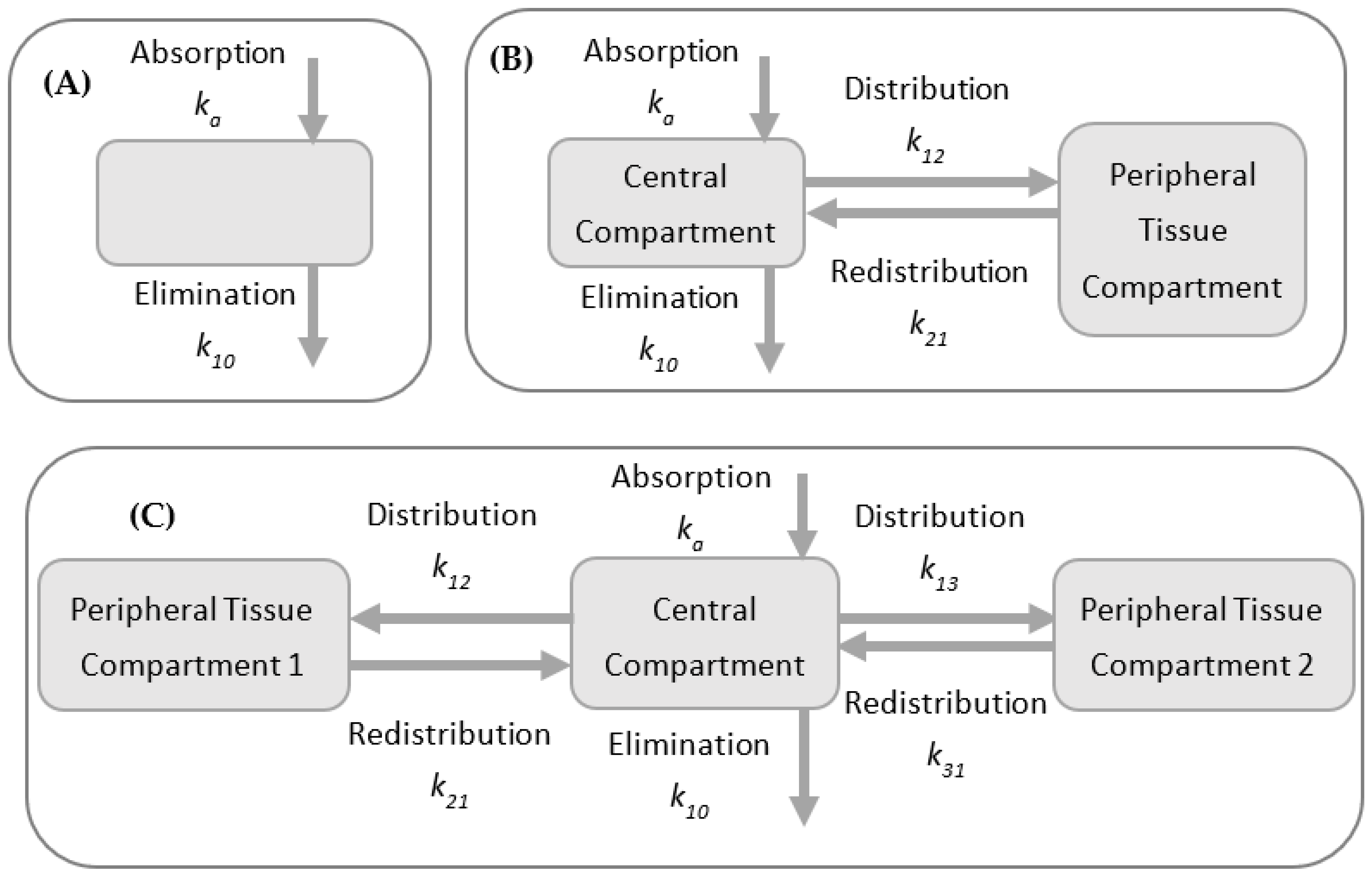
PK models are often used to describe the plasma or relevant tissue–drug concentration through time and are built using compartments as “building blocks” with increasing complexity, from noncompartmental models and models with two or three compartments (Figure 4), to more intricate models, such as whole-body or physiologically based pharmacokinetic (PBPK) models and population PK models [22,23].
2.1. Noncompartmental PK Analysis (NCA)
The most elementary PK information can be provided by NCA and relies on simple algebraic equations to analyze peak concentration and elimination half-life and estimate PK parameters. As NCA does not rely on compartments, there are no physiological assumptions or parameterizations required. The organism is considered as one single homogenous compartment. As such, NCA is a much faster and cost-efficient method. Despite the numerous advantages of compartmental models, NCA, generally applied to first-order (linear) models, is typically favored for characterizing PK within a single study, including during the steps to make dose escalation decisions. This method is also convenient for the characterization of new drug products, helping guide development at various stages [24,25].
2.2. Compartmental Models
Different body organs and tissues can be defined by compartmental models, kinetically interconnected [26]. Typically, a central compartment representing plasma is linked to one or two peripheral compartments via rate constants. Although more complex than NCA, these models allow for more variability since certain assumptions are made to build and parameterize the PK model. Though these models generally do not hold any physiological meaning, they can provide important PK descriptors such as clearance and volume of distribution and, thus, effective drug half-life or “residence” time.
2.3. Physiologically Based Pharmacokinetic Models (PBPK)
Using similar mathematical frameworks and a series of differential equations, PBPK models have more compartments, parameterized with physiological knowledge of specific organs or tissues and flow rates connecting the system. These dynamic models can predict most PK attributes and the concentration-time profile after drug administration. PBPK models can be used for a wide variety of purposes and applications and present numerous advantages compared to other methods since PBPK models account for sequential metabolism and permeability limited processes [27,28,29,30,31]. After obtaining data from multiple sources (for example, from physicochemical or biochemical experiments and in vitro or in vivo pharmacological or toxicological assays), PBPK models can be used collectively to analyze and integrate this information. Of particular clinical interest, these integrative systems can predict drug and metabolites’ concentrations in different body sites/organs. Thus, PBPK models can assist dosage optimization and adjustment and explore different routes of administration, predicting drug exposure. Furthermore, these models can integrate the physiological data of preclinical species, help with allometric scaling, and be parametrized for specific individuals or populations (for example, by biological sex; children, adults, the elderly; non-pregnant and pregnant women; and patients with cirrhosis).
Sager et al. conducted a systematic review of publications between 2008 and May 2014 related to PBPK models [29]. Searching the PubMed database for papers that included the terms “PBPK” and “physiologically based pharmacokinetic model”, a total of 366 articles were analyzed regarding the models’ development and applications. These publications have been steadily increasing, from 9 papers in 2008 to 94 in 2014. The most common applications were drug–drug interaction (DDI) studies (28%), interindividual variability and general clinical pharmacokinetics predictions (23%), absorption kinetics (12%), and age-related changes in pharmacokinetics (10%). For FDA regulatory filings, models were primarily used for DDI predictions (60%), pediatrics (21%), and absorption predictions (6%).
2.4. Population PK
While individual PK studies are the best approach when rapid processing of PK parameters is needed or when complete individual PK profiles are to be defined, population PK (PopPK) analysis and modeling approaches are of value to study variability in drug concentrations within a population of patients receiving clinically relevant doses of a drug of interest. This method requires concentration–time data from multiple individuals and can incorporate covariate information such as age, sex, weight, race, renal/hepatic function, and data about concomitant medications that can lead to DDIs. PopPK aims to identify the most impacting covariates, meaning the measurable pathophysiologic factors that influence the dose–concentration relationship the most and their extent. User-friendly PopPK software has been developed to support all stages of drug development and surveillance because assessing the sources of PK variability can be essential for drug safety and efficacy, and appropriate dosages can be selected for a given population or subgroup with information granted by PopPK models [32,33]. However, in physiological and pathological conditions, the limitations of PBPK modeling can be associated with comprehensive data about the physiological, biochemical, and physicochemical processes. These data are not available from only one source, and information gaps may exist.
3. PK Prediction in Silico Tools
As previously mentioned, there has been increasing interest and investment in pharmacokinetic studies, including the development of tools and software for predicting PK attributes and modeling and simulating profiles. Some of the available software used for PK studies are GastroPlus™ and Monolix® by Simulations Plus, Inc. (Lancaster, CA, USA), Simcyp®, NONMEM (ICON plc), Phoenix® WinNonlin® and Phoenix® NLME™ by Certara (UK Limited, Sheffield, UK), and PK-Sim® (OSP, Open Systems Pharmacology).
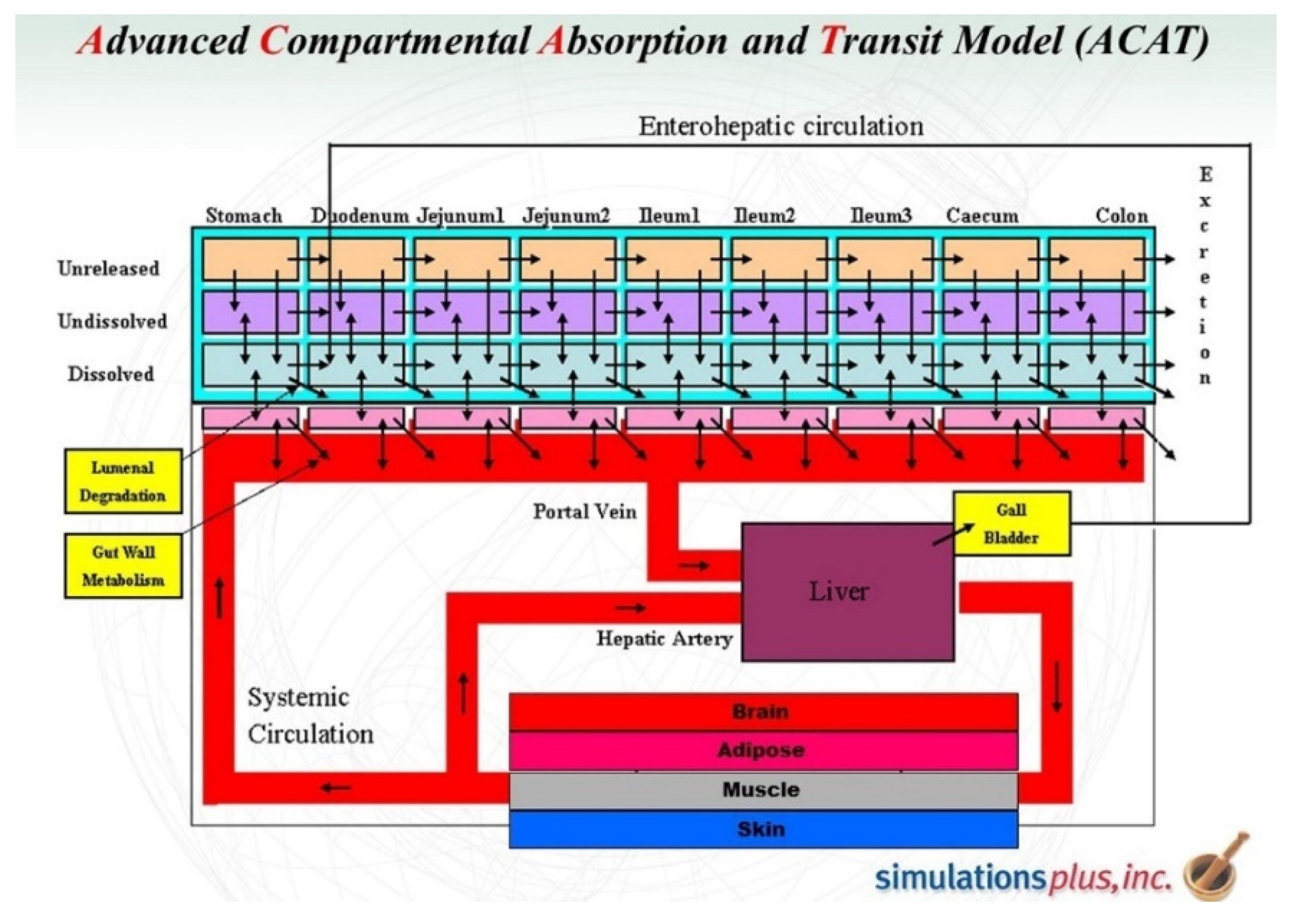
Whereas some of these software packages are generic tools to compute complex mathematical equations and perform compartmental analysis, others, such as GastroPlus™, were specifically designed for PK studies, particularly physiologically based pharmacokinetics (PBPK) and physiologically based biopharmaceutics modeling (PBBM). GastroPlus™’s features and capabilities allow the prediction of drug absorption and disposition, and the simulation of absorption, pharmacokinetics, and pharmacodynamics in humans and many preclinical species, thanks to preinstalled physiological parameters for numerous tissues (such as tissue weights and volumes, perfusion rates, and compartments’ pH) and an integrated advanced compartmental and transit (ACAT™) model (Figure 5). Thus, GastroPlus™ supports model-based drug development and PK assessments in all phases of drug discovery, translational research, and clinical development. This not only improves decision-making throughout clinical drug development but also enables the design and optimization of dosing regimens and formulations, increasing the chances of the drug reaching its target with the desired concentration and maintaining drug plasma concentration within the therapeutic window [27,29,34,35,36,37].
Calculations and simulations rely on the numerical integration of differential equations that coordinate a set of well-characterized physical events that occur and interconnect as a result of diverse physicochemical and biologic phenomena. Many of the currently available software have increasingly intuitive and modern graphical user interfaces and are relatively easy for someone with a background in ADME to learn and use, enabling smooth, rapid transitions from setting up inputs to evaluating results. High-quality and amply customizable PBPK models can thus be easily developed. Simulation studies can be initiated based on a drug’s structure and a small set of physicochemical data to predict the most important parameters in pharmacokinetics (PK), such as the maximum concentration (Cmax) reached in plasma and the liver, time necessary to reach such concentrations (Tmax), half-life (t1/2), fraction absorbed (Fa) and bioavailability (F), and area under the curve (AUC).
Additionally, some of these software programs, including GastroPlus™, not only calculate PK attributes but also draw a graphical representation of concentration over time profiles for quicker interpretation of simulation results and are able to perform parameter sensitivity analysis (PSA) to measure the impact of fluctuations in input parameters and population PK studies.
As previously stated, the scope of applications of PK goes well beyond its central role in the R&D process. In fact, pharmacokinetics is valuable in countless pharmacological evaluations, whether for academic purposes, drug development and clinical research, or in clinical medicine for therapeutic drug monitoring and individualized dosing towards precision medicine.
4. Therapeutic Drug Monitoring (TDM)
Even after the myriad studies and optimizations required to approve a drug with a determined therapeutic application and recommended dosing regimens (label and guidelines), not all drugs will perform as a “one-size-fits-all”. Due to certain pharmacological characteristics of some drugs and drug classes, their dosing regimens will need to be adjusted and customized for each patient [39,40], which can be accomplished through therapeutic drug monitoring (TDM).
This branch of clinical chemistry and clinical pharmacology specializes in measuring circulating drug concentrations to adjust dosing regimens to reach a defined target exposure associated with optimal efficacy and minimal toxicity [41,42]. TDM can be traced back to the late 1960s and the efforts of clinicians to improve patient care and clinical outcome [42]. The cases that required dosage individualization have been extensively reviewed, and TDM is now indicated and recommended for critically ill patients undergoing sufficiently long treatment to justify dosage adjustment efforts and for drugs that have the following pharmacological properties [43]:
- Poorly predictable PK and significant interpatient variability, resulting in a wide range of concentration levels between patients after standard dosage administration;
- Narrow therapeutic window that, combined with interpatient variability, poses a high risk of misdoing. The standard dosage could be subtherapeutic for some patients, but the use of very high standard doses in all patients to ensure overall efficacy is forbidden due to the risk of toxicity [44];
- Consistent concentration exposure and response and/or toxicity (PD) relationships; moreover, effects following changes in drug exposure should be reversible, enabling the definition of a range of concentrations associated with optimal efficacy and minimal toxicity;
- Lack of readily assessable PD markers and quick response to dosage changes;
- Acceptable PK stability, considering within-patient PK variability over time (inter-occasion variability) and assay and/or model-related errors [45].
TDM has proven favorable and recommended for hundreds of therapeutic agents, including anticancer drugs [46], anti-infectives [47], antiretrovirals [48], biologic therapeutic agents [49], and psychotropic agents [50]. Traditionally, clinicians would analyze the results from the TDM and empirically modify a patient’s dosage to approximate circulating concentrations to the identified target therapeutic window. Advantages of this approach include its simple interpretation of the TDM data and undemanding implementation because the adjustment can generally be made based on a mathematical “rule of three”, changing either dose or dosing interval (Dettli rules [51]). Some conventional therapeutic ranges have been extended to nomograms that can assist in the adjustment decision.
Notwithstanding its simplicity and usefulness, traditional TDM holds some limitations. The blood samples to determine drug concentration can only be collected after steady-state is reached, typically meaning dosage will only be adjusted 3–4 days after the beginning of treatment. While this is a suitable time span for many drugs, in the case of infections and antibiotic treatment the PK/PD target should be promptly achieved. Moreover, some antibiotics exhibit nonlinear PK and are concentration- and/or time-dependent. In such cases, dosage adjustment cannot be based on the “rule of three”. Another weakness of this approach is likewise related to sampling because a single sample determination (as Cpeak or Ctrough) is frequently an insufficient indicator of drug exposure. Additionally, the timing of both dose administration and sample collection is a critical factor to ensure accurate interpretation of results and appropriate adjustments [43].
Consequently, dosage adjustments based on traditional TDM are considered a passive procedure because therapeutic ranges are often wide, and due to the interpatient variability and other factors described above, PK/PD targets and successful clinical outcomes may not be achieved. Thus, improvements to this practice are needed.
4.1. Target Concentration Intervention (TCI)
Taking advantage of the increasing computational power and advances in computer sciences, TCI has been introduced as a more adequate approach. The main goal of TCI is to reach the target therapeutic concentration by administering the appropriate dose to each patient. This approach is based on both individual PK data and PD observations that are analyzed to predict the dose required to achieve the target. Moreover, TCI improves clinical outcomes while being cost-effective. Computer-assisted solutions for interpreting TDM results in a clinical setting are now available, and mathematical models have been developed to further inform clinicians’ decisions [52,53].
4.2. Model-Informed Precision Dosing (MIPD)
MIPD has emerged as an integrative approach to precision medicine and is considered the next milestone in medical progress after evidence-based medicine [54]. These mathematical models are built with the input from observational population PK studies to interpret the measured drug concentration and predict personalized dosing beyond a specific approach or technique. When in significant numbers, these studies assemble data on drugs’ average PK parameters and identify the most impacting covariates or individual factors contributing to inter and intraindividual variability. The latter can be accounted for using both parametric and nonparametric approaches [55,56]. The main difference is that in nonparametric approaches, support points are estimated from the clinical data, while parametric approaches use a defined distribution of PK parameters.
The most recurrently recognized covariates include age, body weight, biological sex, and serum creatinine (which are needed to evaluate renal function). Genetic aspects, comorbidities, the patient’s clinical status (such as disease status, renal/hepatic function, biological markers, and treatment tolerance), and comedications also influence PK attributes [57]. These variables and their impact on PK profiles can also be evaluated using PBPK modeling tools because these models can be customized to represent a wide variety of patients’ physiological and pathological characteristics [58,59,60].
Bayesian inference is also a powerful approach to individualizing dosing regimens and is of undeniable value for TDM and MIPD. This approach is well-befitting in clinical settings where few drug concentrations measured in blood samples are available, making it a valuable tool in the effective implementation of therapeutic drug monitoring [43,61,62,63]. The main components of the Bayesian approach are prior distribution, likelihood principle, posterior probabilities, decision rules, and predictive probability. Patients’ measurements are compared with population percentiles and a priori percentiles expected for patients with similar individual characteristics (covariates). Following this, the dosage can be adjusted after Bayesian inference deduction of a posteriori percentiles from the preceding analyses and the patient’s observation.
5. Infections and Antibiotic Therapy
Infections are a serious health threat. They are caused by infectious agents (also called pathogens) such as viruses and microorganisms like bacteria, fungi, parasites, and arthropods. The immune system can fight many infections, but specific medication is often needed, especially since some of these agents are becoming increasingly aggressive. Additionally, severe consequences can arise from the infectious agents reaching the bloodstream and spreading to other locations and from sepsis, a life-threatening condition where tissues and organs are gravely affected by the own body’s response to infection. This project focused on the anti-infective class of antibiotics.
The first antibiotic was penicillin, discovered by Alexander Fleming in 1928. Since then, and more predominantly after the 1940s, antibiotics have revolutionized the treatment of patients with severe bacterial infections, significantly reducing morbidity and mortality. However, these drugs have been overused and are currently one of the most widely, and often injudiciously, prescribed and used therapeutic drugs worldwide, which has led to a bacterial selection of resistant strains.
Nowadays, antibiotic resistance is one of the biggest public health concerns and is a major problem in both hospital environments and outpatient situations. A report by the World Health Organization (WHO) revealed antibiotic resistance is a “serious threat (that) is no longer a prediction for the future, it is happening right now in every region of the world and has the potential to affect anyone, of any age, in any country. Antibiotic resistance–when bacteria change so antibiotics no longer work in people who need them to treat infections–is now a major threat to public health.” [64,65].
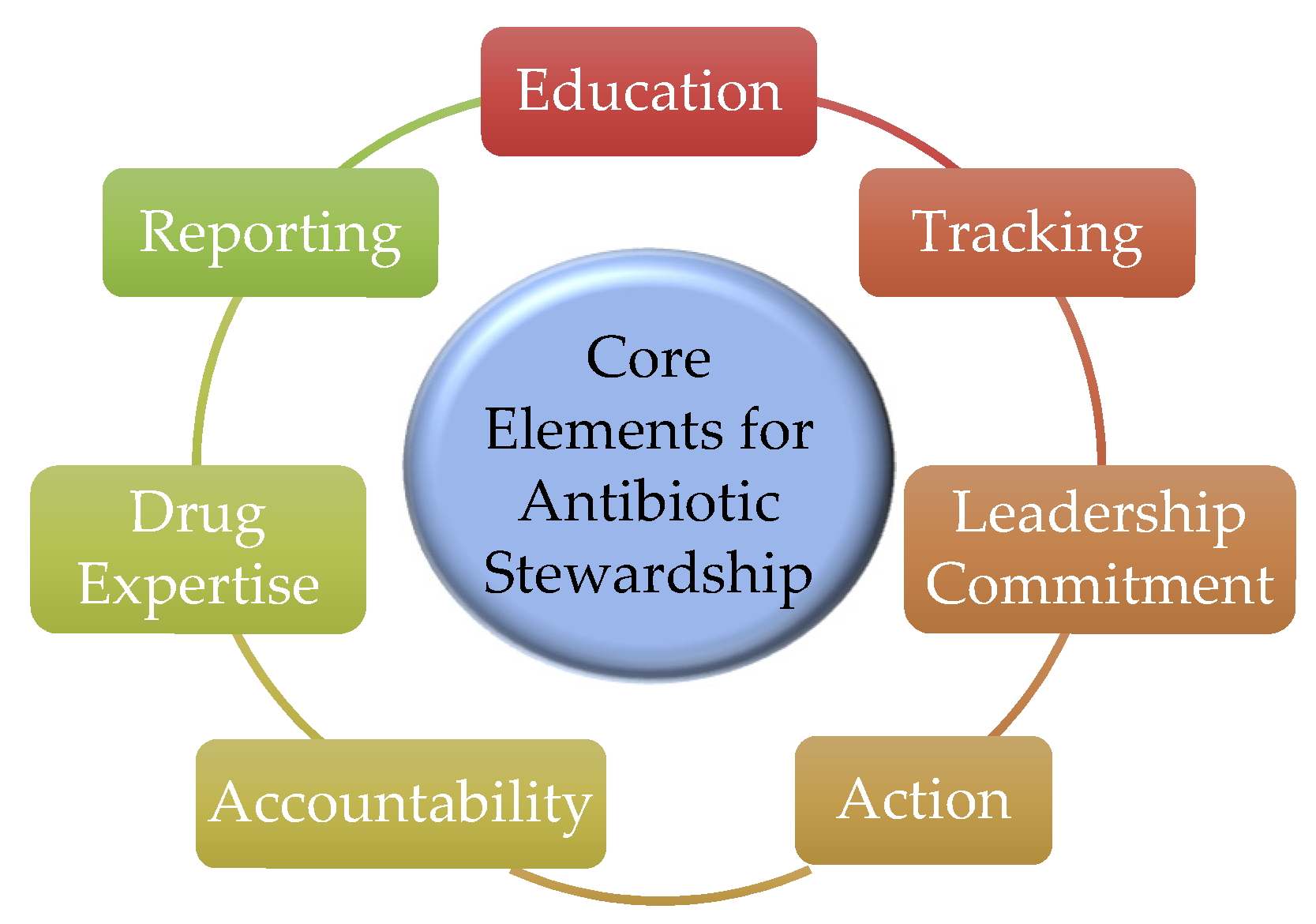
Antibiotic resistance is a major factor supporting the importance of monitoring and optimizing antibiotics use and the implementation of antibiotic stewardship programs (ASP). The Centers for Disease Control and Prevention (CDC) identified seven core elements of antibiotic stewardship in 2014 and recommend that all hospitals have an ASP (Figure 6) [66]. Tracking (monitoring process measures), reporting information on antibiotic use and resistance, and educating clinicians and health care providers are three of these core elements. Optimizing the use of antibiotics leads to the maximization of therapeutic success and will extend the clinical lifespan of currently available antimicrobial agents by limiting the emergence of resistance [67,68].
Antibiotics and the Need for TDM and Dosage Adjustment
As discussed earlier, monitoring patients and adjusting their dosing regimens can be vital to ensure their successful clinical outcome, with minimized side effects. It has been extensively demonstrated that this is crucial in the case of antibiotics, with confirmed beneficial results [69,70,71,72,73,74].
Typically, patients’ health and welfare are supervised by a team of medical professionals, and biological samples, mainly blood, are collected and biochemically analyzed regularly. Therapeutic drug monitoring (TDM) evaluates drug concentrations and other biochemical markers (among which, in the case of many antibiotics that are renally excreted, creatinine is of key importance to assess renal function) to ensure the appropriate dose is administered to patients by continuously recommending dosing adjustments to optimize clinical outcomes without developing severe side effects.
The most frequently monitored antibiotics in inpatients are aminoglycosides (such as amikacin, gentamicin, and tobramycin) and glycopeptide vancomycin, which can be explained by their narrow therapeutic indexes and potential to cause adverse effects, namely nephrotoxicity, particularly in prolonged treatments [75,76,77]. These antibiotics are widely used to treat severe infections caused by Gram-negative (aminoglycosides) and Gram-positive (vancomycin) bacteria.
Though historically, TDM was mostly implemented to prevent toxic adverse effects, mainly for glycopeptides and aminoglycosides, the assessment of trough and peak concentrations is considered diagnostically and therapeutically important and strongly recommended for patients using the aforementioned antibiotics. Since the treatment of serious infections can imply the administration of high doses of antibiotics, an accumulation of these drugs in plasma is recurrent, often leading to toxic effects, and the value of TDM of these antibiotics has been demonstrated [73,74,78,79,80,81,82,83,84,85,86]. Although TDM has been recommended for these antibiotics, it is still not a routine clinical practice for reasons still not systematically reviewed. Not only does this process entails costs, but it is also a massive challenge, particularly for intensive care units, since these patients often present altered PK and significant inter- and intra-individual pharmacokinetic (PK) variability [87].
In silico modeling tools, including PBPK, have proven extended value and can provide additional knowledge to assist clinicians in the process of adjusting therapeutic regimens [88,89,90]. Despite the lack of uniformization, with growing data becoming available, these models can be further refined and contribute to TDM and dosage adjustment processes, evolving towards personalized medicine.
6. Final Remarks
The importance of pharmacokinetics was highlighted and some of its applications explored, from basic research and drugs’ R&D, to optimization of patients’ clinical outcomes in hospital settings and the advancements towards precision medicine. Various methods to predict drug disposition were reviewed, with an emphasis on PBPK.
Approaches such as PBPK, TDM, TCI, and MIPD, which were mentioned in this paper, have extensively demonstrated value and can provide knowledge to assist clinicians in the process of adjusting therapeutic regimens. For cases such as antibiotic therapy, these personalized interventions can be of vital importance.
Author Contributions
Conceptualization, N.V.; methodology, A.F. and N.V.; software, A.F.; validation, N.V., A.F. and R.L.; investigation, A.F. and N.V.; resources, N.V. and R.L.; writing—original draft preparation, A.F.; writing—review and editing, R.L. and N.V.; supervision, N.V.; project administration, N.V.; funding acquisition, R.L. and N.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financed by FEDER–Fundo Europeu de Desenvolvimento Regional through the COMPETE 2020–Operational Programme for Competitiveness and Internationalization (POCI), Portugal 2020, and by Portuguese funds through FCT–Fundação para a Ciência e a Tecnologia, in a framework of CINTESIS, R&D Unit (reference UIDB/4255/2020). N.V. also thanks support from FCT and FEDER (European Union), award number IF/00092/2014/CP1255/CT0004. RL thanks FCT through grant UID/QUI/50006/2019 (LAQV-REQUIMTE).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
A.F. thanks FCT for a doctoral fellowship (PD/BD/135120/2017). The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of the FCT.
Conflicts of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Tozer, T.N.; Rowland, M. Essentials of Pharmacokinetics and Pharmacodynamics; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2015; ISBN 9781451194425. [Google Scholar]
- Fan, J.; de Lannoy, I.A. Pharmacokinetics. Biochem. Pharmacol. 2014, 87, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, M.; Duffus, J.; Templeton, D.M. Glossary of terms used in toxicokinetics (IUPAC Recommendations 2003). Pure Appl. Chem. 2004, 76, 1033–1082. [Google Scholar] [CrossRef]
- Research and Development in the Pharmaceutical Industry. Available online: https://www.cbo.gov/publication/57126 (accessed on 1 July 2021).
- Sertkaya, A.; Wong, H.-H.; Jessup, A.; Beleche, T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials 2016, 13, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. New drugs cost US$2.6 billion to develop. Nat. Rev. Drug Discov. 2014, 13, 877. [Google Scholar] [CrossRef]
- USA Food and Drug Admistration. The Drug Development Process. Available online: https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process (accessed on 1 July 2021).
- Alavijeh, M.; Palmer, A. The pivotal role of drug metabolism and pharmacokinetics in the discovery and development of new medicines. IDrugs 2004, 7, 755–763. [Google Scholar]
- Pappalardo, F.; Russo, G.; Tshinanu, F.M.; Viceconti, M. In silico clinical trials: Concepts and early adoptions. Br. Bioinform. 2019, 20, 1699–1708. [Google Scholar] [CrossRef]
- Marsousi, N.; Desmeules, J.A.; Rudaz, S.; Daali, Y. Usefulness of PBPK Modeling in Incorporation of Clinical Conditions in Personalized Medicine. J. Pharm. Sci. 2017, 106, 2380–2391. [Google Scholar] [CrossRef] [Green Version]
- Michelson, S.; Sehgal, A.; Friedrich, C. In silico prediction of clinical efficacy. Curr. Opin. Biotechnol. 2006, 17, 666–670. [Google Scholar] [CrossRef]
- Kimko, H.; Pinheiro, J. Model-based clinical drug development in the past, present and future: A commentary. Br. J. Clin. Pharmacol. 2015, 79, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Bolger, M.B.; Fraczkiewicz, R.; Lukacova, V. Simulations of Absorption, Metabolism, and Bioavailability. In Drug Bioavailability; Mannhold, R., Kubinyi, H., Folkers, G., van de Waterbeemd, H., Testa, B., Eds.; John Wiley Sons: Hoboken, NJ, USA, 2008; pp. 453–495. [Google Scholar] [CrossRef]
- Huang, Q.; Riviere, J.E. The application of allometric scaling principles to predict pharmacokinetic parameters across species. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1241–1253. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, J.E.; Kim, D.D.; Yoon, I.S. In Vitro-In Vivo extrapolation (IVIVE) for predicting human intestinal absorption and first-pass elimination of drugs: Principles and applications. Drug Dev. Ind. Pharm. 2014, 40, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T. Predictions of the ADMET properties of candidate drug molecules utilizing different QSAR/QSPR modelling approaches. Curr. Drug Metab. 2010, 11, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U.; Bergström, C.A.S. Prediction of ADMET properties. ChemMedChem Chem. Enabling Drug Discov. 2006, 1, 920–937. [Google Scholar]
- Van der Graaf, P.H.; Nilsson, J.; Van Schaick, E.A.; Danhof, M. Multivariate quantitative structure-pharmacokinetic relationships (QSPKR) analysis of adenosine A1 receptor agonists in rat. J. Pharm. Sci. 1999, 88, 306–312. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Tylutki, Z.; Polak, S.; Wiśniowska, B. Top-down, Bottom-up and Middle-out Strategies for Drug Cardiac Safety Assessment via Modeling and Simulations. Curr. Pharmacol. Rep. 2016, 2, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Tsamandouras, N.; Rostami-Hodjegan, A.; Aarons, L. Combining the “bottom up” and “top down” approaches in pharmacokinetic modelling: Fitting PBPK models to observed clinical data. Br. J. Clin. Pharmacol. 2015, 79, 48–55. [Google Scholar] [CrossRef]
- Gabrielsson, J.; Weiner, D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications, 3rd ed.; Taylor & Francis: Abingdon, UK, 2001; ISBN 9789186274924. [Google Scholar]
- Marchenko, O.V.; Katenka, N.V. Quantitative Methods in Pharmaceutical Research and Development: Concepts and Applications; Springer International Publishing: Cham, Switzerland, 2020; ISBN 9783030485559. [Google Scholar]
- Gabrielsson, J.; Weiner, D. Non-compartmental analysis. Methods Mol. Biol. 2012, 929, 377–389. [Google Scholar] [CrossRef]
- Bulitta, J.B.; Holford, N.H.G. Non-Compartmental Analysis. In Wiley Encyclopedia of Clinical Trials; Wiley: Hoboken, NJ, USA, 2007; pp. 1–21. [Google Scholar]
- Chen, B.; Abuassba, A.O.M. Compartmental Models with Application to Pharmacokinetics. Procedia Comput. Sci. 2021, 187, 60–70. [Google Scholar] [CrossRef]
- Parrott, N.; Lave, T. Applications of physiologically based absorption models in drug discovery and development. Mol. Pharm. 2008, 5, 760–775. [Google Scholar] [CrossRef]
- Jones, H.; Rowland-Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e63. [Google Scholar] [CrossRef]
- Sager, J.E.; Yu, J.; Ragueneau-Majlessi, I.; Isoherranen, N. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab. Dispos. 2015, 43, 1823–1837. [Google Scholar] [CrossRef]
- Upton, R.N.; Foster, D.J.R.; Abuhelwa, A.Y. An introduction to physiologically-based pharmacokinetic models. Pediatr. Anesth. 2016, 26, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Nestorov, I. Whole Body Pharmacokinetic Models. Clin. Pharmacokinet. 2003, 42, 883–908. [Google Scholar] [CrossRef] [PubMed]
- Charles, B. Population pharmacokinetics: An overview. Aust. Prescr. 2014, 37, 210–213. [Google Scholar] [CrossRef]
- Ette, E.I.; Williams, P.J. Population Pharmacokinetics I: Background, Concepts, and Models. Ann. Pharmacother. 2004, 38, 1702–1706. [Google Scholar] [CrossRef]
- Di, L.; Feng, B.; Goosen, T.C.; Lai, Y.; Steyn, S.J.; Varma, M.V.; Obach, R.S. A Perspective on the Prediction of Drug Pharmacokinetics and Disposition in Drug Research and Development. Drug Metab. Dispos. 2013, 41, 1975–1993. [Google Scholar] [CrossRef] [Green Version]
- Daga, P.R.; Bolger, M.B.; Haworth, I.S.; Clark, R.D.; Martin, E.J. Physiologically Based Pharmacokinetic Modeling in Lead Optimization. 1. Evaluation and Adaptation of GastroPlus To Predict Bioavailability of Medchem Series. Mol. Pharm. 2018, 15, 821–830. [Google Scholar] [CrossRef]
- Xia, B.; Yang, Z.; Zhou, H.; Lukacova, V.; Zhu, W.; Milewski, M.; Kesisoglou, F. Development of a Novel Oral Cavity Compartmental Absorption and Transit Model for Sublingual Administration: Illustration with Zolpidem. Am. Assoc. Pharm. Sci. J. 2015, 17, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.; Lobato, L.; Vale, N. Clinical pharmacokinetic study of latrepirdine via in silico sublingual administration. Silico Pharmacol. 2021, 9, 29. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A.; et al. PBPK models for the prediction of In Vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, B.A.; Marshall, J.L.; Hartley, M.; Salem, M.E. A paradigm shift from one-size-fits-all to tailor-made therapy for metastatic colorectal cancer. Clin. Adv. Hematol. Oncol. 2016, 14, 116–128. [Google Scholar] [PubMed]
- Gastmeier, P. From “one size fits all” to personalized infection prevention. J. Hosp. Infect. 2020, 104, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Clarke, W.; Dasgupta, A. Clinical Challenges in Therapeutic Drug Monitoring: Special Populations, Physiological Conditions and Pharmacogenomics; Elsevier Science: Amsterdam, The Netherlands, 2016; ISBN 9780128020524. [Google Scholar]
- Kang, J.S.; Lee, M.H. Overview of therapeutic drug monitoring. Korean J. Intern. Med. 2009, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Buclin, T.; Thoma, Y.; Widmer, N.; André, P.; Guidi, M.; Csajka, C.; Decosterd, L.A. The Steps to Therapeutic Drug Monitoring: A Structured Approach Illustrated with Imatinib. Front. Pharmacol. 2020, 11, 177. [Google Scholar] [CrossRef]
- Holford, N.H.G.; Buclin, T. Safe and Effective Variability—A Criterion for Dose Individualization. Ther. Drug Monit. 2012, 34, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Abrantes, J.A.; Jönsson, S.; Karlsson, M.O.; Nielsen, E.I. Handling interoccasion variability in model-based dose individualization using therapeutic drug monitoring data. Br. J. Clin. Pharmacol. 2019, 85, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Widmer, N.; Bardin, C.; Chatelut, E.; Paci, A.; Beijnen, J.; Levêque, D.; Veal, G.; Astier, A. Review of therapeutic drug monitoring of anticancer drugs part two-Targeted therapies. Eur. J. Cancer 2014, 50, 2020–2036. [Google Scholar] [CrossRef]
- Muller, A.E.; Huttner, B.; Huttner, A. Therapeutic Drug Monitoring of Beta-Lactams and Other Antibiotics in the Intensive Care Unit: Which Agents, Which Patients and Which Infections? Drugs 2018, 78, 439–451. [Google Scholar] [CrossRef]
- Maitre, T.; Muret, P.; Blot, M.; Waldner, A.; Duong, M.; Si-Mohammed, A.; Chavanet, P.; Aho, S.; Piroth, L. Benefits and Limits of Antiretroviral Drug Monitoring in Routine Practice. Curr. HIV Res. 2019, 17, 190–197. [Google Scholar] [CrossRef]
- Papamichael, K.; Cheifetz, A.S. Therapeutic drug monitoring in patients on biologics: Lessons from gastroenterology. Curr. Opin. Rheumatol. 2020, 32, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Schoretsanitis, G.; Paulzen, M.; Unterecker, S.; Schwarz, M.; Conca, A.; Zernig, G.; Gründer, G.; Haen, E.; Baumann, P.; Bergemann, N.; et al. TDM in psychiatry and neurology: A comprehensive summary of the consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology, update 2017; a tool for clinicians. World J. Biol. Psychiatry 2018, 19, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Keller, F.; Schröppel, B.; Ludwig, U. Pharmacokinetic and pharmacodynamic considerations of antimicrobial drug therapy in cancer patients with kidney dysfunction. World J. Nephrol. 2015, 4, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Holford, N. Pharmacodynamic principles and target concentration intervention. Transl. Clin. Pharmacol. 2018, 26, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holford, N.; Ma, G.; Metz, D. TDM is dead. Long live TCI! Br. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Keizer, R.J.; ter Heine, R.; Frymoyer, A.; Lesko, L.J.; Mangat, R.; Goswami, S. Model-Informed Precision Dosing at the Bedside: Scientific Challenges and Opportunities. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 785–787. [Google Scholar] [CrossRef]
- Guidi, M.; Csajka, C.; Buclin, T. Parametric Approaches in Population Pharmacokinetics. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Goutelle, S.; Woillard, J.B.; Neely, M.; Yamada, W.; Bourguignon, L. Nonparametric Methods in Population Pharmacokinetics. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Xu, X.S.; Yuan, M.; Zhu, H.; Yang, Y.; Wang, H.; Zhou, H.; Xu, J.; Zhang, L.; Pinheiro, J. Full covariate modelling approach in population pharmacokinetics: Understanding the underlying hypothesis tests and implications of multiplicity. Br. J. Clin. Pharmacol. 2018, 84, 1525–1534. [Google Scholar] [CrossRef] [Green Version]
- Abbiati, R.A.; Manca, D. A modeling tool for the personalization of pharmacokinetic predictions. Comput. Chem. Eng. 2016, 91, 28–37. [Google Scholar] [CrossRef]
- Hartmanshenn, C.; Scherholz, M.; Androulakis, I.P. Physiologically-based pharmacokinetic models: Approaches for enabling personalized medicine. J. Pharmacokinet. Pharmacodyn. 2016, 43, 481–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, M.; Hirani, R.; Tsiu, M.; Kabani, K.; Chaturvedula, A.; Palasik, B. Utility of Physiologically Based Pharmacokinetic Modeling in Point-of-Care Decisions: An Example Using Digoxin Dosing in Continuous Venovenous Hemodiafiltration. Ther. Drug Monit. 2020, 42, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K. Use of Bayesian statistics in drug development: Advantages and challenges. Int. J. Appl. Basic Med. Res. 2012, 2, 3–6. [Google Scholar] [CrossRef]
- Donagher, J.; Barras, M.A. Therapeutic drug monitoring: Using Bayesian methods to evaluate hospital practice. J. Pharm. Pract. Res. 2018, 48, 522–529. [Google Scholar] [CrossRef]
- Drennan, P.; Doogue, M.; van Hal, S.J.; Chin, P. Bayesian therapeutic drug monitoring software: Past, present and future. Int. J. Pharmacokinet. 2018, 3, 109–114. [Google Scholar] [CrossRef]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014; ISBN 9789241564748. [Google Scholar]
- WHO’s First Global Report on Antibiotic Resistance Reveals Serious, Worldwide Threat to Public Health. Available online: https://www.who.int/southeastasia/news/detail/30-04-2014-who-s-first-global-report-on-antibiotic-resistance-reveals-serious-worldwide-threat-to-public-health (accessed on 1 July 2021).
- Pollack, L.A.; Srinivasan, A. Core elements of hospital antibiotic stewardship programs from the Centers for Disease Control and Prevention. Clin. Infect. Dis. 2014, 59 (Suppl. 3), S97–S100. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.A.; Abdul-Aziz, M.H.; Lipman, J.; Mouton, J.W.; Vinks, A.A.; Felton, T.W.; Hope, W.W.; Farkas, A.; Neely, M.N.; Schentag, J.J.; et al. Individualised antibiotic dosing for patients who are critically ill: Challenges and potential solutions. Lancet Infect. Dis. 2014, 14, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Cunha, C.B.; Opal, S.M. Antibiotic Stewardship: Strategies to Minimize Antibiotic Resistance While Maximizing Antibiotic Effectiveness. Med. Clin. N. Am. 2018, 102, 831–843. [Google Scholar] [CrossRef]
- Leekha, S.; Terrell, C.L.; Edson, R.S. General Principles of Antimicrobial Therapy. Mayo Clin. Proc. 2011, 86, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Aziz, M.H.; Alffenaar, J.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.A.; Pea, F.; Sjovall, F.; et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: A Position Paper. Intensive Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef]
- Mabilat, C.; Gros, M.F.; Nicolau, D.; Mouton, J.W.; Textoris, J.; Roberts, J.A.; Cotta, M.O.; van Belkum, A.; Caniaux, I. Diagnostic and medical needs for therapeutic drug monitoring of antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 791–797. [Google Scholar] [CrossRef] [Green Version]
- Wicha, S.G.; Märtson, A.G.; Nielsen, E.I.; Koch, B.C.P.; Friberg, L.E.; Alffenaar, J.W.; Minichmayr, I.K. From Therapeutic Drug Monitoring to Model-Informed Precision Dosing for Antibiotics. Clin. Pharmacol. Ther. 2021, 109, 928–941. [Google Scholar] [CrossRef]
- Choi, R.; Woo, H.I.; Park, H.D.; Lee, S.Y. A nationwide utilization survey of therapeutic drug monitoring for five antibiotics in South Korea. Infect. Drug Resist. 2019, 12, 2163–2173. [Google Scholar] [CrossRef]
- Begg, E.J.; Barclay, M.L.; Kirkpatrick, C.M. The therapeutic monitoring of antimicrobial agents. Br. J. Clin. Pharmacol. 2001, 52 (Suppl. 1), 35s–43s. [Google Scholar] [CrossRef]
- Reeves, D.; Lovering, A.; Thomson, A. Therapeutic drug monitoring in the past 40 years of the Journal of Antimicrobial Chemotherapy. J. Antimicrob. Chemother. 2016, 71, 3330–3332. [Google Scholar] [CrossRef] [Green Version]
- Cunha, B.A. Antibiotic side effects. Med. Clin. N. Am. 2001, 85, 149–185. [Google Scholar] [CrossRef]
- Yılmaz, Ç.; Özcengiz, G. Antibiotics: Pharmacokinetics, toxicity, resistance and multidrug efflux pumps. Biochem. Pharmacol. 2017, 133, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, K.P.; Peckham, D.G.; Conway, S.P.; Smith, C.A.; Bell, J.; Turnidge, J. Therapeutic drug monitoring of once daily tobramycin in cystic fibrosis--caution with trough concentrations. J. Cyst. Fibros. 2007, 6, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.K.; Tang, H.L.; Zhai, S.D. Benefits of therapeutic drug monitoring of vancomycin: A systematic review and meta-analysis. PLoS ONE 2013, 8, e77169. [Google Scholar] [CrossRef] [PubMed]
- Rybak, M.; Lomaestro, B.; Rotschafer, J.C.; Moellering Jr., R.; Craig, W.; Billeter, M.; Dalovisio, J.R.; Levine, D.P. Therapeutic monitoring of vancomycin in adult patients: A consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am. J. Health-Syst. Pharm. 2009, 66, 82–98. [Google Scholar] [CrossRef]
- Jenkins, A.; Thomson, A.H.; Brown, N.M.; Semple, Y.; Sluman, C.; MacGowan, A.; Lovering, A.M.; Wiffen, P.J. Amikacin use and therapeutic drug monitoring in adults: Do dose regimens and drug exposures affect either outcome or adverse events? A systematic review. J. Antimicrob. Chemother. 2016, 71, 2754–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovačević, T.; Avram, S.; Milaković, D.; Špirić, N.; Kovačević, P. Therapeutic monitoring of amikacin and gentamicin in critically and noncritically ill patients. J. Basic. Clin. Pharm. 2016, 7, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Velde, F.; Mouton, J.W.; de Winter, B.C.M.; van Gelder, T.; Koch, B.C.P. Clinical applications of population pharmacokinetic models of antibiotics: Challenges and perspectives. Pharmacol. Res. 2018, 134, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Hodiamont, C.J.; Janssen, J.M.; de Jong, M.D.; Mathôt, R.A.; Juffermans, N.P.; van Hest, R.M. Therapeutic Drug Monitoring of Gentamicin Peak Concentrations in Critically Ill Patients. Ther. Drug Monit. 2017, 39, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Drennan, P.G.; Thoma, Y.; Barry, L.; Matthey, J.; Sivam, S.; van Hal, S.J. Bayesian Forecasting for Intravenous Tobramycin Dosing in Adults with Cystic Fibrosis Using One Versus Two Serum Concentrations in a Dosing Interval. Ther. Drug Monit. 2021, 43, 505–511. [Google Scholar] [CrossRef]
- Möhlmann, J.E.; van Luin, M.; Mascini, E.M.; van Leeuwen, H.J.; de Maat, M.R. Monitoring of tobramycin serum concentrations in selected critically ill patients receiving selective decontamination of the digestive tract: A retrospective evaluation. Eur. J. Clin. Pharmacol. 2019, 75, 831–836. [Google Scholar] [CrossRef]
- Roberts, J.A.; Lipman, J. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit. Care Med. 2009, 37, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Zapke, S.E.; Willmann, S.; Grebe, S.-O.; Menke, K.; Thürmann, P.A.; Schmiedl, S. Comparing Predictions of a PBPK Model for Cyclosporine with Drug Levels from Therapeutic Drug Monitoring. Front. Pharmacol. 2021, 12, 1134. [Google Scholar] [CrossRef]
- Emoto, C.; McPhail, B.T.; Fukuda, T. Clinical applications of physiologically based pharmacokinetic modeling: Perspectives on the advantages and challenges. Ther. Drug Monit. 2020, 42, 157–158. [Google Scholar] [CrossRef]
- Perry, C.; Davis, G.; Conner, T.M.; Zhang, T. Utilization of physiologically based pharmacokinetic modeling in clinical pharmacology and therapeutics: An overview. Curr. Pharmacol. Rep. 2020, 6, 71–84. [Google Scholar] [CrossRef]
Figure 1.
Schematic summary of ADME properties.

Figure 2.
Diversity of applications of PK.

Figure 3.
Outline and summary of the stages of new drugs’ R&D.

Figure 4.
Schematic comparison between models with (A) 1, (B) 2, and (C) 3 compartments; first-order rate transfer constants for drug absorption (ka), movement of drug between compartments (k12, k21, k13, k31), and drug elimination (k10) are indicated.
Figure 4.
Schematic comparison between models with (A) 1, (B) 2, and (C) 3 compartments; first-order rate transfer constants for drug absorption (ka), movement of drug between compartments (k12, k21, k13, k31), and drug elimination (k10) are indicated.

Figure 5.
GastroPlus™ advanced compartmental absorption and transit model (ACAT™). Adapted from [38].
Figure 5.
GastroPlus™ advanced compartmental absorption and transit model (ACAT™). Adapted from [38].

Figure 6.
The core elements for antibiotic stewardship programs.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ferreira, A.; Lapa, R.; Vale, N. PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment. Processes 2021, 9, 2087. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9112087
AMA Style
Ferreira A, Lapa R, Vale N. PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment. Processes. 2021; 9(11):2087. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9112087
Chicago/Turabian StyleFerreira, Abigail, Rui Lapa, and Nuno Vale. 2021. "PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment" Processes 9, no. 11: 2087. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9112087
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.